
Abstract
In the present work, potential protective effects of quercitrin (a phytoestrogen) on Aβ-induced neurotoxicity in cultured rat hippocampal neurons were investigated in comparison with 17β-estradiol. Cell viability, oxidative status, and antioxidative potentials were used as comparative parameters. Co-exposure of cultured neurons to Aβ25–35 with either quercitrin or 17β-estradiol (50–100 μM) for 72 h attenuated Aβ25–35-induced neurotoxicity and lipid peroxidation, but not Aβ25–35-induced ROS accumulation. However, only 17β-estradiol counteracted a reduction in glutathione content and only quercitrin counteracted a reduction in glutathione peroxidase activity. Both compounds displayed no effects on superoxide dismutase activity. A specific estrogen receptor antagonist, ICI 182780, did not abolish neuroprotective effects of quercitrin and 17β-estradiol. These findings suggested that quercitrin and 17β-estradiol attenuated Aβ25–35-induced neurotoxicity in a comparable manner. Underlying neuroprotective mechanisms of both compounds were probably not related to estrogen receptor-mediated genomic mechanisms but might involve with their antioxidant and free radical scavenging properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is characterized by two pathologic hallmarks, extracellular plaques of amyloid-β (Aβ) peptide aggregates and intracellular neurofibrillary tangles [1, 2]. It has been demonstrated that Aβ is toxic to neurons either in vitro [3, 4] or in vivo [3, 5]. Several synthetic Aβ peptides have been used to study the mechanisms of their toxicity in various cell lines [6] and primary neuronal cell cultures [7]. Among Aβ fragments, the Aβ25–35 peptide represents the shortest fragment of Aβ processed in vivo by brain proteases and exhibits significant levels of molecular aggregation, retaining toxicity of the full length peptide [7]. Recently, a study of Aβ25–35 in organotypic hippocampal slice cultures revealed that this peptide induced neural injury in a similar pattern to Aβ1–42 [8], thereby reinforcing the use of Aβ25–35 as a convenient tool for the investigation of neurotoxic mechanisms involved in AD [4].
Several mechanisms by which Aβ and its aggregates mediated toxicity have been proposed such as the disruption of calcium homeostasis [9], consequently inducing the accumulation of free radicals and/or reactive oxygen species (ROS), and eventually leading to lipid and protein oxidation. After the exposure of rat PC12 and primary cortical cultures to Aβ25–35, a marked reduction in cell survival and activities of glutathione peroxidase (GPx) and catalase was observed along with an increase of malondialdehyde (MDA) production [10]. Additionally, Aβ25–35 significantly decreased superoxide dismutase (SOD) and increased GPx activities in astrocyte cultures [11], decreased glutathione (GSH) levels in mixed hippocampal cultures [12], decreased SOD and GSH with increasing lipid peroxidation in peripheral lymphocytes [13] and decreased SOD and GPx as well as MDA in hippocampal cultures [14]. These findings suggest that Aβ25–35 is not only a potent inducer of lipid peroxide, but also alterations of the antioxidant enzyme activity.
Neurotoxic effects of Aβ are at least in part mediated by free radicals, and some antioxidants such as vitamin E [15] and estradiol [16] have been proved to rescue cells from Aβ-mediated apoptosis. Therefore, therapeutic efforts aimed at scavenging oxygen free radicals or reducing their formation may be beneficial in AD. There has been a substantial amount of clinical research showing that estrogen replacement therapy (ERT) could reduce the risk and delay the onset of AD. However, the use of estrogen, especially long-term use, as a treatment for AD is limited. For this reason, other estrogenic agents with fewer side effects, such as phytoestrogens, have received considerable attention as potential alternatives to ERT. Recently, there has been considerable interest in neuroprotective effects of flavonoids, especially with respect to their mode of action as antioxidants. Neuroprotective effects of several phytoestrogens on Aβ-induced toxicity have been reported such as kaempferol in PC12 neuroblastroma cells and rat cortical neurons [17, 18], apigenin in rat cortical neurons [18], genistein in hippocampal neurons [19] and SH-SY5Y human neuroblastoma cells [20].
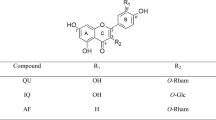
Among a wide variety of phytoestrogens, quercitrin, a glycoside form of quercetin (Fig. 1) exhibited a scavenging property and lipid peroxidation inhibition in rat brain homogenate [21] but not in microglial cells [22]. Quercitrin might be more potent antioxidant and neuroprotective agent than quercetin due to its high bioavailability in the digestive track [23, 24]. Therefore, the purpose of this study is to investigate potential neuroprotective effects of quercitrin on Aβ-induced neurotoxicity in cultured hippocampal neurons, as well as its possible mechanisms that mediate the in vitro neuroprotection. A well recognized estrogenic compound, 17β-estradiol, was tested in parallel as a comparative agent.

Chemical structures of quercitrin and 17β-estradiol
Materials and Methods
Materials
Chemicals
Equine serum and fetal bovine serum (FBS) were purchased from Hyclone (Utah, USA). Superoxide dismutase assay kit and glutathione peroxidase assay kit were purchased from Cayman Chemical Co. (USA). Estrogen receptor antagonist (ICI 182780) was purchased from Tocris Cookson, Inc. (Ellisville, MO, USA). All other chemicals, tissue culture media, supplements, and assay kits were obtained from Sigma–Aldrich Chemical Co. (St. Louis, MO, USA).
Experimental Animals
Timed-pregnant Wistar albino rats of gestation day 14 were purchased from the National Laboratory Animal Center, Nakornpathom, Thailand. They were individually housed in plastic cages with wood shavings as bedding in the animal room of Faculty of Pharmaceutical Sciences, Chulalongkorn University, under controlled environmental conditions (room temperature 25 ± 1°C with 12-h light/dark cycle, humidity of approximately 60%). All pregnant rats were fed with commercial rodent chow and tap water ad libitum until the operation day.
Methods
Neuronal Cell Culture
Primary cultures of hippocampal neurons were prepared from 18-day-old Wistar rat fetuses as previously described [25]. They were cultured at 37°C for 24 h and the medium was changed to serum-free medium with chemically defined supplements and then used in the experiments. Our control cultures at 4 DIV were approximately 90% neuronal cells and the remaining cells (about 10%) were mostly astrocytes as determined by immunohistochemical techniques (unpublished data). Animal handling and experimental protocols with animals were approved by the Animal Ethics Committee of Faculty of Pharmaceutical Sciences, Chulalongkorn University (Bangkok, Thailand, No. 134/2004).
Aβ25–35-Induced Neurocytotoxicity
Aβ25–35 stock solution was made in sterile distilled water at a concentration of 1 mg/ml and stored at −20°C. An aliquot of the stock solution was incubated at 37°C for 72 h to aggregate the peptide before use [26]. Cultured hippocampal neurons were exposed to various concentrations of Aβ25–35 (1–20 μM) for 72 h. After exposure, cell survival and cell death was analyzed by MTT reduction and LDH release assays.
Attenuation of Aβ25–35-Induced Neurocytotoxicity by Quercitrin and 17β-Estradiol
Quercitrin and 17β-estradiol were dissolved in dimethylsulfoxide (DMSO) and diluted with sterile distilled water as stock solutions. Cultured hippocampal neurons were exposed to the combination of Aβ25–35 (5 μM) and either quercitrin or 17β-estradiol (0.1, 1, 10, 50 and 100 μM) for 72 h. Thereafter, assessment of neurocytotoxicity was carried out.
Involvement of estrogenic effects was testified by using ICI 182780, a specific estrogen receptor antagonist. Cultured hippocampal neurons were pretreated with 1 μM ICI 182780 for 2 h before the addition of Aβ25–35 with either quercitrin or 17β-estradiol as previously mentioned. Assessment of neurocytotoxicity was carried out as well.
Assessment of Neurocytotoxicity
Cell viability. Mitochondrial metabolic activity, an indicator for cell viability, was determined by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reduction assay which was a modified colorimetric method from that of Mosmann [27]. Briefly, MTT stock solution (5 mg/ml) was added to the culture medium (a final concentration of 100 μg/ml). After 1 h of incubation at 37°C, the medium was aspirated, and 200 μl of DMSO was added to dissolve the dark blue formazan crystal. The absorbance was measured at 570/620 nm using an Anthos Labtec HT2 microplate reader. Results were expressed as the percentage of MTT reduction.
Cell death. Lactate dehydrogenase (LDH) release, an indicator of cell death, was determined by an In Vitro Toxicity Assay Kit with a procedure according to the manufacturer’s instructions. Briefly, medium LDH was assayed in 100 μl aliquots of culture medium and cellular LDH was measured in 100 μl aliquots of cell lysate. The reaction was initiated by adding 50 μl of assay mixture, left at room temperature for 30 min, and then terminated by adding 50 μl of 0.5 N HCl. The absorbance was determined at 490/690 nm using an Anthos Labtec HT2 microplate reader. LDH release into the medium was expressed as the percentage of total LDH.
Lipid peroxidation. Malondialdehyde (MDA), a by-product of lipid peroxidation, was quantitated using a modified thiobarbituric acid reactive substance (TBARS) procedure from that of Ohkawa et al. [28]. Cultured neurons were lysed with 160 μl of 2% sodium dodecyl sulfate for 30 min. The lysates from two culture wells were pooled and adjusted with 2% sodium dodecyl sulfate to 1 ml and serially added with 50 μl of 4% butylated hydroxytoluene in ethanol, 1 ml of 10% phosphotungstic acids in 0.5 M sulfuric acid and 1.5 ml of 0.7% thiobarbituric acid. The reaction mixtures were boiled at 95°C for 60 min. After cooling, the mixtures were extracted with n-butanol and centrifuged at 3,500 rpm for 10 min. The fluorescence of n-butanol layer was measured at the excitation wavelength of 515 nm and the emission wavelength of 553 nm using Jasco Model FS 777 spectrofluorometer. In this experiment, 1,1,3,3-tetraethoxypropane (TEP) was used as an external standard. Lipid peroxidation was expressed in term of MDA equivalence (1 M TEP = 1 M MDA).
Levels of reactive oxygen species (ROS). The accumulation of ROS was determined by analyzing 2′,7′-dichlorofluorescein (DCF) fluorescence as described previously [29]. Briefly, cells were washed twice with cooled PBS and incubated with 50 μM of DCFH-DA at 37°C for 45 min. Loaded cells were washed with cooled PBS and subsequently scraped into PBS. The cell suspension was pelleted by centrifugation at 5,000×g at 4°C and then the cell pellet was resuspended in cooled PBS. The fluorescent intensity was quantified with a Perkin-Elmer VICTOR3 Wallac 1420 microplate reader using excitation and emission wavelengths of 485 and 535 nm, respectively.
Glutathione content. Total glutathione (GSH) was determined by using enzymatic cycling method that modified from that of Tietz [30]. In brief, pooled cultured neurons from two culture wells were extracted with 300 μl of 1% (w/v) sulfosalicylic acid and centrifuged. Twenty microlitre of the supernatant was transferred to a 96-well plate and then volume was adjusted to 100 μl. After that, 100 μl of reaction mixture was added to each well. The formation of 2-nitro-5 benzoic acid was monitored at the wavelength of 405 nm every 10 s for 2 min using an Anthos Labtec HT2 microplate reader. The slope of initial rate of reaction was used for calculating GSH content from a standard curve derived from known amounts of GSH.
Superoxide dismutase activity. Superoxide dismutase (SOD) was determined by using the Cayman Chemical SOD assay kit. Briefly, pooled cultured neurons from two culture wells were harvested and lysed in cold buffer and then centrifuged at 1,500×g for 5 min at 4°C. The assay of SOD activity in the supernatant was performed according to the manufacturer’s protocol. The xanthine/xanthine oxidase system was used to generate the superoxide anion which reduced a tetrazolium salt to formazan dye. The SOD activity removed the superoxide anion and produced an inhibition of the tetrazolium salt reduction which was monitored at 450 nm with a Perkin-Elmer VICTOR3 Wallac 1420 microplate reader. One unit of SOD activity was defined as the amount of enzyme need to exhibit 50% dismutation of superoxide radical. The specific activity was expressed as units per milligram protein.
Glutathione peroxidase activity. Glutathione peroxidase (GPx) was determined by using the Cayman Chemical GPx assay kit. In brief, pooled cultured neurons from two culture wells were harvested and lysed in cold buffer and then centrifuged at 10,000×g for 15 min at 4°C. The assay of GPx activity in the supernatant was performed according to the manufacturer’s protocol. GPx activity was measured indirectly by a coupled reaction with glutathione reductase (GR). Oxidized glutathione (GSSG), produced upon reduction of an organic hydroperoxide by GPx, is recycled to its reduced state by GR and NADPH. The oxidation of NADPH to NADP+ is accompanied by a decrease in absorbance at 340 nm which was monitored every minute at 340 nm using a Perkin–Elmer VICTOR3 Wallac 1420 microplate reader. One unit of GPx activity was defined as the amount of enzyme that catalyzed the oxidation of 1.0 nmol of NADPH to NADP+/min at 25°C and specific activity is represented as units per milligram protein.
Protein content. Protein content was determined by Bradford reagent [31] using bovine serum albumin as a protein standard.
Statistical Analysis
Data were expressed as mean ± SEM of values obtained from at least three separate independent cultures, each performed in duplicate. Differences among mean values of various groups were tested using one-way analysis of variance (ANOVA) followed by pairwise comparisons between groups with Scheffe’s post hoc analysis. P values less than 0.05 were considered to be significant.
Results
Aβ25–35-Induced Neurocytotoxicity
Exposure to 1–20 μM of Aβ25–35 for 72 h was apparently cytotoxic to cultured hippocampal neurons in a concentration-dependent manner as considered from analyses of cell viability and cell death (data not shown). Neurons with typical appearances of irregular shape and fragmented neurites were clearly seen after exposure to higher concentrations of Aβ25–35. Approximately 50% of cell viability was seen after an exposure to 5 μM Aβ25–35 for 72 h. Therefore, this experimental setting was arbitrary adopted as the neurotoxic insult in our in vitro cellular model of neurodegeneration.
Attenuation of Aβ25–35-Induced Neurocytotoxicity by Quercitrin and 17β-Estradiol
After the exposure of cultured hippocampal neurons to either quercitrin or 17β-estradiol at increasing concentrations up to 100 μM for 72 h, both compounds at all concentrations tested did not induce significant changes in the viability of cultured neurons (data not shown). We then employed concentrations of 0.1–100 μM as a nontoxic concentration range of quercitrin and 17β-estradiol in the study of their neuroprotective effects.
Cultured hippocampal neurons were exposed to 5 μM Aβ25–35 for 72 h in combination with various concentrations of either quercitrin or 17β-estradiol prior to determination of cell viability. As shown in Fig. 2, co-exposure with higher concentrations of quercitrin, 50 and 100 μM, increased the percentage of cell viability (estimated by MTT reduction) from 55.62 ± 2.98% in only Aβ25–35-treated controls to 71.43 ± 2.26 and 76.09 ± 2.81%, respectively (P < 0.01). In a similar manner, co-exposure with 50 and 100 μM of 17β-estradiol also rescued cell viability from 54.15 ± 3.38% in only Aβ25–35-treated controls to 78.27 ± 5.07 and 81.83 ± 4.71%, respectively (P < 0.05).

Protective effects of co-exposure with quercitrin or 17β-estradiol on Aβ25–35-induced decrease in cell viability in cultured hippocampal neurons. Cultured neurons were treated with 5 μM Aβ25–35 in combination with different concentrations (0.1–100 μM) of quercitrin or 17β-estradiol for 72 h. After co-exposure, cell viability was determined by MTT reduction assay. Data are expressed as mean ± SEM of values from four independent experiments. ** P < 0.01 compared with control cultures, # P < 0.05, ## P < 0.01 compared with only Aβ25–35-treated cultures. There are no significant differences in neuroprotection between treatment with quercitrin and treatment with 17β-estradiol (independent sample Student t-test)
Potential neuroprotective effects of quercitrin and 17β-estradiol at higher concentrations were convincingly supported by the analysis of cell death (Fig. 3). In all experimental conditions, the magnitude of cell death was correlated with the percentage of cell viability in a complementary manner.

Protective effects of co-exposure with quercitrin or 17β-estradiol on Aβ25–35-induced cell death in cultured hippocampal neurons. Cultured neurons were treated with 5 μM Aβ25–35 in combination with quercitrin or 17β-estradiol (50 and 100 μM) for 72 h. After co-exposure, cell death was determined by LDH release assay. Data are expressed as mean ± SEM of values from three independent experiments. ** P < 0.01 compared with control cultures, # P < 0.05, ## P < 0.01 compared with only Aβ25–35-treated cultures. There are no significant differences in neuroprotection between treatment with quercitrin and treatment with 17β-estradiol (independent sample Student t-test)
Possible Mechanism(s) of Neuroprotective Effects of Quercitrin and 17β-Estradiol
Antioxidant-Related Mechanisms
As shown in Fig. 4, exposure to 5 μM Aβ25–35 alone for 72 h increased cellular lipid peroxidation to 148.22 ± 3.31% of controls (P < 0.001). Co-exposure with 50 and 100 μM of either quercitrin or 17β-estradiol significantly reduced that increment in a concentration-related manner. Exposure with either of these two test compounds alone did not cause significant changes in cellular lipid peroxidation (data not shown). Therefore, attenuation of Aβ25–35-induced neurotoxicity by higher concentrations of quercitrin and 17β-estradiol was apparently associated with decreased generation of lipid peroxidation in cultured neurons.

Beneficial effects of co-exposure with quercitrin or 17β-estradiol on Aβ25–35-induced lipid peroxidation in cultured hippocampal neurons. Cultured neurons were treated with 5 μM Aβ25–35 in combination with quercitrin or 17β-estradiol (50 and 100 μM) for 72 h. After co-exposure, levels of lipid peroxidation were determined by TBARS assay. Data are expressed as mean ± SEM of values from four independent experiments. *** P < 0.001 compared with control cultures, # P < 0.05 compared with only Aβ25–35-treated cultures
Exposure of cultured neurons to 5 μM Aβ25–35 gradually increased intracellular ROS levels with a peak level around 18 h of exposure (165.71 ± 13.19% of controls, P < 0.01) and apparently declined thereafter. However, both quercitrin and 17β-estradiol at concentrations of 50 and 100 μM had no significant effects on Aβ25–35-induced ROS accumulation (Fig. 5). Exposure with either of these two test compounds alone did not cause significant changes in ROS levels (data not shown). These results revealed that apparent neuroprotective effects of quercitrin and 17β-estradiol were not associated with a decrease in ROS generation.

Effects of co-exposure with quercitrin or 17β-estradiol on Aβ25–35-induced ROS accumulation in cultured hippocampal neurons. Cultured neurons were treated with 5 μM Aβ25–35 (Aβ) in combination with 50 and 100 μM of quercitrin (Q50, Q100) or 17β-estradiol (E50, E100) for different durations (6, 12, 18 and 24 h). After co-exposure, cellular ROS level was quantified by using DCFH-DA and a spectrofluorometer. Data are expressed as mean ± SEM of values from three independent experiments. ** P < 0.01 compared with control cultures
Due to a long exposure time, quercitrin and 17β-estradiol might alter intracellular antioxidant defense systems of cultured hippocampal neurons including GSH, SOD and GPx. Exposure to 5 μM Aβ25–35 alone for 72 h caused a marked reduction of cellular GSH contents to 34.58 ± 5.53% of controls (P < 0.001) (Fig. 6). Co-exposure to Aβ25–35 in combination with 50 and 100 μM 17β-estradiol augmented cellular GSH contents to 58.4 ± 4.7 and 64.69 ± 5.42% of controls, respectively, which were significantly different from that of only Aβ25–35-treated groups (P < 0.05 and P < 0.01, respectively). Exposure with either of quercitrin or 17β-estradiol alone did not cause significant changes in GSH contents (data not shown). Co-exposure with quercitrin, however, marginally and insignificantly recovered GSH contents.

Effects of co-exposure with quercitrin or 17β-estradiol on Aβ25–35-induced diminution in GSH contents in cultured hippocampal neurons. Cultured neurons were treated with 5 μM Aβ25–35 in combination with quercitrin or 17β-estradiol (50 and 100 μM) for 72 h. After co-exposure, total cellular GSH content was measured. Data are expressed as mean ± SEM of values from four independent experiments. *** P < 0.001 compared with control cultures, # P < 0.05, ## P < 0.01 compared with only Aβ25–35-treated cultures
Regarding the SOD activity, exposure of cultured hippocampal neurons to 5 μM Aβ25–35 for 72 h slightly and insignificantly decreased SOD activity as compared to controls (data not shown). In addition, no significant changes were found in all co-exposure conditions of Aβ25–35 with either quercitrin or 17β-estradiol.
Regarding the GPx activity, exposure to 5 μM Aβ25–35 alone for 72 h significantly decreased GPx activity by approximately 20% of controls (Fig. 7). Co-exposure with 50 and 100 μM of quercitrin induced significant increases in GPx activity from 136.35 ± 4.75 U/mg protein in only Aβ25–35-treated cultures to 167.88 ± 6.13 U/mg protein and 170.32 ± 4.61 U/mg protein, respectively (P < 0.05). However, no significant changes in the activity of GPx enzyme were found when cultured neurons were co-exposed to Aβ25–35 and 17β-estradiol. Exposure with either of quercitrin or 17β-estradiol alone did not cause significant changes in the GPx activity as well (data not shown).

Effects of co-exposure with quercitrin or 17β-estradiol on Aβ25–35-induced changes in glutathione peroxidase activity in cultured hippocampal neurons. Cultured neurons were treated with 5 μM Aβ25–35 in combination with quercitrin or 17β-estradiol (50 and 100 μM) for 72 h. After co-exposure, cellular GPx activity was measured. Data are expressed as mean ± SEM of values from four independent experiments. *P < 0.05 compared with control cultures, # P < 0.05 compared with only Aβ25–35-treated cultures
Estrogen Receptor-Related Mechanisms
Neuroprotective effects of quercitrin and 17β-estradiol might mediate through estrogen receptor-dependent mechanisms. This hypothesis was tested by pre-incubating cultured neurons with ICI 182780, a specific estrogen receptor antagonist, for 2 h prior to the addition of Aβ25–35 in combination with either quercitrin or 17β-estradiol and further incubated for 72 h. Exposure to 5 μM Aβ25–35 alone significantly decreased cell viability to approximately 63% of controls. Quercitrin and 17β-estradiol at a concentration of 50 μM recovered this decreased cell viability up to approximately 81 and 83% of controls, respectively (P < 0.01). It was notable that ICI 182780 at 1 μM, a concentration that effectively blocked estrogen receptors without affecting cell viability, was unable to antagonize neuroprotective effects of both quercitrin and 17β-estradiol (Fig. 8). These results suggested that protective efficacy of quercitrin and 17β-estradiol against Aβ25–35-induced neurocytotoxicity, under our experimental conditions, might not be dependent on activation of conventional estrogen receptors.

Ineffectiveness of a specific estrogen receptor antagonist to block protective effect of quercitrin or 17β-estradiol against Aβ25–35-induced neurotoxicity in cultured hippocampal neurons. Cultured neurons were pretreated with 1 μM of ICI 182780 (ICI) for 2 h and further treated with 5 μM Aβ25–35 in combination with quercitrin or 17β-estradiol (50 and 100 μM) for another 72 h. After co-exposure, cell viability was measured by MTT reduction assay. Data are expressed as mean ± SEM of values from four independent experiments. *** P < 0.001 compared with control cultures, ## P < 0.01 compared with only Aβ25–35-treated cultures
Discussion
In the present study, we employed immature cultured hippocampal neurons exposed to Aβ25–35 as an in vitro neurodegenerative paradigm because of its higher sensitivity and vulnerability to oxidative damages and the relevance to the pathogenesis of AD [32, 33]. Our results showed that Aβ25–35 decreased the viability of cultured hippocampal neurons in a concentration-dependent manner which are consistent with previous studies [34, 35].
Our study revealed that quercitrin which is a member of phytoestrogens, at a higher concentration range of 50–100 μM, attenuated Aβ25–35-induced neurocytotoxicity in comparable to a natural estrogen, 17β-estradiol. There have been several lines of evidence supporting neuroprotective effects of 17β-estradiol against Aβ-induced toxicity [3, 16, 36, 37] whereas supporting evidence for neuroprotective effects of quercitrin has been inadequate. It is most likely that our work offers the first in vitro experimental evidence on neuroprotective effects of quercitrin against Aβ-induced neurotoxicity in cultured hippocampal neurons.
Cellular injury during AD may result from ROS as well as from impaired cellular repair mechanisms following oxidative injury. Several mechanisms have been proposed whereby Aβ may increase ROS generation. Apart from generating ROS directly in solution, Aβ may also interact with a number of biological systems and increase the rate of ROS production through modification or stimulation of intrinsic pathways [38], e.g., causing damage to the mitochondrial respiratory chain [39]. In our study, these mechanisms might induce a gradual increase in intracellular ROS levels with a peak level around 18 h of exposure. With sustained increase of ROS generation, various cellular defense mechanisms came into play thereby lessening ROS levels. In spite of attenuating the increased lipid peroxidation induced by 72 h exposure to Aβ25–35, quercitrin and 17β-estradiol showed marginal benefit on intracellular ROS increment during 24 h exposure to Aβ25–35. As considered from the nature of assay methods used, quercitrin and 17β-estradiol seem to be more effective against membrane-derived nonpolar free radicals (TBARS assay) than cytosol-derived polar free radicals (DCF fluorescence assay). Alternatively, this discrepancy may be due to the difference in exposure time of cultured neurons to Aβ25–35.
A role of oxidative stress in AD is supported by increased levels of thiobarbituric acid-reactive substance, a measure of lipid peroxidation [40, 41]. The neurotoxicity of Aβ25–35 is mediated through lipid peroxidation, leading to oxidative damage of synaptic terminals and eventually to neuronal death [42]. It has been demonstrated in vitro that 17β-estradiol protected cells from lipid peroxidation caused by Aβ25–35 [43]. Recently, quercitrin was found to prevent lipid peroxidation induced by pro-oxidant agents in rat brain homogenates, suggesting that quercitrin exhibited a scavenger and antioxidant role involving the negative modulation of the Fenton reaction [21]. In accordance with previous studies, the present study suggested that Aβ25–35-induced neurocytotoxicity by increasing cellular lipid peroxidation. After co-incubation, both quercitrin and 17β-estradiol showed protective effects against Aβ25–35-induced lipid peroxidation via their antioxidant and free radical scavenging properties.
Cells possess a variety of defense mechanisms and repair systems against ROS. This can sometimes be inadequate, leading to oxidative stress in which the production of ROS overwhelms antioxidant defenses of the organism. Quercitrin and 17β-estradiol may facilitate cellular defense systems (such as GSH levels, SOD and GPx activities) to counteract increased ROS levels. In this study, Aβ25–35 alone or Aβ25–35 in combination with either quercitrin or 17β-estradiol had no apparent effects on SOD enzyme activity in cultured hippocampal neurons. One major index of oxidative stress is the GSH level which has been described in a variety of model systems in AD brains and cells exposed to Aβ [44, 45]. However, the total brain levels of GSH appeared to be unaffected in AD [45], whereas GPx and glutathione reductase were found to be elevated in different brain regions [40] or unchanged [41]. In our study, Aβ25–35 significantly decreased the GSH content and GPx activity in cultured hippocampal neurons, in line with a previous report that exposure to Aβ for 24 h caused GSH depletion in hippocampal neurons [12]. However, co-incubation with 17β-estradiol attenuated Aβ25–35-induced GSH depletion whereas quercitrin did not show this beneficial effect. Meanwhile, co-incubation with quercitrin, but not 17β-estradiol, revealed a significant increase in GPx activity. Based on these findings, quercitrin might induce upregulation of antioxidant enzymes including GPx. However, there has been no published report on the induction effect of quercitrin on this antioxidant enzyme.
One of the defense mechanisms that protect the cell from oxidative damage is the dismutation of superoxide anion radical to hydrogen peroxide by SOD which is further converted to water by GPx, using GSH as a substrate. The generated hydrogen peroxide can be converted in spontaneous reaction catalyzed by metals such as iron (Fenton reaction) to the highly reactive hydroxyl radical, reacting with various biological macromolecules including polyunsaturated fatty acids by which initiating the process of lipid peroxidation [46]. In our study, a clear-cut enhancement of GPx activity that caused by quercitrin may conceal an expected increase in GSH levels. As a result, this facilitated defense mechanism shifts the conversion of hydrogen peroxide to water thereby reducing the formation of highly reactive hydroxyl radical, and eventually leading to decreased lipid peroxidation.
Estrogens coordinate cellular mechanisms involved in the development and differentiation of various neuronal populations, modulation of synaptic plasticity and neuronal excitability, induction of neuronal survival and axonal outgrowth, and neurogenesis in adult [47]. Increasing evidence supports the role of estrogen as neuroprotective compound that can act dependently or independently of estrogen receptors. Estrogen-mediated neuroprotection may involve the activation of intracellular estrogen receptors [36, 48, 49]. The exact role of estrogen receptors in estrogen-induced neuroprotection is still controversial. It was reported that ICI 182780, a specific estrogen receptors antagonist, antagonized estrogen effects in cortical neurons and 5H-SY5Y cells [50, 51] but not in cortical, hippocampal and SK-N-SH cells [43, 52, 53]. However, our findings demonstrated that ICI 182780 did not block quercitrin- or 17β-estradiol-mediated neuroprotection against Aβ25–35, suggesting that neuroprotective effects of both compounds may not involve cytoplasmic or nuclear estrogen receptor-mediated mechanisms. In accordance with our study, high concentrations of 17β-estradiol necessary for cellular protection in some studies suggest a hormone receptor-independent effect, probably mediated by the antioxidant activity [43, 54].
The endoplasmic reticulum (ER) plays a leading role in lipid and protein synthesis. In parallel with protein synthesis at ER-bounded ribosomes, membrane-spanning or water soluble proteins are folded by a chaperone in ER. However, under many insulting conditions, e.g., heat shock, dysregulation of calcium dynamics or cellular redox control, proteins are misfolded or unfolded (ER stress) which leads to the aggregation of unusual proteins and apoptosis [55]. ER stress has been attracting considerable attention because it relates to many disorders including neurodegenerative diseases, diabetes, arteriosclerosis, inflammation, and liver or kidney damage [56].
In cultured hippocampal neurons and organotypic hippocampal slice cultures, Aβ-induced neurotoxicity might interact with ER stress resulting in potentiated neuronal cell death which could be protected by S-allyl-l-cysteine, an organosulfur compound purified from aged garlic extract [8, 57]. Recently, it was found that quercetin, a typical flavonoid ubiquitously present in fruits and vegetables, suppressed the ER stress caused by calcium dynamics dysregulation in cultured intestinal epithelial cells via the inhibition of PI3K [58]. It is conceivable that quercitrin, a closely structure-related flavonoid to quercetin, might share this particular property which contributes to its neuroprotective activity in cultured neurons. However, there has been no direct supporting evidence available so far.
It may be apprehensive that we employed a high concentration range of test compounds which may be unreasonable in practical situations. The key point of this study is the experimental finding of potential neuroprotective effects of quercitrin in an in vitro model of neurodegeneration. Nevertheless, the relevance of such finding to therapeutic applications is comprehensible because various pharmacological aspects, in addition to drug concentrations or doses, determine the efficacy and safety of drug therapy in humans. Incidentally, more detailed pharmacokinetic and pharmacodynamic studies of quercitrin are needed to justify this crucial application.
In conclusion, we firstly illustrate the potential neuroprotective effect of quercitrin, a member of phytoestrogens, on a neuronal cell culture model of neurodegeneration. This beneficial effect may involve inhibition of lipid peroxidation and induction of GPx activity. Underlying neuroprotective mechanisms of quercitrin might not be related to estrogen receptor-mediated genomic mechanisms, but apparently may involve with its antioxidant and free radical scavenging properties. Currently, the exact mechanisms of quercitrin-induced neuroprotection and its potential therapeutic applications in neurodegenerative disorders are still unclear and warrant further investigation.
References
Selkoe DJ, Schenk D (2003) Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 43:545–584
Kar S, Slowikowski SPM, Westaway D et al (2004) Interactions between β-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatr Neurosci 29:427–441
Khan AA, Mao XO, Banwait S et al (2007) Neuroglobin attenuates β-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo. Proc Natl Acad Sci USA 104:19114–19119
Frozza RL, Horn AP, Hoppe JB et al (2009) A comparative study of β-amyloid peptides Aβ1–42 and Aβ25–35 toxicity in organotypic hippocampal slice cultures. Neurochem Res 34:295–303
Lesné S, Koh MT, Kotilinek L et al (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440:352–357
Jang J-H, Surh Y-J (2005) β-Amyloid-induced apoptosis is associated with cyclooxygenase-2 up-regulation via the mitogen activated protein kinase-NFκB signaling pathway. Free Radic Biol Med 38:1604–1613
Abe K, Saito H (2000) Amyloid β neurotoxicity not mediated the mitogen-activated protein kinase cascade in cultured rat hippocampal and cortical neurons. Neurosci Lett 292:1–4
Kosuge Y, Koen Y, Ishige K et al (2003) S-allyl-l-cysteine selectively protects cultured rat hippocampal neurons from amyloid β-protein- and tunicamycin-induced neuronal death. Neuroscience 122:885–895
Korol TY, Korol SV, Kostyuk EP et al (2008) β-Amyloid-induced changes in calcium homeostasis in cultured hippocampal neurons of the rat. Neurophysiology 40:9–12
Xiao XQ, Wang R, Han YF et al (2000) Protective effects of huperzine A on β-amyloid25–35 induced oxidative injury in rat pheochromocytoma cells. Neurosci Lett 286:155–158
Kim H, Bang OY, Jung MW et al (2001) Neuroprotective effects of estrogen against beta-amyloid toxicity are mediated by estrogen receptors in cultured neuronal cells. Neurosci Lett 302:58–62
Keelan J, Allen NJ, Antcliffe D et al (2001) Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J Neurosci Res 66:873–884
Jayakumar R, Murali J, Koteeswari D et al (2004) Cytotoxic and membrane perturbation effects of a novel amyloid forming model peptide poly (leucine-glutamic acid). J Biochem 136:457–462
Dong YL, Zuo PP, Li Q et al (2007) Protective effects of phytoestrogen α-zearalanol on beta amyloid25–35 induced oxidative damage in cultured rat hippocampal neurons. Endocrinology 32:206–211
Dhitavat S, Orti D, Rogers E et al (2005) Folate, vitamin E, and acetyl-l-carnitine provide synergistic protection against oxidative stress resulting from exposure of human neuroblastoma cells to amyloid-beta. Brain Res 1061:114–117
Nilsen J, Chen S, Irwin RW et al (2006) Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci 7:74–87
Roth A, Schaffner W, Hertel C (1999) Phytoestrogen kaempferol (3, 4′, 5, 7-tetrahydroxyflavone) protects PC12 and T47D cells from beta-amyloid-induced toxicity. J Neurosci Res 57:399–404
Wang CN, Chi CW, Lin YL et al (2001) The neuroprotective effects of phytoestrogens on amyloid β protein-induced toxicity are mediated by abrogating the activation of caspase cascade in rat cortical neurons. J Biol Chem 276:5287–5295
Zeng H, Chen Q, Zhao B (2004) Genistein ameliorates β-amyloid peptide (25–35)-induced hippocampal neuronal apoptosis. Free Radic Biol Med 36:180–188
Bang OY, Hong HS, Kim DH et al (2004) Neuroprotective effect of genistein against beta amyloid-induced neurotoxicity. Neurobiol Dis 16:21–28
Wagner C, Fachinetto R, Dalla Corte CL et al (2006) Quercitrin, a glycoside form of quercetin, prevents lipid peroxidation in vitro. Brain Res 1107:192–198
Kraus B, Wolff H, Heilmann J et al (2007) Influence of Hypericum perforatum extract and its single compounds on amyloid-β mediated toxicity in microglial cells. Life Sci 81:884–894
Hollman PC, de Vries JH, Van Leeuwen SD et al (1995) Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers. Am J Clin Nutr 62:1276–1282
Morand C, Manach C, Crespy V et al (2000) Quercetin 3-O-beta-glucoside is better absorbed than other quercetin forms and is not present in rat plasma. Free Radic Res 33:667–676
Unchern S, Nagata K, Saito H (1997) Selective cytotoxicity of piperine on cultured rat hippocampal neurons in comparison with cultured astrocytes: the possible involvement of lipid peroxidation. Biol Pharm Bull 20:958–961
Pike C, Overman MJ, Cotman CW (1995) Amino terminal detection enhanced aggregation of beta amyloid peptide in vitro. J Biol Chem 270:23895–23898
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358
Bastianetto S, Zheng WH, Quirion R (2000) The Ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by beta-amyloid. Eur J Neurosci 12:1882–1890
Tietze F (1969) Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 27:502–522
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72:248–254
Jiang X, Mu D, Manabat C et al (2004) Differential vulnerability of immature murine neurons to oxygen-glucose deprivation. Exp Neurol 190:224–232
Shirai K, Mizui T, Suzuki Y et al (2006) Differential effects of x-irradiation on immature and mature hippocampal neurons in vitro. Neurosci Lett 399:57–60
Behl C, Skutella T, Lezoualch F et al (1997) Neuroprotection against oxidative stress by estrogens: structure–activity relationship. Mol Pharmacol 51:535–541
Yagyu K, Kitagawa K, Irie T et al (2001) Amyloid proteins inhibit Cl–ATPase activity in cultured rat hippocampal neurons. J Neurochem 78:569–576
Fitzpatrick JL, Mize AL, Wade CB et al (2002) Estrogen-mediated neuroprotection against beta-amyloid toxicity requires expression of estrogen receptor alpha or beta and activation of MAPK pathway. J Neurochem 82:674–682
Quintanilla RA, Muñoz FJ, Metcalfe MJ et al (2005) Trolox and 17β-estradiol protect against amyloid β-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J Biol Chem 280:11615–11625
Canevari L, Abramov AY, Duchen MR (2004) Toxicity of amyloid β peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem Res 29:637–650
Benzi G, Moretti A (1995) Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol Aging 16:661–674
Lovell MA, Ehmann WD, Butler SM et al (1995) Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 45:1594–1601
Marcus DL, Thomas C, Rodriguez C et al (1998) Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp Neurol 150:40–44
Butterfield DA, Lauderback CM (2002) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic Bio Med 32:1050–1060
Bae YH, Hwang JY, Kim YH et al (2000) Antioxidative neuroprotection by estrogens in mouse cortical cultures. J Korean Med Sci 15:327–336
Cecchi C, Latorraca S, Sorbi S et al (1999) Glutathione level is altered in lymphoblasts from patients with familial Alzheimer’s disease. Neurosci Lett 275:152–154
Abramov AY, Canevari L, Duchen MR (2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23:5088–5095
Reiter RJ (1995) Oxidative processes and antioxidative defense mechanisms in the aging brain. FASEB J 9:526–533
McEwen B (2002) Estrogen actions throughout the brain. Recent Prog Horm Res 57:357–384
Dubal DB, Shughrue PJ, Wilson ME et al (1999) Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci 19:6385–6393
Sawada H, Ibi M, Kihara T et al (2000) Mechanisms of antiapoptotic effects of estrogens in nigral dopaminergic neurons. FASEB J 14:1202–1214
Wilson ME, Dubal DB, Wise PM (2000) Estradiol protects against injury-induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Res 873:235–242
Lee SY, Andoh T, Murphy DL et al (2003) 17β-Estradiol activates ICI 182, 780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. FASEB J 17:947–948
Howard SA, Brooke SM, Sapolsky RM (2001) Mechanisms of estrogenic protection against gp 120-induced neurotoxicity. Exp Neurol 168:385–391
Wang X, Dykens JA, Perez E et al (2006) Neuroprotective effects of 17β-estradiol and nonfeminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH cells. Mol Pharmacol 70:395–404
Behl C, Widmann M, Trapp T et al (1995) 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Comm 216:473–482
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115:2656–2664
Yoshida H (2007) ER stress and diseases. FEBS J 274:630–658
Imai T, Kosuge Y, Ishige K et al (2007) Amyloid β-protein potentiates tunicamycin-induced neuronal death in organotypic hippocampal slice cultures. Neuroscience 147:639–651
Natsume Y, Ito S, Satsu H et al (2009) Protective effect of quercetin on ER stress caused by calcium dynamics dysregulation in intestinal epithelial cells. Toxicology 258:164–175
Acknowledgments
This study was supported partly by the Graduate Research Funds from the Graduate School, Chulalongkorn University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rattanajarasroj, S., Unchern, S. Comparable Attenuation of Aβ25–35-Induced Neurotoxicity by Quercitrin and 17β-Estradiol in Cultured Rat Hippocampal Neurons. Neurochem Res 35, 1196–1205 (2010). https://doi.org/10.1007/s11064-010-0175-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-010-0175-6