
Abstract
We established a model of Alzheimer’s disease in vitro by exposing primary hippocampal neurons of neonatal Wistar rats to the β-Amyloid peptide fragment 25–35, Aβ25–35. We then observed the effects of genistein, a type of soybean isoflavone, on Aβ25–35-incubated hippocampal neuron viability, and the electrophysiological properties of voltage-gated sodium channels (NaV) and potassium channels (KV) in the hippocampal neurons. Aβ25–35 exposure reduced the viability of hippocampal neurons, decreased the peak amplitude of voltage-activated sodium channel currents (INa), and significantly reduced INa at different membrane potentials. Moreover, Aβ25–35 shifted the activation curve toward depolarization, shifted the inactivation curve toward hyperpolarization, and increased the time constant of recovery from inactivation. Aβ25–35 exposure significantly shifted the inactivation curve of transient outward K+ currents (IA) toward hyperpolarization and increased its time constant of recovery from inactivation. In addition, Aβ25–35 significantly decreased the peak density of outward-delayed rectifier potassium channel currents (IDR) and significantly reduced IDR value at different membrane potentials. We found that genistein partially reversed the decrease in hippocampal neuron viability, and the alterations in electrophysiological properties of NaV and KV induced by Aβ25–35. Our results suggest that genistein could inhibit Aβ25–35-induced neuronal damage with changes in the electrophysiological properties of NaV and KV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is one of the most frequent causes of dementia, and its main clinical symptoms include progressive memory loss accompanied by cognitive impairment and character changes (Graham et al. 2017). A few pathogenesis mechanisms that underlie these neuropathological changes include β-amyloid (Aβ) aggregation and tau hyperphosphorylation. Additionally, inflammatory processes, oxidative stress, and mitochondrial dysfunction have also been studied in neuropathologies (Blennow et al. 2006). The dominant hypothesis to explain these observations is the ‘amyloid cascade hypothesis’, which supposes that deposition of Aβ in the brain is a crucial step in the pathogenesis of AD (Klafki et al. 2006). Other than acetylcholinesterase inhibitors and memantine, which have passed the FDA approval for AD management and show some symptomatic improvement in some Alzheimer’s patients (Anand et al. 2014; Scarpini et al. 2003), there are currently no medications that slow the disease progression in AD (Klafki et al. 2006). Therefore, novel therapeutic strategies for AD are in great demand.
Flavonoids have been recognized to have potential neuroprotective roles (Frandsen and Narayanasamy 2017; Sharma et al. 2007). Soy isoflavones, or flavonoids from soybean, namely phytoestrogens, can affect estrogen-mediated processes (Molteni et al. 1995). High soybean diets clearly improve short-term and long-term memory (File et al. 2001). The protective effect of genistein, the most active component of soy isoflavones, against Aβ-induced neurological damage has been reported. Pretreatment with genistein significantly inhibits Aβ25–35-induced injury via the PKC signaling pathway in PC12 cells (Luo et al. 2012). Moreover, genistein has been reported to reverse the Aβ1–40-induced damage of short-term spatial memory (Bagheri et al. 2011), and it also inhibits the aggregation of exogenous Aβ1–40 in rat hippocampus (Bagheri et al. 2012). The mechanisms of genistein-mediated inhibition of Aβ-induced neuronal damage, however, still need to be clarified.
Ion channels are considered as vital proteins for maintaining neuronal functions, since functional impairment of neurons is often accompanied by abnormal activity of ion channels (Calabresi et al. 1995; Du et al. 2008). Consequently, ion channels have become attractive drug-targets for the treatment of nervous system diseases.
Voltage-gated sodium channels (NaV) are required to generate and propagate action potential (AP), and are indispensable in excitable cells (Yu and Catterall 2003). Dysfunctional NaV have been perceived to be correlated with AD. The levels of NaV1.1 subtype in the cortex of AD patients as well as in human amyloid precursor protein (hAPP) transgenic mice are decreased, and this reduction results in abnormal conditions of network activity and cognition in hAPP mice (Verret et al. 2012). After treatment with Aβ1–42, voltage-dependent sodium current density and expression of NaV and NaV1.6 subtype in cortical neurons increased significantly, and a similar trend was observed in the expression of NaV1.6 in APP/PS1 mice (Wang et al. 2016). Moreover, the current density of INa in CA1 pyramidal neurons was observed to be depressed in APP/PS1 mice (Brown et al. 2011) as well as in wild-type aged mice (Randall et al. 2012). Based on these finding, NaV has become a potential target for new drugs to treat AD.
Voltage-gated potassium channels (KV) are closely related to neuronal excitability and are necessary for cell survival (Shah and Aizenman 2014). In rat hippocampal neurons, several types of KV, such as the outward-delayed rectifier potassium channels and transient outward potassium channels, have been recognized (Mitterdorfer and Bean 2002). Dysfunctional KV are also relevant in pathology of AD. In the hippocampus, incubation with Aβ25–35 reduced the gene expression levels of KV2 as well as KV3 (Mayordomo-Cava et al. 2015). In hAPP mice, dendrites of hippocampal neurons showed overexcitability associated with depletion of KV4.2 channel subunits (Hall et al. 2015). Additionally, the expression and function of KV3.4 channel subunits was enhanced both in primary cultured astrocytes exposed to Aβ and in astrocytes of Tg2576 mice, a transgenic animal model for AD (Boscia et al. 2017), indicating that KV is also a potential target for new drugs that treated AD.
In the present research, we studied the effects of genistein on cell viability, and electrophysiological properties of voltage-gated sodium and potassium channels in Aβ25–35-incubated hippocampal neurons. We hope to understand whether genistein can rescue the Aβ25–35-induced cell death and the involved mechanism.
Materials and Methods
Chemicals and Animals
Dulbecco’s modified Eagle medium (DMEM)/F12 + GlutamaxTM-1, fetal bovine serum (FBS) and B27 supplements were purchased from Gibco, Invitrogen (NY, USA). HEPES was purchased from Gen-View Scientific Inc. (FL, USA). Antibiotics (penicillin and streptomycin), cytosine arabinoside (Ara-c), genistein, poly-l-lysine, tetraethylammonium-chloride (TEA-Cl), 4-aminopyridine (4-AP), and tetrodotoxin (TTX) were obtained from Sigma-Aldrich (MO, USA). 3-(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was purchased from AMRESCO, Inc. (Solon, OH, USA). Mouse anti-neuronal class III-tubulin (Tuj1) antibody and Cy-3 conjugated goat anti-mouse IgG secondary antibody were purchased from Beyotime Biotechnology Co., Ltd (Nanjing, China).
The neonatal Wistar rats were purchased from the Laboratory Animal Center of Academy of Military Medical Sciences. The Ethics Committee of Nankai University have ratified the experimental program.
Hippocampal Neurons Culture and Neuron Identification via Immunofluorescence
Hippocampal neurons were prepared from neonatal Wistar rats (1 day old) based on methods from a previous study by our laboratory (Ma et al. 2016). Briefly, the hippocampus isolated from the euthanized rats were placed in Hank’s balanced salt solution dissociation buffer and incubated with 0.125% trypsin for 20 min at 37 °C following treatment with trypsin inhibitor. Finally, the cells were plated into poly-l-lysine-coated culture dishes at a density of 1.0 × 105 to 5.0 × 105 cells/cm2 in DMEM/F12 + GlutamaxTM-1. We then added 2% B27, 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, and cultured the cells in an incubator (Sanyo, Japan) at 37 °C under 5% CO2. After incubation for 12 h, the culture medium was changed to DMEM/F12 + GlutamaxTM-1, and an additional 2% B27, 100 μg/mL streptomycin, and 100 U/mL penicillin were added after 12 h. Non-neuronal cell division was inhibited by adding 3 μM Ara-c for 24 h, after 48 h of culture. Half of the culture medium was replenished every 3 days. The cultures were maintained for 7 days prior to the experimental treatment.
To identify the purity of hippocampal neurons, they were fixed in 4% paraformaldehyde for 30 min and then washed with PBS three times. The hippocampal neurons were permeabilized by 0.25% Triton X-100 for 10 min and then washed with PBS three times. Next the hippocampal neurons were incubated with 10% BSA and 10% horse serum for 30 min at room temperature. Then, we incubated the hippocampal neurons with primary antibody [mouse anti-neuronal class III-tubulin (Tuj1) antibody (1:250)] overnight at 4 °C and then washed them with PBS three times. Subsequently, we incubated the hippocampal neurons with secondary antibody [Cy-3 conjugated goat anti-mouse IgG (H + L) (1:500)] for 1 h at room temperature, followed by three PBS washes. Then, the cell nucleus were re-dyed with DAPI. After the PBS washes, we observed the cover slips under fluorescence microscopy. After counting the number of Tuj1-positive hippocampal neurons and the number of cells in 12 randomly selected views from each cover slip (20 × and 40 × objectives), the percentage of Tuj1-positive hippocampal neurons versus the total neuronal number was calculated and averaged.
Aβ25–35 Treatment and Experimental Group Design
In this study, we designed three groups including the control group, Aβ25–35 exposure (AD model) group, and Aβ25–35 exposure plus genistein treatment (Aβ + genistein) group. The most appropriate concentrations of Aβ25–35 and genistein were confirmed by applying MTT assay before the experimental treatment (data not shown). Based on the results, treatment with 20 μM Aβ25–35 for 3 h (in the whole-cell patch-clamp recordings) or 24 h (in the MTT assay) was selected to establish the AD cellular model. Following the AD model, the Aβ + genistein group was established by applying genistein (10 μM) with Aβ25–35.
Measurement of Cell Viability Using the MTT Assay
We used 96-well plates to culture hippocampal neurons for this experiment. Following the different treatment, we added 10 μL 5 mg/mL MTT per well. After incubation for 4 h, the culture medium was discarded and 150 μL DMSO per well was added. We used a Beauty Diagnostic Microplate Reader to measure the absorbance at 570 nm. The results are shown as a percentage of viable hippocampal neurons versus the control group.
Whole-Cell Patch-Clamp Recordings of Voltage-Activated Potassium and Sodium Currents
After the different treatments, voltage-activated sodium and potassium channel currents in the hippocampal neurons were recorded by Multiclamp 700B amplifier and DigiData 1440A digitizer at 23–25 °C and analyzed using pClamp 10.1 (Axon Instruments, San Jose, CA, USA). The glass electrodes had a tip resistance of 3–6 MΩ. We suctioned suitably until establishing a giga seal and then automatically compensated pipette resistance and capacitance. Then, we used suitable “zap” to break the patch membrane for formation of the whole-cell voltage-clamp configuration, followed by series resistance compensation. We recorded the currents only when membrane resistance was greater than 800 MΩ, using Multi-Clamp Commander P/N subtraction.
To record the voltage-gated sodium channel currents, we prepared the internal solution and external solution, respectively. The former solution comprised 130 mM CsCl, 1 mM MgCl2·6H2O, 10 mM EGTA, 20 mM TEA-Cl, 10 mM HEPES, and 3 mM Na2 ATP·3H2O. We then adjusted the pH to 7.3 with CsOH. The latter solution included 5 mM KCl, 125 mM NaCl, 2 mM MgCl2·6H2O, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES. Eventually we adjusted the pH to 7.4 with NaOH. Additionally, we added 4 mM 4-AP and 20 mM TEA-Cl to block K+ currents and 200 mM CdCl2 to block Ca2+ currents.
To record the voltage-activated potassium channel currents, we compounded the internal solution and external solution, respectively. The former solution contained 140 mM KCl, 1 mM MgCl2·6H2O, 10 mM HEPES, 10 mM EGTA, and 4 mM Na2ATP·3H2O. Finally, we regulated the pH to 7.3 with KOH. The later solution was formed with 5.4 mM KCl, 145 mM NaCl, 2 mM MgCl2·6H2O, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES. Eventually we regulated the pH to 7.4 with NaOH. Additionally, Ca2+ currents and Na+ currents were blocked by, respectively, adding 200 mM CdCl2 and 1 mM TTX. Moreover, IK was separated by adding 4 mM 4-AP and IA was separated by adding 20 mM TEA-Cl.
It is worth mentioning that we counted the density of INa, IA, and IDR in the hippocampal neurons with current amplitude (pA)/Cm to weaken the effect of the differences in neuronal size.
Data Analyses
We employed the Clampfit 10.3(Axon Instruments, San Jose, CA, USA), Origin 8.5, SPSS 20.0 to analyze experimental results which are shown as mean ± SEM. In addition, we used one-way ANOVA to analyze the statistical significance among multiple groups. p < 0.05 represents significance, and p < 0.01 represents extreme significance.
The method to obtain the activation curves was as follows, we used the formula G = I/(Vm − Vr) to calculate the conductance (G) at each test potential. G was divided by the maximum conductance value (Gmax). We then fitted the G/Gmax − V curves by the Boltzmann equation G = Gmax/{1 + exp [(Vm − V1/2)/k]} (V1/2, the voltage at which G is half-maximal; k, the slope factor).
Results
Genistein Inhibited Aβ25–35-Induced Hippocampal Neurons Death
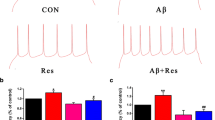
After confirming 92.13 ± 1.06% of Tuj1-positive hippocampal neurons in the cultured neurons, the morphology of hippocampal neurons was observed in the three treatment groups. In the control group, the cultured hippocampal neurons showed normal morphology. Unlike the control group, the Aβ25–35-treated group showed morphological changes in hippocampal neurons, which included shrunken and incomplete neuronal membranes with decreased numbers of neurons, which exhibited reduced attachment to the culture dish. We observed that genistein treatment partly rescued Aβ25–35-induced morphological changes in the hippocampal neurons (Fig. 1a).

Effects of genistein on the survival status of hippocampal neurons treated with Aβ25–35. a Representative status of hippocampal neurons under micrograph upon different treatments; internal scale, 100 µm. b Effects of genistein on the viability of hippocampal neurons subjected to Aβ25–35 treatment. **p < 0.01 compared with the control group; ##p < 0.01 compared with the Aβ group; n = 10
The viability of hippocampal neurons after Aβ25–35 treatment was 64.94 ± 0.68%, which was dramatically lower than that of the control neurons (Aβ vs. control, p < 0.01). However, genistein significantly reversed the reduction induced by Aβ25–35 exposure (Aβ + genistein vs. Aβ; p < 0.01) (Fig. 1b).
Effects of Genistein on the Electrophysiological Properties of Voltage-Gated Sodium Channels in Aβ25–35-Treated Hippocampal Neurons
Figures 2, 3 and 4 show the properties of voltage-gated sodium channels in hippocampal neurons subjected to different treatments.

Effects of genistein on the amplitudes and activation properties of NaV in hippocampal neurons subjected to Aβ25–35 treatments. a Typical traces of INa in the hippocampal neurons (on the left) and the protocol (on the right). b The amplitudes of the INa in different treatments. cI − V curves of the NaV in different treatments. d Voltage-dependent activation curves of NaV in different treatments. e Kinetic parameters (V1/2 and k) of the activation curves of NaV in different treatments. *p < 0.05 and **p < 0.01 compared with the control group; #p < 0.05 and ##p < 0.01 compared with the Aβ group; n = 9 for the control and Aβ groups; n = 12 for the Aβ + genistein group

Effects of genistein on the inactivation properties of NaV in hippocampal neurons subjected to Aβ25–35 treatments. a Typical traces of NaV inactivation currents in the hippocampal neurons (on the right) and the protocol (on the left). b The inactivation curves of NaV in different treatments. c Kinetic parameters (V1/2 and k) of the inactivation curves for NaV in different treatments. *p < 0.05 and **p < 0.01 compared with the control group; #p < 0.05 compared with the Aβ group; n = 7 for the control group; n = 8 for the Aβ and Aβ + genistein groups

Effects of genistein on the recovery of NaV from inactivation in hippocampal neurons subjected to Aβ25–35 treatments. a Typical recovery traces of NaV in the hippocampal neurons (on the left) and the protocol (on the right). b The recovery curves of NaV in different treatments. c Kinetic parameters [τ (ms)] of the recovery curves for NaV in different treatments. *p < 0.05 and **p < 0.01 compared with the control group; #p < 0.05 compared with the Aβ group; n = 8
As shown in Fig. 2a, representative traces of INa in the hippocampal neurons were elicited by the protocol. As shown in Fig. 2b, Aβ25–35 treatment resulted in a significant decrease in peak INa density in the hippocampal neurons [Aβ (− 33.30 ± 6.04 pA/pF) versus the control (− 84.39 ± 10.08 pA/pF), p < 0.01], but genistein treatment markedly reversed the Aβ25–35-induced decrease (Aβ + genistein vs. the Aβ, p < 0.01). Figure 2c shows that Aβ25–35 treatment significantly reduced INa at different membrane potentials, as shown in the I–V curve (Aβ vs. control, p < 0.05); genistein markedly reversed the Aβ25–35-induced effects on INa [Aβ + genistein vs. Aβ, p < 0.01)]. After treatment with Aβ25–35, the activation curve of NaV was positively shifted, V1/2 was trending up (Aβ vs. control, p > 0.05), and k significantly increased (Aβ vs. control, p < 0.01), whereas genistein treatment significantly reversed the Aβ25–35-induced effects (Aβ + genistein vs. the Aβ, p < 0.01, Fig. 2d and e).
Figure 3a shows the typical traces of NaV inactivation currents in the hippocampal neurons as well as the detailed protocol. We used the Boltzmann equation to find the inactivation curves. The inactivation curve of NaV was negatively shifted, V1/2 was significantly decreased (Aβ vs. control, p < 0.05), and k was trending up (Aβ vs. control, p > 0.05) after treatment with Aβ25–35, whereas genistein treatment partly reversed the Aβ25–35-induced effects (Fig. 3b and c).
Figure 4a shows the representative recovery traces of NaV in the hippocampal neurons as well as the detailed protocol. The method was as follows. First, we fitted the duration using the mono-exponential equation: I/Imax = 1 − exp (−△t/τ) (τ, the time constant). We observed that τ was trending up after treatment with Aβ25–35 (Aβ vs. control, p > 0.05). However, genistein treatment partly reversed the Aβ25–35-induced increase in the time course of NaV recovery (Fig. 4b and c).
Effects of Genistein on the Electrophysiological Properties of Transient Outward Potassium Channels in Aβ25–35-Treated Hippocampal Neurons
Figures 5, 6 and 7 show the electrophysiological properties of the transient outward potassium channels in the hippocampal neurons of three different groups.

Effects of genistein on the amplitudes and activation properties of transient outward potassium channels in hippocampal neurons subjected to Aβ25–35 treatments. a Typical traces of IA in the hippocampal neurons (on the left) and the protocol (on the right). b The amplitudes of the IA in different treatments. cI–V curves of transient outward potassium channels in different treatments. d Voltage-dependent activation curves of transient outward potassium channels in different treatments. e Kinetic parameters (V1/2 and k) of the activation curves for transient outward potassium channels in different treatments. *p < 0.05 compared with the control group; ##p < 0.01 and #p < 0.05 compared with the Aβ group; n = 9

Effects of genistein on the inactivation properties of transient outward potassium channels in the hippocampal neurons subjected to Aβ25–35 treatments. a Typical traces of transient outward potassium channels inactivation currents in the hippocampal neurons (on the right) and the protocol (on the left). b The inactivation curves of transient outward potassium channels in different treatments. c Kinetic parameters (V1/2 and k) of the inactivation curves for transient outward potassium channels in different treatments. **p < 0.01 and *p < 0.05 compared with the control group; ##p < 0.01, and #p < 0.05 compared with the Aβ group; n = 10

Effects of genistein on the recovery of transient outward potassium channels from inactivation to resting state in the hippocampal neurons subjected to Aβ25–35 treatments. a Typical recovery traces of the transient outward potassium channels in the hippocampal neurons (on the left) and the protocol (on the right). b The recovery curves of transient outward potassium channels in different treatments. c Kinetic parameters [τ (ms)] of the recovery curves for transient outward potassium channels in different treatments. *p < 0.05 compared with the control group; #p < 0.05 compared with the Aβ group; n = 11 for the control group, n = 7 for the Aβ group and n = 8 for the Aβ + genistein group
As shown in Fig. 5a, the representative traces of IA in the hippocampal neurons were obtained using the protocol. As shown in Fig. 5b, the peak IA density was trending up after treatment with Aβ25–35 [Aβ (145.84 ± 21.59 pA/pF) vs. control (132.14 ± 15.80 pA/pF), p > 0.05], whereas genistein significantly reversed the Aβ25–35-induced increase (Aβ + genistein vs. Aβ, p < 0.05). Figure 5c shows that IA at different membrane potentials were trending up after treatment with Aβ25–35, which can be observed in the I–V curve (Aβ vs. control, p > 0.05); whereas genistein administration reversed the Aβ25–35-induced increase (Aβ + genistein vs. Aβ, p < 0.05). The activation curves (I/Imax − V) were fitted using the Boltzmann equation, and the results are shown in Fig. 5d and e. The activation curve was positively shifted, and V1/2 (Aβ vs. control, p > 0.05) and k (Aβ vs. control, p > 0.05) were trending up after treatment with Aβ25–35, whereas genistein treatment significantly inhibited the Aβ25–35-induced effects (Aβ + genistein vs. Aβ, p < 0.05 or p < 0.01).
Figure 6a shows the representative traces of transient outward potassium channel inactivation currents from the hippocampal neurons and the detailed protocol. Inactivation curves of transient outward potassium channels were also obtained using the Boltzmann equation. Aβ25–35 treatment significantly shifted the inactivation curve toward hyperpolarization and significantly decreased V1/2 (Aβ vs. control, p < 0.01) and k (Aβ vs. control, p < 0.05). However, genistein treatment significantly suppressed the Aβ25–35-induced effects (Aβ + genistein vs. Aβ, p < 0.05 or p < 0.01; Fig. 6b and c).
Figure 7a shows the representative recovery traces of transient outward potassium channels in the hippocampal neurons and the detailed protocol. We fitted the duration with the mono-exponential equation. Aβ25–35 treatment significantly increased τ (Aβ vs. control, p < 0.05). However, genistein significantly reduced the Aβ25–35-induced increase in the time course for the recovery (Aβ + genistein vs. Aβ, p < 0.05; Fig. 7b and c).
Effects of Genistein on the Electrophysiological Properties of Outward-Delayed Rectifier Potassium Channels in Aβ-Treated Hippocampal Neurons
Figure 8 shows the electrophysiological properties of the outward-delayed rectifier potassium channels in the hippocampal neurons of three different groups.

Effects of genistein on the properties of outward-delayed rectifier potassium channels in hippocampal neurons subjected to Aβ25–35 treatments. a Representative traces of IDR in the hippocampal neurons (on the left) and the protocol (on the right). b The amplitudes of IDR in different treatments. cI–V curves for outward-delayed rectifier potassium channels in different treatments. d Voltage-dependent activation curves for outward-delayed rectifier potassium channels in different treatments. e Kinetic parameters (V1/2 and k) of the activation curves for IDR in different treatments. **p < 0.01 and *p < 0.05 compared with the control group; #p < 0.05 compared with the Aβ group; n = 9
Figure 8a shows the typical traces of IDR in the hippocampal neurons as well as the detailed protocol. Aβ25–35 treatment significantly decreased the peak IDR density in the hippocampal neurons [Aβ (38.35 ± 3.84 pA/pF) vs. control (105.57 ± 14.16 pA/pF), p < 0.01]. However, the addition of genistein significantly inhibited the Aβ25–35-induced effects (Aβ + genistein vs. Aβ, p < 0.05; Fig. 8b). Aβ25–35 treatment significantly reduced IDR at different membrane potentials, which can be observed in the I–V curves (Aβ vs. control, p < 0.05). Moreover, genistein markedly inhibited Aβ25–35-induced effects (Aβ + genistein vs. Aβ, p < 0.05; Fig. 8c). The activation curves (I/Imax − V) were fitted using the Boltzmann equation, and the results are shown in Figs. 8d and e. The activation curve was negatively shifted, and V1/2 (Aβ vs. control, p > 0.05) and k (Aβ vs. control, p > 0.05) was trending down after treatment with Aβ25–35. Genistein treatment partly rescued the Aβ25–35-induced effects (Aβ + genistein vs. Aβ, p > 0.05).
Discussion
In the present study, we established an in vitro model of AD by incubating the hippocampal neurons of neonatal Wistar rats with Aβ25–35, and then observed the effects of genistein, a soybean isoflavone, on the viability and the electrophysiological properties of NaV and KV in the Aβ25–35-incubated hippocampal neurons. The results showed that Aβ25–35 decreased cell viability and led to neuronal injury. Aβ25–35 exposure also altered the electrophysiological properties of NaV and KV, including a marked decrease in the activity of voltage-gated sodium channels, delayed rectifier potassium channels, and a slight increase in the activity of transient outward potassium channels. However, genistein treatment partially reversed these effects induced by Aβ25–35 exposure. These results revealed that genistein inhibited neuronal damage with the changes in the electrophysiological properties of NaV and KV in Aβ25–35 exposed hippocampal neurons.
Voltage-gated sodium channels chiefly determine neuronal excitability: to be more specific, they are crucial in depolarization in excitable cells (Yang et al. 2010b). The hippocampal and cortical networks exhibit spontaneous network hyperexcitability in hAPP transgenic mice (Palop et al. 2007). Age-dependent Aβ overproduction leads to a significantly short AP waveform, which correlates with decreased current density of INa (Brown et al. 2011; Tamagnini et al. 2015). The levels of NaV1.1 are decreased in the cortex of hAPP mice and AD patients; further study speculated that the reduction was related to abnormalities in network activity and cognitive dysfunction in hAPP mice and possibly in AD patients (Verret et al. 2012). Additionally, INa density in CA1 pyramidal neurons was found to be reduced both in APP/PS1 mice (Brown et al. 2011) and in wild-type aged mice (Randall et al. 2012). In the present study, Aβ25–35 markedly decreased peak INa density and significantly reduced INa at different membrane potentials. Moreover, Aβ25–35 positively shifted the activation curve. Furthermore, V1/2 was trending up, and the k values increased, suggesting that Aβ25–35 exposure decreased the sensitivity of NaV activation positive shifting as well as the activation rate. In terms of the inactivation curves, Aβ25–35 promoted NaV inactivation and negatively shifted them. Moreover, the V1/2 of the inactivation curve markedly decreased, and the value of k of the activation curve was trending up, suggesting that Aβ25–35 leads to inactivation of NaV more easily, while the rate of inactivation was trending down. Furthermore, the recovery curves of INa and the respective time constants indicated that the time constant was trending up after treatment with Aβ25–35, suggesting that Aβ25–35 postponed the recovery of NaV from inactivation. In a nutshell, Aβ25–35 reduced the activity of NaV in hippocampal neurons. Furthermore, we speculate that alterations in the electrophysiological properties of NaV may contribute to a decrease in the viability of hippocampal neurons. Genistein treatment largely reversed the Aβ25–35-caused changes in the electrophysiological properties of NaV. Hence, we conclude that genistein inhibits Aβ25–35-induced neuronal death with changes in the electrophysiological properties of NaV. Genistein-mediated inhibition of INa through PTK-dependent pathways in rabbit ventricular myocytes has been previously reported (Wang et al. 2003); however, the question of whether genistein reversed the alteration of voltage-gated sodium channels in Aβ25–35-treated hippocampal neurons through PTK-dependent pathways needs further research.
Voltage-gated potassium channels also determine several neuronal properties, such as repolarization, firing frequency, and neuronal excitability (Yang et al. 2010a).
The KV4.x family encodes for a majority of transient outward potassium channels. Preincubation of rat cerebellar granule neurons as well as HEK293 cells expressing KV4.2 subunits with Aβ1–40 significantly increases the IA density and KV4.2 mRNA levels (Kerrigan et al. 2008). Similarly, treatment with Aβ1–40 and Aβ1-42 increases IA as well as the expression of KV4.2 and KV4.3 subunits in cerebellar granule neurons (Plant et al. 2006). The KV3.4 subunit is also a component of transient outward potassium channels (Weiser et al. 1994). It has been reported that treatment with Aβ1-42 increases the levels of KV3.4, as well as the IA amplitude; moreover, neuronal apoptosis is related to the increase in KV3.4 (Pannaccione et al. 2007). In the early stages of AD, the increase in KV3.4 changes the potassium current in neurons and leads to changes in synaptic activity that might be involved in the observed neurodegeneration (Angulo et al. 2004). In the present study, we observed that the peak IA density and IA at different membrane potentials were trending up after treatment with Aβ25–35. The current activation was positively shifted after treatment with Aβ25–35. Moreover, the V1/2 and k of the activation curve were trending up, suggesting that the sensitivity and rate of transient outward potassium channel activation were trending down after treatment with Aβ25–35. In terms of inactivation curves, Aβ25–35 exposure promoted the inactivation of transient outward potassium channels. Furthermore, the V1/2 and k of the inactivation curve decreased significantly following treatment with Aβ25–35, suggesting that Aβ25–35 treatment can inactivate the transient outward potassium channels at an increased rate. In terms of recovery curves of IA, Aβ25–35 significantly increased the time constant, suggesting that it postponed the recovery from the inactivation. In brief, Aβ25–35 slightly increased the activity of transient outward potassium channels in hippocampal neurons. Furthermore, we speculate that the decrease in the viability of hippocampal neurons may be correlated with alterations in electrophysiological properties of transient outward potassium channels.
Researchers have focused on the effect of Aβ on delayed rectifier rectifier potassium currents (IDR). Exposure to Aβ25–35 or Aβ1-42 enhanced IDR and shifted the activation curve toward hyperpolarized in cultured cortical neurons (Yu et al. 1998). The Kv3.1 subunit belongs to the delayed rectifier potassium channels (Weiser et al. 1994). Age-dependent Aβ overproduction leads to narrowing of AP waveforms, and narrow AP width correlates with an increased expression of KV3.1 channels (Tamagnini et al. 2015; Wykes et al. 2012). One study indicated that Aβ25–35 could significantly inhibit IDR in hippocampal neurons (Yin et al. 2017). In our study, we found that Aβ25–35 significantly decreased the peak current density of IDR and significantly reduced IDR at different membrane potentials. The activation curve negatively shifted after treatment with Aβ25–35. Furthermore, the V1/2 and k of activation curve were trending down, suggesting that the sensitivity and rate of outward delayed rectifier potassium channel activation were trending up after treatment with Aβ25–35. In short, treating the hippocampal neurons with Aβ25–35 decreased the activity of delayed rectifier potassium channels in hippocampal neurons. Moreover, we speculate that the decrease in the viability of hippocampal neurons may be correlated with alterations in the electrophysiological properties of delayed rectifier potassium channels.
Based on the above results, Aβ25–35 differentially affected the electrophysiological properties of transient outward potassium channels and delayed rectifier potassium channels in hippocampal neurons. Although Aβ25–35 exposure produced different effects on the electrophysiological properties of transient outward potassium channels and delayed rectifier potassium channels, genistein partially reversed the Aβ25–35-induced alteration. Hence, we concluded that genistein inhibits Aβ25–35-induced neuronal death with changes in the electrophysiological properties of KV, including transient outward potassium channels and delayed rectifier potassium channels in Aβ25–35 treated hippocampal neurons. One study in mouse Schwann cells showed that genistein decreases the tyrosine phosphorylation of KV1.4 as well as KV1.5 and KV2.1, which, respectively, code for transient outward potassium channels and delayed-rectifier potassium channel alpha subunits, thereby reducing the amplitude of IA and IDR (Peretz et al. 1999). However, genistein-mediated reversal of electrophysiological properties of transient outward potassium channels and delayed rectifier potassium channels in Aβ25–35-treated hippocampal neurons through PTK-dependent pathways requires further research.
Here we will discuss the limitation of voltage-clamp technique and existed issue on space clamp in our experiment. The voltage-clamp technique was first used to require quantitative description of the ionic channels (Hodgkin and Huxley 1952). Generally, voltage-clamp recordings can only be properly analyzed in isopotential structures. However, the cell membrane in most excitable cells such as neurons, muscle cells, and glandular cells are not in isopotential structures, so problems that occur upon incomplete space clamp have been addressed extensively (Armstrong and Gilly 1992; Major 1993; Spruston et al. 1993). Typically, incomplete space clamp may distort the recorded currents, rendering accurate analysis impossible in voltage-clamp experiments. Researchers have presented numerical algorithms that corrects the space clamp errors (Castelfranco and Hartline 2004; Schaefer et al. 2003). Not unexpectedly, the issue of “space clamp” may emerge in our recording of the hippocampal neurons based on the delay in the activation of the Na+ currents. In present study, we set series resistance compensation to reduce the negative effects of incomplete space clamp referred on the previous research (Bekkers 2000), and we performed more than seven neurons recording in each treatment. We hope that the changes in electrophysiological properties approximately be a consequence of experimental treatment, not simply be a consequence of experimental errors. Certainly, finer recording methods such as applying nucleated patches might accurately reflect the impact of experimental treatment on electrophysiological properties.
Conclusion
Aβ25–35-induced neuronal death may be correlated with the alterations in electrophysiological properties of NaV and KV, including a marked decrease in the activities of NaV and delayed rectifier potassium channels, and a slight increase in the activity of transient outward potassium channels in the hippocampal neurons. In conclusion, we suggest that genistein may inhibit Aβ25–35-induced neuronal death with changes in the electrophysiological properties of NaV and KV in Aβ25–35-treated hippocampal neurons.
References
Anand R, Gill KD, Mahdi AA (2014) Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology 76:27–50
Angulo E, Noe V, Casado V, Mallol J, Gomez-Isla T, Lluis C, Ferrer I, Ciudad CJ, Franco R (2004) Up-regulation of the Kv3.4 potassium channel subunit in early stages of Alzheimer’s disease. J Neurochem 91:547–557
Armstrong CM, Gilly WF (1992) Access resistance and space clamp problems associated with whole-cell patch clamping. Methods Enzymol 207:100–122
Bagheri M, Joghataei MT, Mohseni S, Roghani M (2011) Genistein ameliorates learning and memory deficits in amyloid beta((1-40)) rat model of Alzheimer’s disease. Neurobiol Learn Mem 95:270–276
Bagheri M, Roghani M, Joghataei MT, Mohseni S (2012) Genistein inhibits aggregation of exogenous amyloid-beta(1-40) and alleviates astrogliosis in the hippocampus of rats. Brain Res 1429:145–154
Bekkers JM (2000) Properties of voltage-gated potassium currents in nucleated patches from large layer 5 cortical pyramidal neurons of the rat. J Physiol Lond 525:593–609
Blennow K, de Leon MJ, Zetterberg H (2006) Alzheimer’s disease. Lancet 368:387–403
Boscia F, Pannaccione A, Ciccone R, Casamassa A, Franco C, Piccialli I, de Rosa V, Vinciguerra A, Di Renzo G, Annunziato L (2017) The expression and activity of K(V)3.4 channel subunits are precociously upregulated in astrocytes exposed to Ab oligomers and in astrocytes of Alzheimer’s disease Tg2576 mice. Neurobiol Aging 54:187–198
Brown JT, Chin J, Leiser SC, Pangalos MN, Randall AD (2011) Altered intrinsic neuronal excitability and reduced Na + currents in a mouse model of Alzheimer’s disease. Neurobiol Aging 32(11):2109.e1-14
Calabresi P, Pisani A, Mercuri NB, Bernardi G (1995) On the mechanisms underlying hypoxia-induced membrane depolarization in striatal neurons. Brain 118(Pt 4):1027–1038
Castelfranco AM, Hartline DK (2004) Corrections for space-clamp errors in measured parameters of voltage-dependent conductances in a cylindrical neurite. Biol Cybern 90:280–290
Du H, Li M, Yang P (2008) Effects of 3-benzidino-6-phenylpyridazine, as an acetylcholinesterase inhibitor, on outward potassium current in acutely isolated rat hippocampal pyramidal neurons. Toxicol Lett 181:104–111
File SE, Jarrett N, Fluck E, Duffy R, Casey K, Wiseman H (2001) Eating soya improves human memory. Psychopharmacology 157:430–436
Frandsen JR, Narayanasamy P (2017) Neuroprotection through flavonoid: enhancement of the glyoxalase pathway. Redox Biol 14:465–473
Graham WV, Bonito-Oliva A, Sakmar TP (2017) Update on Alzheimer’s disease therapy and prevention strategies. Annu Rev Med 68:413–430
Hall AM, Throesch BT, Buckingham SC, Markwardt SJ, Peng Y, Wang Q, Hoffman DA, Roberson ED (2015) Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J Neurosci 35:6221–6230
Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117:500–544
Kerrigan TL, Atkinson L, Peers C, Pearson HA (2008) Modulation of ‘A’-type K + current by rodent and human forms of amyloid beta protein. NeuroReport 19:839–843
Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J (2006) Therapeutic approaches to Alzheimer’s disease. Brain 129:2840–2855
Luo S, Lan T, Liao W, Zhao M, Yang H (2012) Genistein inhibits a beta(25-35)-induced neurotoxicity in PC12 cells via PKC signaling pathway. Neurochem Res 37:2787–2794
Ma XL, Zhang F, Wang YX, He CC, Tian K, Wang HG, An D, Heng B, Liu YQ (2016) Genistein inhibition of OGD-induced brain neuron death correlates with its modulation of apoptosis, voltage-gated potassium and sodium currents and glutamate signal pathway. Chem Biol Interact 254:73–82
Major G (1993) Solutions for transients in arbitrarily branching cables: III. Voltage clamp problems. Biophys J 65:469–491
Mayordomo-Cava J, Yajeya J, Navarro-Lopez JD, Jimenez-Diaz L (2015) Amyloid-beta((25-35)) modulates the expression of GirK and KCNQ channel genes in the hippocampus. PLoS ONE 10:e0134385
Mitterdorfer J, Bean BP (2002) Potassium currents during the action potential of hippocampal CA3 neurons. J Neurosci 22:10106–10115
Molteni A, Brizio-Molteni L, Persky V (1995) In vitro hormonal effects of soybean isoflavones. J Nutr 125:751S–756S
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A et al (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55:697–711
Pannaccione A, Boscia F, Scorziello A, Adornetto A, Castaldo P, Sirabella R, Taglialatela M, Di Renzo GF, Annunziato L (2007) Up-regulation and increased activity of K(V)3.4 channels and their accessory subunit MinK-Related peptide 2 induced by amyloid peptide are involved in apoptotic neuronal death. Mol Pharmacol 72:665–673
Peretz A, Sobko A, Attali B (1999) Tyrosine kinases modulate K + channel gating in mouse Schwann cells. J Physiol 519:373–384
Plant LD, Webster NJ, Boyle JP, Ramsden M, Freir DB, Peers C, Pearson HA (2006) Amyloid beta peptide as a physiological modulator of neuronal ‘A’-type K + current. Neurobiol Aging 27:1673–1683
Randall AD, Booth C, Brown JT (2012) Age-related changes to Na + channel gating contribute to modified intrinsic neuronal excitability. Neurobiol Aging 33:2715–2720
Scarpini E, Scheltens P, Feldman H (2003) Treatment of Alzheimer’s disease: current status and new perspectives. Lancet Neurol 2:539–547
Schaefer AT, Helmstaedter M, Sakmann B, Korngreen A (2003) Correction of conductance measurements in non-space-clamped structures: 1. Voltage-gated K + channels. Biophys J 84:3508–3528
Shah NH, Aizenman E (2014) Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl Stroke Res 5:38–58
Sharma V, Mishra M, Ghosh S, Tewari R, Basu A, Seth P, Sen E (2007) Modulation of interleukin-1 beta mediated inflammatory response in human astrocytes by flavonoids: implications in neuroprotection. Brain Res Bull 73:55–63
Spruston N, Jaffe DB, Williams SH, Johnston D (1993) Voltage- and space-clamp errors associated with the measurement of electrotonically remote synaptic events. J Neurophysiol 70:781–802
Tamagnini F, Novelia J, Kerrigan TL, Brown JT, Tsaneva-Atanasova K, Randall AD (2015) Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front Cell Neurosci 9:372
Verret L, Mann EO, Hang GB, Barth AMI, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I et al (2012) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149:708–721
Wang YG, Wagner MB, Kumar R, Cheng J, Joyner RW (2003) Inhibition of fast sodium current in rabbit ventricular myocytes by protein tyrosine kinase inhibitors. Pflug Arch Eur J Phy 446:485–491
Wang X, Zhang XG, Zhou TT, Li N, Jang CY, Xiao ZC, Ma QH, Li S (2016) Elevated neuronal excitability due to modulation of the voltage-gated sodium channel Nav1.6 by A beta(1-42). Front Neurosci 10:94
Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B (1994) Differential expression of Shaw-related K + channels in the rat central nervous system. J Neurosci 14:949–972
Wykes R, Kalmbach A, Eliava M, Waters J (2012) Changes in the physiology of CA1 hippocampal pyramidal neurons in preplaque CRND8 mice. Neurobiol Aging 33:1609–1623
Yang JJ, Tian YT, Yang Z, Zhang T (2010a) Effect of melamine on potassium currents in rat hippocampal CA1 neurons. Toxicol Vitro 24:397–403
Yang JJ, Yang Z, Zhang T (2010b) Action potential changes associated with impairment of functional properties of sodium channels in hippocampal neurons induced by melamine. Toxicol Lett 198:171–176
Yin H, Wang H, Zhang H, Gao N, Zhang T, Yang Z (2017) Resveratrol attenuates a beta-induced early hippocampal neuron excitability impairment via recovery of function of potassium channels. Neurotox Res 32:311–324
Yu FH, Catterall WA (2003) Overview of the voltage-gated sodium channel family. Genome Biol 4:1–7
Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW (1998) Enhancement of outward potassium current may participate in beta-amyloid peptide-induced cortical neuronal death. Neurobiol Dis 5:81–88
Funding
This study was funded by the Natural Science Foundation of Tianjin City (15JCYBJC24500), the National Natural Science Foundation of China (No. 31272317) and the 111 Project (B08011).
Author information
Authors and Affiliations
Contributions
All authors had full access to all research results and are responsible for the accuracy of the data. WYX performed all the experiments; XZH, JX, LLX, AD, WHG, HB assisted with parts of the experiments. LYQ designed and supervised the experiments. WYX and LYQ contributed to the writing and critical revision of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors have no conflict of interest.
Ethical Approval
All animal care and experimental programs were conducted according to standard ethical guidelines (National Institutes of Health Guide to the use of Laboratory Animals) and approved by the Institutional Animal Care and use Committee of Nankai University. All efforts were made to minimize the number of mice used and their suffering.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, Yx., Xia, Zh., Jiang, X. et al. Genistein Inhibits Aβ25–35-Induced Neuronal Death with Changes in the Electrophysiological Properties of Voltage-Gated Sodium and Potassium Channels. Cell Mol Neurobiol 39, 809–822 (2019). https://doi.org/10.1007/s10571-019-00680-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-019-00680-w