
Abstract
“Modern” medicine and pharmacology require an effective medical drug with a single compound for a specific disease. This seams very scientific but usually has unavoidable side effects. For example, the chemical therapy to cancer can totally damage the immunological ability of the patient leading to death early than non-treatment. On the other hand, natural antioxidant drugs not only can cure the disease but also can enhance the immunological ability of the patient leading to healthier though they usually have several compounds or a mixture. For the degenerative disease such as Alzheimer’s disease (AD) and Parkinson’s disease (PD), natural antioxidant drugs are suitable drugs, because the pathogenesis of these diseases is complex with many targets and pathways. These effects are more evidence when the clinic trial is for long term treatment. The author reviews the studies on the protecting effects of natural antioxidants on neurons in neurodegenerative diseases, especially summarized the results about protective effect of green tea polyphenols on neurons against apoptosis of cellular and animal PD models, and of genestine and nicotine on neurons against Aβ—induced apoptosis of hippocampal neuronal and transgenic mouse AD models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is a common character for neurodegenerative diseases: all of them [such as Parkinson’s disease (PD) and Alzheimer’s disease (AD)] are connected with oxidative stress-induced neuronal apoptosis. It is well known that it is difficult for the patients to be cured because there is no any effective drug for curing these diseases until now. Is there any way to prevent these diseases? From this review we may find a positive answer: Antioxidants should have preventing and even therapeutic effects on these diseases.
Oxidative Stress in AD and PD
It is undoubted that extra free radical results in biological injure heavily. Free radicals peroxidize membrane lipids [1] and oxidize proteins [2], resulting in damage of plasma membrane and cross-linking of cytoskeletal proteins. In addition, free radicals damage RNA [3], and DNA [4, 5]. In brain, the high metabolic rate, the low concentration of glutathione and antioxidant enzyme catalase, and the high proportion of polyunsaturated fatty acids, make the brain tissue particularly susceptible to oxidative damage [6, 7]. Oxidative stress, an imbalance toward the prooxidant side of the prooxidant/antioxidant homeostasis, occurs in several brain neurodegenerative disorders. Among these neurodegenerative brain disorders are those in which protein aggregation is observed in hippocampus of AD brain and in substantia nigra of PD brain.
Oxidative Stress and AD
Accumulated data demonstrated that oxidative damage occurs in AD brain [5]. Aβ peptide has been proven to produce hydrogen peroxide (H2O2) through metal ion reduction, with concomitant release of thiobarbituric acid-reactive substances (TBARS), a process probably mediated by formation of hydroxyl radicals [8, 9]. The cytotoxicity of Aβ fibrils also implicated to be relative to an oxidative mechanism. Aβ fibrils-induced H2O2 was detected by several laboratories [10–12]. There is considerable evidence consistent with the importance of oxidative stress in the pathology of AD (for recent reviews, see literatures [13–15]). Evidence supporting the notion of free radical oxidative stress in AD brain includes: increased redox-active metal ions in AD brain; increased lipid peroxidation detected by decreased levels of polyunsaturated fatty acids and increased levels of the lipid peroxidation products, acrolein, TBARS, and neuroprostanes; increased protein oxidation; increased oxidation of DNA and RNA; decreased activity of oxidatively prone enzymes, such as glutamine synthetase (GS).
Oxidation of proteins normally is caused by free radicals, and this process, from a chemical thermodynamics standpoint, is an exothermic event. Oxidative reactions of peptides are mediated mainly by the hydroxyl radical. There are two possible oxidative pathways that can occur in backbone oxidation and side-chain oxidation [1]. Backbone oxidation is initiated by carbon abstraction of hydrogen by the free radical leading to the formation of a carbon-centered radical. In the presence of oxygen, this radical is converted to a peroxyl radical. This can lead to the formation of an alkoxyl radical and subsequent hydroxylation of the peptide backbone. The oxidation of amino acid side-chains greatly depends on their structure. An important oxidative process with profound functional and structural consequences involves the irreversible nitration of tyrosine residues by peroxynitrite (ONOO−) [16].The levels of protein oxidation in membrane systems can be indirectly monitored by use of electron paramagnetic resonance (EPR) spin-labeling techniques [17].
Oxidative Stress and PD
Parkinson’s disease is a progressive neurodegenerative disorder and the hallmark of this disease is selective loss of dopaminergic neurons in the substantia nigra pars compacta [18]. Recently, the death of dopaminergic neurons has been reported to occur by apoptosis [19–21]. Oxidative stress has been widely believed to be an important pathogenetic mechanism of neuronal apoptosis in PD [22]. Overproduction of reactive oxygen (ROS) species can lead to oxidative damage in the brain of PD, as shown by increased lipid peroxidation and DNA damage in the substantia nigra. Increased protein oxidation is also apparent in many areas of the brain, while substantia nigra is particularly vulnerable [23]. Under physiological conditions, 6-OHDA is rapidly and nonenzymatically oxidized by molecular oxygen to form H2O2 and the corresponding p-quinone [24]. The former can react with iron (II) to form the reactive and damaging hydroxyl free radical. The latter then undergoes an intramolecular cyclization followed by a cascade of oxidative reactions resulting in the formation of an insoluble polymeric pigment related to neuromelanin [25, 26].
Natural Antioxidants Protect Neurons Against PD and AD
Nerve cell death from oxidative stress has been implicated in a variety of pathologies, including stroke, trauma, and diseases such as AD and PD. It should be reasonable to use antioxidants to prevent the diseases caused by oxidative stress. Here we include several natural antioxidants, such as flavonoids, polyphenols, and nicotine in protecting neuron against AD and PD.
Flavonoids
Flavonoids isolated from plants are well investigated as natural antioxidants and widely used to treat patients as effective components of many medical drugs in clinics. To determine the potential protective mechanisms of flavonoids in cell death, the mouse hippocampal cell line HT-22 was used and it was found that exogenous glutamate inhibits cystine uptake and depletes intracellular GSH, leading to the accumulation of ROS and an increase in Ca2+ influx, which ultimately causes neuronal death. Many, but not all, flavonoids protect HT-22 cells and rat primary neurons from glutamate toxicity as well as from five other oxidative injuries. Three structural requirements of flavonoids for protection from glutamate are the hydroxylated C3, an unsaturated C ring, and hydrophobicity. It was also found three distinct mechanisms of protection. These include increasing intracellular GSH, directly lowering levels of ROS, and preventing the influx of Ca2+ despite high levels of ROS. These data show that the mechanism of protection from oxidative insults by flavonoids is highly specific for each compound [27].
Ginkgo-Biloba extract (EGb), which contains flavonoids, ginkgolides and biolobalids, has been reported to protect the brain against hypoxic damage and inhibit ROS formation in cerebellar neurons. We investigated the protective effects of EGb on dissociated cortex neurons from damage caused by ROS using spin label and molecular techniques. It was found that the order parameter (S), rotational correlation time (τ) and the ratio of strong immobilized component to weak immobilized component (S/W) of the cell membrane treated with hydroxyl radical were higher than those of control (P < 0.05), indicating that the membrane fluidity attacked by free radical was lower than that of control. With increase of EGb concentration (5–50 μg/ml), a dose dependent membrane fluidity increased in hydrophobic areas of the membrane. EGb also protected the change of the protein conformation on the membrane caused by free radical. The lactic dehydrogenase activity and cell apoptosis attacked by hydroxyl radical were higher than that of control (P < 0.05). EGb (50 μg/ml) was found to have protective effect on the cells attacked by free radicals [28].
The protective effect of EGb761 and its active constitutes against apoptosis were also examined and the results showed that hydroxyl radicals generated by Fenton reaction induced apoptosis in cerebellar granule cells, which was associated with decrease in Bcl-2, mRNA level and increase in the protein levels and caused the changes in the sulfhydryl group binding sites on the membrane proteins and its different constitutes showed different effects, The total flavonoid components of EGb761 and a mixture of flavonoids and terpens protected cerebellar granule cell from oxidative damage and apoptosis induced by hydroxyl radicals. Total terpens of EGb761 did not protect against apoptosis. Flavonoids and terpens did show a synergic effect in this regard [29, 30].
Green Tea Polyphenols
Green tea polyphenols and their major constituents, such as EGCG, have diverse pharmacological activities, such as antimutagenic and anticarcinogenic effects. It is believed that these beneficial effects of green tea polyphenols are due to their potent antioxidative properties. In fact, it was demonstrated that green tea polyphenols serve as powerful antioxidants against free radicals such as DPPH radicals [31], superoxide anion [32–34], lipid free radicals and hydroxyl radicals [35, 36]. In the central nervous system, there is also some evidence to show that oral administration of green tea polyphenols and flavonoid-related compounds has preventive effects on iron-induced lipid peroxide accumulation and age-related accumulation of neurotoxic lipid peroxides in rat brain [37, 38].
Tea catechins (TC) are usually expected as scavengers of free radicals but different components have different functions. We studied the effect of TC on the PC 12 exposed to 6-OHDA and it demonstrated TC could protect PC12 cells against apoptosis caused by 6-OHDP. We investigated the effects of exposure of PC12 cells to 6-OHDA alone or associated with pre-treatment of TC. Exposure of PC12 cells to 6-OHDA induced a concentration-dependent decrease in cell viability determined by MTT assay and apoptosis of PC12 cells observed by flow cytometry, fluorescence microscopy and DNA fragmentation technique. TC displayed significantly inhibitory effects against PC12 cell death. EGCG and ECG were more effective than TC but EGC, EC and (+)-C were less effective [39]. 6-OHDA-induced apoptosis was greatly inhibited by green tea polyphenols at 200–400 μM. From 50 to 400 μM, the protective effects increased with the concentrations and EGCG was better than green tea polyphenols at the same concentrations. From the data of flow cytometry, the apoptotic cells were inhibited greatly by 200–400 μM of green tea polyphenols and EGCG (the inhibitory ratios of green tea polyphenols were 83.1 and 84.8%; of EGCG were 88.3 and 90.3%, respectively). The nuclear changes characteristic of apoptosis were disappeared, especially in the EGCG protected PC 12 cells. The DNA ladder also disappeared in the channels of 200–400 μM of green tea polyphenols and EGCG [40].
We here also investigate the protective mechanisms of green tea polyphenols on SH-SY5Y cells against apoptosis induced by the pro-parkinsonian neurotoxin 6-OHDA. Green tea polyphenols rescued the changes in condensed nuclear and apoptotic bodies, attenuated 6-OHDA-induced early apoptosis, prevented the decrease in mitochondrial membrane potential, and suppressed accumulation of ROS and of intracellular free Ca2+. Green tea polyphenols also counteracted the 6-OHDA-induced nitric oxide increase and overexpression of nNOS and iNOS, and decreased the level of protein-bound 3-nitrotyrosine (3-NT). In addition, green tea polyphenols inhibited the autooxidation of 6-OHDA and scavenged oxygen free radicals in a dose- and time-dependent manner. Our results show that the protective effects of green tea polyphenols on SH-SY5Y cells are mediated, at least in part, by controlling the ROS-NO pathway [41].
Nitric oxide and related pathways are thought to play an important role in the pathogenesis of PD. Immunohistochemistry, terminal deoxynucleotidyl transferase-mediated dUTP Nick End Labeling assay, electron spin resonance pin trapping, enzyme linked immunosorbent assay, and molecular biological methods were used to investigate the effects of green tea polyphenols in an anilateral 6-hydroxydopamine (6-OHDA)-treated rat model of PD in our recent study. Green tea polyphenols treatment dose-dependently protected dopaminergic neurons by preventing from midbrain and striatal 6-OHDA-induced increase in (1) both ROS and NO levels, (2) lipid peroxidation, (3) nitrite/nitrate content, (4) inducible nitric oxide synthase, and (5) protein-bound-nitro-tyrosine. Moreover, Green tea polyphenols treatment dose-dependently preserved the free radical scavenging capability of both the midbrain and the triatum. These results support the in vivo protection of green tea polyphenols against 6-OHDA and suggest that green tea polyphenols treatment might represent a neuroprotective treatment of PD [42].
In an animal study Mandel group demonstrated neuroprotective property of green tea extract and ECG in MPTP treated mice model of PD. MPTP neurotoxin caused dopamine neuron loss in substantia nigra concomitant with a depletion in striatal dopamine and tyrosine hydroxylase protein levels. Pretreatment with either green tea extract or ECG prevented these effects. In addition, the neurotoxin caused an elevation in striatal antioxidant enzymes SOD (240%) and catalase (165%) activities, both effects being prevented by ECG. ECG itself also increased the activities of both enzymes in the brain. The neuroprotective effects are not likely to be caused by inhibition of MPTP conversion to its active metabolite 1-methyl-4-phenylpyridinium by monoamine oxidase-B, as both green tea and ECG are very poor inhibitors of this enzyme in vitro (770 mg/ml and 660 mM, respectively). Brain penetrating property of polyphenols, as well as their antioxidant and iron-chelating properties may make such compounds an important class of drugs to be developed for treatment of neurodegenerative diseases where oxidative stress has been implicated [43].
They demonstrated highly potent antioxidant-radical scavenging activities of green tea (GT) and black tea (BT) extracts on brain mitochondrial membrane fraction, against iron-induced lipid peroxidation. Both extracts were shown to attenuate the neurotoxic action of 6-OHDA-induced neuronal death. 6-OHDA activated the iron dependent inflammatory redox sensitive NF-κB in PC12 and SH-SY5Y cells, respectively. Immunofluorescence and electromobility shift assays showed increased nuclear translocation and binding activity of NF-κB after exposure to 6-OHDA in SH-SY5Y cells, with a concomitant disappearance from the cytoplasm. Introduction of GT extract before 6-OHDA inhibited both NF-κB nuclear translocation and binding activity induced by this toxin in SH-SY5Y cells. Neuroprotection was attributed to the potent antioxidant and iron chelating actions of the polyphenolic constituents of tea extracts, preventing nuclear translocation and activation of cell death promoting NF-κB [44].
They also demonstrated that EGCG restored the reduced PKC and extracellular signal-regulated kinases (ERK1/2) activities caused by 6-OHDA toxicity. However, the neuroprotective effect of EGCG on cell survival was abolished by pretreatment with PKC inhibitor GF 109203X. Because EGCG increased phosphorylated PKC, they suggest that PKC isoenzymes are involved in the neuroprotective action of EGCG against 6-OHDA. In addition, gene expression analysis revealed that EGCG prevented both the 6-OHDA-induced expression of several mRNAs, such as Bax, Bad, and Mdm2, and the decrease in Bcl-2, Bcl-w, and Bcl-xL. These results suggest that the neuroprotective mechanism of EGCG against oxidative stress induced cell death includes stimulation of PKC and modulation of cell survival/cell cycle genes [45].
Rezai-Zadeh et al. studied the modulation of EGCG on amyloid precursor protein (APP) cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. They found that EGCG reduced Aβ generation in both murine neuron-like cells (N2a) transfected with the human “Swedish” mutant APP and in primary neurons derived from Swedish mutant APP-overexpressing mice. In concert with these observations, they found that EGCG markedly promoted cleavage of the α-C-terminal fragment of APP and elevated the N-terminal APP cleavage product, soluble APP-α. These cleavage events are associated with elevated α-secretase activity and enhanced hydrolysis of TNF α-converting enzyme, a primary candidate α-secretase. As a validation of these findings in vivo, they treated Tg APPsw transgenic mice overproducing Aβ with EGCG and found decreased Aβ levels and plaques associated with promotion of the nonamyloidogenic α-secretase proteolytic pathway. These data raise the possibility that EGCG dietary supplementation may provide effective prophylaxis for AD [46].
Isoflavone
Genistein, the most active component of soy isoflavone, has been investigated to have affinity to estrogen receptor [47], antioxidation [48, 49], increases cellular reduced glutathione [50], inhibits protein tyrosine kinase (PTK) [51], and has other physiological functions [52]. The existing data strongly suggest that the soy isoflavones have a protective action against several chronic diseases such as atherosclerosis [53], the diseases associated with postmenopausal estrogen deficiency, and hormone-dependent breast and prostate cancers [54]. Recently, some researchers found that genistein showed neuroprotection. Andersen [55] reported that genitein protected rat brain synatosome from insult induced by Aβ25–35. Genistein, a phytoestrogen, which is capable to cross the blood–brain barrier [56], has been reported to have antioxidation from insult of UV [57] and of chemicals [48].
We studied the neuroprotective effect of genistein against Aβ25–35-induced apoptosis in cultured hippocampal neurons [58]. It was found exposure to aged Aβ for 24 h increased the DCF fluorescence intensity twofolds relative to controls. The increase in DCF fluorescence intensity was eliminated by 63% when co-treated with genistein at 40 μM and aged Aβ, while genistein at 0.1 μM decreased about 18% of the production of ROS induced by Aβ. Therefore, genistein at high concentration has stronger antioxidative activities in comparison with the low concentration in protection of the neuronal cell death induced by Aβ. Treatment with 25 μg/ml aged Aβ for 24 h decreased the viability of hippocampal neuronal cells about 43.3% compared to controls. Genistein at the concentrations of 0.1 and 40 μM rescued aged Aβ-induced decrease of viability rate by 7.2 and 13.9%, respectively. The ER antagonist, ICI182,780, significantly blocked the protective effect of genistein at the concentration of 0.1 μM while had little effect on genistein at the concentration of 40 μM. ICI182,780 itself did not affect aged Aβ-induced decrease of cell viability. In addition, no difference was seen on the cells viability when the cells were treated with genistein alone compared to controls. Treatment with 25 μg/ml aged Aβ for 24 h decreased the viability of hippocampal neuronal cells about 43.3% compared to controls. It can concluded that Aβ-induced apoptosis, and all these phenotypes induced by Aβ are reverted by genistein. Our results further show that at nanomolar level, genistein protects neurons from Aβ25–35-induced damages largely via the estrogen receptor (ER)-mediated pathway and at micromolar level, the neuroprotective effect of genistein is mainly mediated by its antioxidative properties.
The neuroprotective effect of estrogen prompted us to determine potential therapeutic impact of soy-derived estrogenic compounds. Transgenic C. elegans, that express human Aβ, were fed with soy derived isoflavones genistein, daidzein and glycitein (100 μg/ml) and then examined for Aβ-induced paralysis and the levels of reactive oxygen species. Among the three compounds tested, only glycitein alleviated Aβ expression-induced paralysis in the transgenic C. elegans. This activity of glycitein correlated with a reduced level of hydrogen peroxide in the transgenic C. elegans. In vitro scavenging effects of glycitein on three types of reactive oxygen species confirmed its antioxidant properties. Furthermore, the transgenic C. elegans fed with glycitein exhibited reduced formation of Aβ. These findings suggest that a specific soy isoflavone glycitein may suppress Aβ toxicity through combined antioxidative activity and inhibition of Aβ deposition, thus may have therapeutic potential for prevention of Aβ associated neurodegenerative disorders [59].
Nicotine
Smokers have less risk to catch degenerative diseases such as AD and PD that are both correlated with enhanced oxidative stress. We studied the scavenging effect of nicotine on free radicals by electron spin resonance techniques. It was found that the scavenging effect of nicotine on hydroxyl radicals and superoxide free radicals was higher than that of vitamin C. It was also found that nicotine could scavenge free radicals in gas-phase cigarette smoke. The data indicate that nicotine may be a potential antioxidant [60].
Nicotine as a major component of cigarette smoke has been suggested to have a protective effect for neurons against Aβ neurotoxicity. Our study demonstrates that nicotine protected cultured hippocampal neurons against the Aβ-induced apoptosis. Nicotine effectively inhibits apoptosis in hippocampal cultures caused by Aβ treatment and increase of caspase activity induced by Aβ. Measurements of cellular oxidation and intracellular free Ca2t showed that nicotine suppressed Aβ-induced accumulation of free radical and increase of intracellular free Ca2t. Cholinergic antagonist mecamylamine inhibited nicotine-induced protection against Aβ-induced caspase-3 activation and ROS accumulation. The data show that the protection of nicotine is partly via nicotinic receptors. Our results suggest that nicotine may be beneficial in retarding the neurodegenerative diseases such as AD [61].
Although nicotine has been associated with a decreased risk of developing PD, the underlying mechanisms are still unclear. By using isolated brain mitochondria, we found that nicotine inhibited N-methyl-4-phenylpyridine (MPPt) and calcium-induced mitochondria high amplitude swelling and cytochrome c release from intact mitochondria. Intra-mitochondria redox state was also maintained by nicotine, which could be attributed to an attenuation of mitochondria permeability transition. Further investigation revealed that nicotine did not prevent MPPt or calcium-induced mitochondria membrane potential loss, but instead decreased the electron leak at the site of respiratory chain complex I. In the presence of mecamylamine hydrochloride, a nonselective nicotinic acetylcholine receptor inhibitor, nicotine significantly postponed mitochondria swelling and cytochrome c release induced by a mixture of neurotoxins (MPPt and 6-OHDA) in SH-SY5Y cells, suggesting that there is a receptor-independent nicotine-mediated neuroprotective effect of nicotine. These results show that interaction of nicotine with mitochondria respiratory chain together with its antioxidant effects should be considered in the neuroprotective effects of nicotine [62].
Nicotine reduces β-amyloidosis and has a beneficial effect against AD, but the underlying mechanism is not clear. The abnormal interactions of Aβ with metal ions such as copper and zinc are implicated in the process of Aβ deposition in AD brains. In one of our studies, we investigated the effect of nicotine on metal homeostasis in the hippocampus and cortex of APPV717I (London mutant form of APP) transgenic mice. A significant reduction in the metal contents of copper and zinc in senile plaques and neuropil is observed after nicotine treatment. The densities of copper and zinc distributions in a subfield of the hippocampus CA1 region are also reduced after nicotine treatment. We further studied the mechanism of nicotine-mediated effect on metal homeostasis by using SH-SY5Y cells overexpressing the Swedish mutant form of human APP (APPsw). Nicotine treatment decreases the intracellular copper concentration and attenuates Aβ-mediated neurotoxicity facilitated by the addition of copper, and these effects are independent of the activation of nicotinic acetylcholine-receptor. These data suggest that the effect of nicotine on reducing β-amyloidosis is partly mediated by regulating metal homeostasis [63].
In our study we show that nicotine decreases accumulation of Aβ in the cortex and hippocampus of APP (V717I) transgenic mice. Nicotine prevents activation of NF-κB and c-Myc by inhibiting the activation of MAP kinases (MAPKs). As a result, the activity of iNOS and the production of NO are down-regulated. RNA interference experiments show that the above nicotine-mediated process requires α7 nAChR. Nicotine decreases Aβ via the activation of α7nAChRs through MAPK, NF-κB, and c-myc pathways. Nicotine also inhibits apoptosis and cell cycle progression in this mouse line. The dissected signaling pathway of nicotine-mediated neuroprotection in the study provides a mechanistic basis for the potential development of drug targets for treating AD [64].
There are presently initiatives to develop the treatment strategies in AD that effectively lower the Aβ load in brain. Nordberg et al. treated transgenic mice carrying APPsw, which develop brain Aβ deposits, with nicotine in drinking fluid from 9 to 14.5 months of age (5.5 months). A significant reduction in Aβ(1–42) positive plaques by more than 80% was observed in the brains of nicotine treated compared to sucrose treated transgenic mice. In addition, there was a selective reduction in extractable amyloid β peptides in nicotine treated mice; cortical insoluble Aβ(1–40) and Aβ(1–42) peptide levels were lower by 48 and 60%, respectively, whilst there was no significant change in soluble Aβ(1–40) or Aβ(1–42) levels. The expression of glial fibrillary acidic protein was not affected by nicotine treatment. These results indicate that nicotine may effectively reduce Aβ peptide aggregation in brain and that nicotinic drug treatment may be a novel protective therapy in AD [65].
Ten days treatment with nicotine reduced insoluble Aβ peptides by 80% in the cortex of 9-month-old APPsw mice, which is more than that observed in 14.5-month-old mice following nicotine treatment for 5.5 months. A reduction in Aβ associated with cerebral vessels was observed in addition to that deposited as paranchymal plaques after 5.5 months treatment. The diminution in Aβ peptides observed was not accompanied by changes in brain α, β or γ secretase-like activities, NGF (nerve growth factor) or BDNF (brain-derived neurotrophic factor) protein expression measured in brain homogenates. A significant increase in sAPP was observed after nicotine treatment of SY5Y neuroblastoma cells that could be blocked by the nicotine antagonist mecamylamine. Attenuation of elevated [125]-α-bungarotoxin binding (α7) in APPsw mice was observed after 5.5 months nicotine treatment. Both these observations suggest that the reduction in insoluble Aβ by nicotine might be in part mediated via the α7 nicotine receptor [66].
Nicotinic cholinergic receptor stimulation induces neuroprotection against glutamate cytotoxicity by its inhibitory action on NO-formation. However, there are different reports about the effect of nicotine on the generation of NO. It was reported nicotine evoked NO release in the rat hippocampal slice and NOS expression in the ventromedial hypothalamic nucleus increased by nicotine treatment. However, it was found that nicotine administration was effective in limiting the enhancement on NOS expression following food restriction in other reports [67, 68]. While a mismatch result was reported about the effect of nicotine on NO production and NOS expression determined between biochemical and histological method was obtained. The contradictory indicates that there is no conclusion and its mechanism is not clear. We studied the production of NO free radicals in the transgenic mice and the effect of nicotine and the primary result showed that nicotine may be involved in protection of the brain against AD by regulation of NO and ROS.
Conclusion
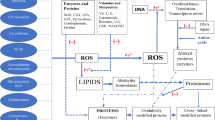
From above results, we can find the pathogenesis of the degenerative disease such as AD and PD is complex with many targets and pathways, it is difficult for the modern medicine with a single target and single pathway to cure these diseases but it is suitable for natural antioxidant drugs with multiple components, functions, targets and pathways (as summarized about effects of green tea polyphenols on PD in Fig. 1 and nicotine on AD in Fig. 2). It is difficult to cure these patients after they have got neurodegenerative diseases such as AD and PD, therefore, the best strategy is to prevent people from neurodegenerative diseases. This review suggests that some natural antioxidants can prevent neurodegeneration, which may open a new way against the oxidative stress related disease. It is easy to take the natural antioxidants from drinking tea, soybean milk and eating fresh fruits and vegetables everyday.

A hypothetical model diagramming for the potential targets of GTP or EGCG are suggested in cell signaling pathways affected by 6-OHDA-induced oxidative stress. (1) Direct inhibition of oxidized 6-OHDA and/or scavenging of ROS; (2) inhibiting the negative effect of 6-OHDA on both PKC and ERK1/2, and EGCG can direct phosphorylative activation of PKC; (3) attenuating NF-κB translocation to the nucleus, and inhibiting it activation; (4) modulating the expression of cell death and cell cycle genes; (5) modulating the intracellular NO level and inhibiting the generation of peroxynitrite

The schematic drawing shows that nicotine abolishes AD through the activation of α7nAChRs and MAPK pathways. In AD brains, Aβ directly stimulates inflammation and the activation of MAPK pathway leads to the inflammation. The inflammation process in turn aggravates the deposition and accumulation of Aβ in the brain. In the down stream, the inflammation increases C-myc, causes the impairments of the apoptosis and cell cycle control, and ultimately leads to neurodegeneration. Nicotine inhibits the inflammation by reducing the productions of NF-κB and NO, which are mediated by the MAPK pathway. The modulation of the MAPK pathway includes modification of B-Raf, ERK and P38 MAPK activities through the activation of the α7 nAchR. By regulating the inflammation-related pathways, nicotine ultimately decreases the expression and deposition of Aβ in the brain. In addition, nicotine regulates the activities of the downstream cell cycle signals including C-Myc, cyclin D1 and CDK4 and the apoptotic proteins including anti-apoptosis Bcl-2, Bax and caspase-3
References
Butterfield DA, Kanski J (2001) Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech Ageing Dev 122:945–962. doi:10.1016/S0047-6374(01)00249-4
Stadtman ER (1990) Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic Biol Med 9:315–325. doi:10.1016/0891-5849(90)90006-5
Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S et al (1999) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci 19:1959–1964
Gabbita SP, Lovell MA, Markesbery WR (1998) Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem 71:2034–2040
Mecocci PL, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36:747–750. doi:10.1002/ana.410360510
Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF et al (1996) Oxidative damage in Alzheimer’s disease. Nature 382:120–121. doi:10.1038/382120b0
Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL et al (1998) Amyloid-β deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 70:2212–2215
Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein E, Scarpa RC et al (1999) The amyloid-b-peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38:7609–7616. doi:10.1021/bi990438f
Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR et al (1999) Cu(II) potentiation of Alzheimer Ab neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 74:37111–37116. doi:10.1074/jbc.274.52.37111
Behl C, Davis JB, Lesley R, Schubert D (1994) Hydrogen peroxide mediates amyloid b protein toxicity. Cell 77:817–827. doi:10.1016/0092-8674(94)90131-7
Markesbery WR, Carney JM (1999) Oxidative alterations in Alzheimer’s disease. Brain Pathol 9:133–146
Markesbery WR (1997) Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 23:134–147. doi:10.1016/S0891-5849(96)00629-6
Christen Y (2000) Oxidative stress and Alzheimer disease. Am J Clin Nutr 71:621S–629S
Smith MA (2000) Oxidative stress in Alzheimer’s disease. Biochim Biophys Acta 1502:139–144
Varadarajan S (2000) Review: Alzheimer’s amyloid-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol 130:184–208. doi:10.1006/jsbi.2000.4274
Beckman JS (1996) Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol 9:836–844. doi:10.1021/tx9501445
Xin W-J, Zhao B-L, Zhang J-Z (1984) Studies on the property of sulfhydryl groups binding sites on the lung normal cell and cancer cell membrane of Chinese hamster with maleimide spin labels. Sci Sin [B] 28:1008–1014
Hirsch EC, Faucheux B, Damier P, Mouatt-Prigent A, Agid Y (1997) Neuronal vulnerability in Parkinson’s disease. J Neural Transm 50:79–88
Mochizuki H, Goto K, Mori H, Mizuno Y (1996) Histochemical detection of apoptosis in Parkinson’s disease. J Neurol Sci 137:120–123. doi:10.1016/0022-510X(95)00336-Z
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J et al (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12:25–31
Tompkins MM, Basgall EJ, Zamrini E, Hill WD (1997) Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigra neurons. Am J Pathol 150:119–131
Halliwall B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59:1609–1623. doi:10.1111/j.1471-4159.1992.tb10990.x
Jenner P, Olanow CW (1998) Understanding cell death in Parkinson’s disease. Ann Neurol 44(Suppl1):S72–S84
Soto-Otero R, Méndez-Álvarez E, Hermida-Ameijeiras Á, Muñoz-Patiño AM, Labandeira-Garcia JL (2000) Autoxidation and neurotoxicity of 6-hydrodopamine in the presence of some antioxidants: potential implication in relation to the pathogenesis of Parkinson’s disease. J Neurochem 74:1605–1612. doi:10.1046/j.1471-4159.2000.0741605.x
Graham D, Tiffany SM, Bell WR Jr, Gutknecht WF (1978) Autoxidation versus covalent binding of quinines as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol Pharmacol 14:644–653
Kuehl FA, Egan RW (1980) Prataglandins, arachidonic acid and inflammation. Science 210:978–984. doi:10.1126/science.6254151
Ishge K, Schubert D, Sagara Y (2001) Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med 30:433–446. doi:10.1016/S0891-5849(00)00498-6
Ni Y-C, Zhao B-L, Hou J-W, Xin W-J (1996) Protection of cerebellar neuron by Ginkgo-biloba extract against apoptosis induced by hydroxyl radicals. Neuronsci Lett 214:115–118. doi:10.1016/0304-3940(96)12897-4
Chen C, Wei T-T, Gao Z, Zhao B-L, Hou J-W, Xu H-B et al (1999) Different effects of the constituents of Egb-761 on apoptosis in rat cerebellar granule cells induced by hydroxyl radicals. Biochem Mol Biol Int 47:397–405
Xin W-J, Wei T-T, Chen C, Ni Y-C, Zhao B-L, Hou J-W (2000) Mechanisms of apoptosis in rat cerebellar granule cells induced by hydroxyl radicals and effects of Egb761 and its constitutes. Toxicology 148:103–110. doi:10.1016/S0300-483X(00)00200-6
Nanjo F, Goto K, Seto R, Suzuki M, Sakai M, Hara Y (1996) Scavenging effect of tea catechins and their derivatives on 1,1-diphenyl-2-picrylhdrazyl radical. Free Radic Biol Med 21:895–902. doi:10.1016/0891-5849(96)00237-7
Zhao B-L, Li X-J, He R-G, Cheng S-J, Xin W-J (1989) Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys 14:175–181
Guo Q, Zhao B-L, Li M-F, Shen S-R, Xin W-J (1996) Studies on protective mechanisms of four components of green tea polyphenols (GTP) against lipid peroxidation in synaptosomes. Biochim Biophys Acta 1304:210–222
Guo Q, Zhao B-L, Hou J-W, Xin W-J (1999) ESR study on the structure-antioxidant activiity relationship of tea catechins and their epimers. Biochim Biophys Acta 1427:13–23
Zhao B-L, Guo Q, Xin W-J (2001) Free radical scavenging by green tea polyphenols. Methods Enzymol 335:217–231. doi:10.1016/S0076-6879(01)35245-X
Nie G-J, Wei T-T, Zhao B-L (2001) Polyphenol protection of DNA against damage. Methods Enzymol 335:232–231244. doi:10.1016/S0076-6879(01)35246-1
Inanami O, Watanabe Y, Syuto B, Nakano M, Tsuji M, Kuwabara M (1998) Oral administration of (−) catechin protects against ischemia-reperfusion-induced neuronal death in the gerbil. Free Radic Res 29:359–365. doi:10.1080/10715769800300401
Yoneda T, Hiramatsu M, Skamoto N, Togasaki K, Komatsu M, Yamaguchi K (1995) Antioxidant effects of “β catechin”. Biochem Mol Biol Int 35:995–1008
Nie GJ, Jin C-F, Zhao B-L (2002) Distinct effects of tea catechins on 6-hydroxydopamine-induced apoptosis in PC12 cells. Arch Biochem Biophys 397:84–90. doi:10.1006/abbi.2001.2636
Nie GJ, Cao YL, Zhao BL (2002) Protective effects of green tea polyphenols and their major component, (−)-epigallocatechin-3-gallate (EGCG), on 6-hydroxyldopamine-induced apoptosis in PC12 cells. Redox Rep 7:170–177. doi:10.1179/135100002125000424
Guo S-H, Bezard E, Zhao B-L (2005) Protective effect of green tea polyphenols on the SH-SY5Y cells against 6-OHDA induced apoptosis through ROS-NO pathway. Free Radic Biol Med 39:682–695. doi:10.1016/j.freeradbiomed.2005.04.022
Guo S, Yan J, Bezard E, Yang T, Yang X, Zhao B (2007) Protective effects of green tea polyphenols in the 6-OHDA rat model of Parkinson’s disease through inhibition of ROS-NO pathway. Biol Psychiatry 62:1353–1362. doi:10.1016/j.biopsych.2007.04.020
Levites Y, Weinreb O, Maor G, Youdim MBH, Mandel S (2001) Green tea polyphenol epigallocatechin-3-gallate prevents MPTP induced dopaminergic neurodegeneration. J Neurochem 78:1073–1082. doi:10.1046/j.1471-4159.2001.00490.x
Levites Y, Youdima MBH, Mao G, Mandel S (2002) Attenuation of 6-OHDA-induced nuclear factor-NF-kB activation and cell death by tea extracts in neuronal cultures. Biochem Pharmacol 63:21–29. doi:10.1016/S0006-2952(01)00813-9
Levites Y, Amit T, Youdim MBH, Mandel S (2002) Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol-epigallocatechin 3-gallate neuroprotective action. J Biol Chem 77:30574–30580. doi:10.1074/jbc.M202832200
Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou H, Jeanniton D et al (2005) Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci 25:8807–8814. doi:10.1523/JNEUROSCI.1521-05.2005
Zhang Y, Zhao BL (2003) Green tea polyphenols enhance sodium nitroprusside induced neurotoxicity in human neuroblastoma SH-SY5Y cells. J Neurochem 86:1189–1200
Kurzer MS, Xu X (1997) Dietary phytoestrogens. Annu Rev Nutr 17:353–381. doi:10.1146/annurev.nutr.17.1.353
Chan W-H, Yu J-S (2000) Inhibition of UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermal carcinoma A431 cells by genistein. J Cell Biochem 78:73–84. doi :10.1002/(SICI)1097-4644(20000701)78:1<;73::AID-JCB7>;3.0.CO;2-P
Johnson KL, Vaillant F, Lawen A (1996) Protein tyrosine kinase inhibitors prevent didemnin B-induced apoptosis in HL-60 cells. FEBS Lett 383:1–5. doi:10.1016/0014-5793(96)00203-7
Qiong G, Gerald R, Hadi M, Stefan W, Lester P (2002) ESR and cell culture studies on free radical-scavenging and antioxidant activities of isoflavonoids. Toxicol 179:171–180. doi:10.1016/S0300-483X(02)00241-X
Ohigashi T, Ueno M, Nonnnaka S, Nakanoma T, Furukawa Y, Deguchi N et al (2000) Tyrosin kinase inhibitors reduce bcl-2 expression and induced apoptosis in androgen-depent cells. Am J Physiol 278:C66–C72
Helen K, Hong L, Lin L, John G (2000) Attenuation of neurodegeneration-relevant modifications of brain proteins by dietary soy. Biofactors 12:243–250
Clarkson TB, Anthony MS, Williams JK, Honore EK, Cline JM (1997) The potential of soybeen phytoestrogens for postmenopausal hormone replacement therapy. Proc Soc Exp Biol Med 217:365–368
Lamartiniere CA, Fritz WA (1998) Genistein: chemoprevention, in vivo mechanism of action, and bioavaliability. Korea Soybean Dig 15:60–80
Andersen JM, Myhre O, Fonnum F (2003) Discussion of the role of the extracellular signal-regulated kinase-phospholipase A2 pathway in production of reactive oxygen species in Alzheimer’s disease. Neurochem Res 28:319–326. doi:10.1023/A:1022389503105
Chang HC, Churchwell MI, Delclos KB, Newbold RR, Doerge DR (2000) Mass spectrometric determination of genistein tissue distribution in diet-exposed Sprague–Dawley rats. J Nutr 130:1963–1970
Zeng HY, Chen Q, Zhao B-L (2004) Genistein ameliorated β-amyloid peptide-induced hippocampal neuronal apoptosis. Free Radic Biol Med 36:180–188. doi:10.1016/j.freeradbiomed.2003.10.018
Gutierrez-Zepeda A, Santell R, Wu R, Brown M, Wu Y, Khan I et al (2005) Soy isoflavone glycitein protects against beta amyloid-induced toxicity and oxidative stress in transgenic Caenorhabditis elegans. BMC Neurosci 6(54):1–9
Liu Q, Tao Y, Zhao B-L (2003) ESR study on scavenging effect of nicotine on free radicals. Appl Magn Reson 24:105–112
Liu Q, Zhao B-L (2004) Nicotine attenuates β-amyloid peptide induced neurotoxicity, free radical and calcium accumulation in hippocampal neuronal cultures. Br J Pharmacol 141:746–754. doi:10.1038/sj.bjp.0705653
Xie Y, Bezard E, Zhao B-L (2005) Unraveling the receptor- independent neuroprotective mechanism in mitochondria. J Biol Chem 37:32405–32412. doi:10.1074/jbc.M504664200
Zhang J, Liu Q, Liu N, Li F, Chen Q, Qin C, Zhu H, Huang Y, Zhao B-L (2006) Nicotine reduces β-amyloidosis by regulating metal homeostasis. FASEB J 20:1212–1214. doi:10.1096/fj.05-5214fje
Liu Q, Zhang J, Zhu H, Qin C, Chen Q, Zhao B-L (2007) Dissecting the signalling pathway of nicotine-mediated neuroprotection in a mouse Alzheimer disease model. FASEB J 21:61–73. doi:10.1096/fj.06-5841com
Nordberg A, m-Lindahl EH, Lee M, Johnson M, Mousavi M, Perry RHE, Bednar I, Court J (2002) Chronic nicotine treatment reduces b-amyloidosis in the brain of a mouse model of Alzheimer’s disease. J Neurochem 81:655–658. doi:10.1046/j.1471-4159.2002.00874.x
Hellstrom-Lindahl E, Court J, Keverne J, Svedberg M, Lee M, Marutle A et al (2004) Nicotine reduces Aβ in the brain and cerebral vessels of APPsw mice. Eur J Neurosci 19:2703–2710. doi:10.1111/j.0953-816X.2004.03377.x
Kaiser S, Wonnacott S (1998) Nicotinic receptor modulation of neurotransmitter release. In: Arneric SP, Brioni JD (eds) Neuronal nicotinic receptors: pharmacology and therapeutic opportunities. Wiley, New York, pp 141–159
Shimohama S, Greenwald DL, Shafron DH, Akaike A, Maeda T, Kaneko S (1998) Nicotinic receptor-mediated protection against β-amyloid neurotoxicity. Brain Res 779:359–363. doi:10.1016/S0006-8993(97)00194-7
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (29935080), 973(2006CB500706), Key Laboratory of Mental Health, Chinese Academy of Sciences, the E-Institutes of Shanghai Municipal Education Commission (Project No. E-04010). I sincerely thank professor Jiankang Liu for his suggestions and comments to this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue in honor of Dr. Akitane Mori.
Rights and permissions
About this article
Cite this article
Zhao, B. Natural Antioxidants Protect Neurons in Alzheimer’s Disease and Parkinson’s Disease. Neurochem Res 34, 630–638 (2009). https://doi.org/10.1007/s11064-008-9900-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-008-9900-9