
Abstract
The predominant molecular symptom of aging is the accumulation of altered gene products. Moreover, several conditions including protein, lipid or glucose oxidation disrupt redox homeostasis and lead to accumulation of unfolded or misfolded proteins in the aging brain. Alzheimer’s and Parkinson’s diseases or Friedreich ataxia are neurological diseases sharing, as a common denominator, production of abnormal proteins, mitochondrial dysfunction and oxidative stress, which contribute to the pathogenesis of these so called “protein conformational diseases”. The central nervous system has evolved the conserved mechanism of unfolded protein response to cope with the accumulation of misfolded proteins. As one of the main intracellular redox systems involved in neuroprotection, the vitagene system is emerging as a neurohormetic potential target for novel cytoprotective interventions. Vitagenes encode for cytoprotective heat shock proteins (Hsp) Hsp70 and heme oxygenase-1, as well as thioredoxin reductase and sirtuins. Nutritional studies show that ageing in animals can be significantly influenced by dietary restriction. Thus, the impact of dietary factors on health and longevity is an increasingly appreciated area of research. Reducing energy intake by controlled caloric restriction or intermittent fasting increases lifespan and protects various tissues against disease. Genetics has revealed that ageing may be controlled by changes in intracellular NAD/NADH ratio regulating sirtuin, a group of proteins linked to aging, metabolism and stress tolerance in several organisms. Recent findings suggest that several phytochemicals exhibit biphasic dose responses on cells with low doses activating signaling pathways that result in increased expression of vitagenes encoding survival proteins, as in the case of the Keap1/Nrf2/ARE pathway activated by curcumin and NAD/NADH-sirtuin-1 activated by resveratrol. Consistently, the neuroprotective roles of dietary antioxidants including curcumin, acetyl-l-carnitine and carnosine have been demonstrated through the activation of these redox-sensitive intracellular pathways. Although the notion that stress proteins are neuroprotective is broadly accepted, still much work needs to be done in order to associate neuroprotection with specific pattern of stress responses. In this review the importance of vitagenes in the cellular stress response and the potential use of dietary antioxidants in the prevention and treatment of neurodegenerative disorders is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The brain has a large potential oxidative capacity but a limited ability to counteract oxidative stress [1–3]. Within the cell, reactive oxygen species (ROS) are physiologically present at minimal concentration as by-products of aerobic metabolism as well as second messengers in many signal transduction pathways and, in normal conditions, there is a steady-state balance between pro-oxidants and antioxidants which is necessary to ensure optimal efficiency of antioxidant defenses [4–7]. However, when the rate of free radical generation exceeds the capacity of antioxidant defenses, oxidative stress ensues with consequential severe damage to DNA, proteins and lipids [8–10] (Fig. 1). Oxidative stress has been implicated in mechanisms leading to neuronal cell injury in various pathological states of the brain, including neurodegenerative disorders such as Alzheimer’s disease (AD) [11–15]. Recently the term “nitrosative stress” has been used to indicate the cellular damage elicited by nitric oxide and its congeners peroxynitrite, N2O3, nitroxyl anion and nitrosonium (all can be indicated as reactive nitrogen species or RNS) [16–18].

Reactive oxygen species as second messengers. ROS at minimal concentration may act as second messengers in many signal transduction pathways and, under normal conditions, a steady-state balance between pro-oxidants and antioxidants exists. Conversely, when the rate of free radical generation exceeds the capacity of antioxidant defenses, oxidative and/or nitrosative stress ensues with consequential severe damage to DNA, protein and lipids. Among the cellular pathways involved in protection against oxidative and nitrosative stress, antioxidant enzymes such as superoxide dismutase, glutathione (GSH)-peroxidase, GSH-S-transferase, GSSG reductase and the vitagenes heat shock protein 70 (Hsp70), heme oxygenase-1 (HO-1), thioredoxin reductase (TrxR) and sirtuins (SIRTs) actively operate counteracting deleterious consequences of free radical damage. GSSG, oxidized glutathione
From a molecular point of view, in the central nervous system (CNS) cells are able to fight against oxidant stress using many resources, including vitamins (A, C and E), bioactive molecules (glutathione, thioredoxin, flavonoids), lipoic acid, enzymes (heat shock protein-32, superoxide dismutase, catalase, glutathione peroxidases, thioredoxin reductase, etc) and redox sensitive protein transcriptional factors (AP-1, NF-κB, Nrf2, HSF, etc) [19–21]. The heat shock proteins (Hsps) are one of the more studied defense system active against cellular damage (Fig. 1).
The idea of the pervasive nature of free radicals has been firmly entrenched in the minds of scientists ever since the group of Britton Chance [22] developed the basic biochemical techniques to show that in the resting state 2% of all oxygen consumed by cells is converted into ROS rather than water. McCord and Fridovich first described superoxide dismutase thus suggesting a physiological role of superoxide [23]. Although, there is now an appreciation that the physiological generation of ROS is likely to be an order of magnitude less, their impact on biomolecules has been amply documented. In response to this assault, the cell has developed a number of antioxidant defence systems such as superoxide dismutase, the peroxidases, the glutathione redox cycle with its associated constitutive enzymes as well as glutathione itself, whose concentration is higher in the cell than that of glucose [22]. Therefore, the cell has become well equipped to cope with the normal production of reactive species. There is growing evidence that the continuous presence of a small stimulus such as low concentrations of ROS is in fact able to induce the expression of antioxidant enzymes and other defence mechanisms. The basis for this phenomenon may be encompassed by the concept of hormesis [24], which can be characterized as a particular dose–response relationship in which a low dose of a substance is stimulatory and a high dose is inhibitory. In this context, radicals may be considered to be beneficial since they act as signals to enhance defences rather than deleterious as they are when cells are exposed to high levels of ROS. On the other hand when in excess can, over long term, disrupt redox homeostasis, impose oxidative stress and subsequently lead to a dramatic loss of molecular fidelity which is the major cause for accumulation of unfolded or misfolded proteins in brain cells. Alzheimer’s (AD), Parkinson’s (PD), and Huntington’s diseases, but also amyotrophic lateral sclerosis and Friedreich ataxia belong to the so called “protein conformational diseases” and affect several millions of aged people in all the world [21]. Cells have evolved mechanisms such as the unfolded protein response, where chaperones can rescue misfolded proteins by breaking up aggregates and assisting the refolding process, while proteins that cannot be rescued by refolding are delivered to the proteasome by other chaperones to be recycled [25]. In general, an unfolded protein response conformational diseases are conditions that arise from the dysfunctional aggregation of proteins in non-native conformations. This often is associated with multiple metabolic derangements that result in the excessive production of ROS and oxidative stress [25]. The ability of a cell to deal with ROS and RNS requires the activation of pro-survival pathways as well as the production of molecules endowed with anti-oxidant and anti-apoptotic activities. Among the cellular pathways involved in protection against oxidative and nitrosative stress, the heat shock proteins family plays a key role, in particular in brain cells. Hsp70, Hsp60, Hsp27 and ubiquitin are functional chaperones triggering a cascade of intracellular cytoprotective events [6, 7, 19–22, 26]. Heme oxygenase-1 (HO-1), also referred to as Hsp32, belongs to the Hsp family and protects brain cells from oxidative stress by degrading toxic heme into free iron, carbon monoxide and biliverdin [21, 26–28]. This latter is then reduced by biliverdin reductase (BVR) into bilirubin (BR), a linear tetrapyrrole with antioxidant properties; very recently, BR has been shown to effectively counteract also nitrosative stress, due to its ability to bind and inactivate NO and RNS [29–32].
In this paper we describe the recent discoveries about the biochemical changes occurring in the CNS when brain cells are exposed to chronic oxidative insult, as well as the function of members of the vitagene system, such as HO-1, thioredoxin and sirtuins in modulating the onset and progression of major neurodegenerative diseases such as, AD, PD or FRDA. In addition, the key role played by the heat shock response, particularly HO-1 and Hsp70 pathways, as well as sirtuins as potential target for nutritional interventions are discussed. Although the notion that stress proteins are neuroprotective is broadly accepted, still much work needs to be done in order to associate neuroprotection with specific patterns of stress responses. Emerging evidence underscores the high potential of the vitagene system as a target for new neuroprotective strategies, especially those aimed at minimizing deleterious consequences associated with oxidative stress, such as in neurodegenerative disorders and brain aging. We review here also the evidence for the role of some dietary antioxidants in modulating redox-dependent mechanisms leading to up-regulation of vitagenes in brain, and hence potentiate brain stress tolerance.
Vitagenes
Vitagenes are a group of genes strictly involved in preserving cellular homeostasis during stressful conditions. The vitagene family is composed of the heat shock proteins (Hsp) HO-1/Hsp32, Hsp70, by the thioredoxin system [19–21] and by sirtuin proteins (Fig. 2). Among these genes HO-1 is receiving considerable attention because of its major role in counteracting both oxidative and nitrosative stress. In fact, HO-1 induction is one of the early events in the cell response to stress. Heme oxygenase-1 exerts protective role, by degrading the intracellular levels of pro-oxidant heme and by producing biliverdin, the precursor of bilirubin, this latter being an endogenous molecule with potent antioxidant and antinitrosative features [21, 26–32] (Fig. 3).

The cellular stress response mediated by vitagenes and the concept of hormesis. Hormesis represents an evolutionary-based adaptive responses to environmentally induced disruptions in homeostasis. Cellular signaling pathways and molecular mechanisms that mediate hormetic responses typically involve antioxidant enzymes and transcription factors such as Nrf-2 and NF-κB. As a result, cells increase their production of cytoprotective and restorative proteins including growth factors, phase 2 and antioxidant enzymes, and protein chaperones. Under conditions of oxidative/nitrosative stress, there is an increased formation of ROS or RNS. They play a key role in the pathogenesis of free radical-induced diseases, such as neurodegenerative disorders. Polyphenols are strong inducers of vitagenes heme oxygenase-1 (HO-1), heat shock protein 70 (Hsp70), thioredoxin reductase and sirtuins. Vitagenes, either individually or by acting in concert, contribute to counteract the NF-κB-dependent ROS/RNS-mediated damage thus originating a neuroprotective response. NF-κB inhibition might also have profound impact on cancerogenesis, modulating tumor cell survival and proliferation. BV, biliverdin; BVR, biliverdin reductase; BR, bilirubin; c-FLIP: cellular FLICE-like inhibitory protein; IAP-1: inhibitor of apoptosis protein 1; IAP-2: inhibitor of apoptosis protein 2; XIAP: X-linked inhibitor of apoptosis protein; 5-LOX: 5-lipooxygenase; COX-2: cyclooxygenase-2; IL-6: interleukin-6; MMP-9: matrix metalloproteinase-9; Epo: erythropoietin; VEGF: Vascular Endothelial Growth Factor; ROS: Reactive oxygen species; iNOS: inducible nitric oxide synthase; TNF: tumor necrosis factor; HIF-1: Hypoxia Inducible Factor-1; NF-κB: nuclear factor κB; Hsp70: heat shock protein 70; TRXr: thioredoxin reductase; HO-1: heme oxygenase-1

Vitagenes and the pathway of cellular stress response. Environmental stressors (metabolic or physical stress, heavy metals, cytokines, etc.) or pathophysiological conditions of oxidant antioxidant balance perturbation (inflammation, neuronal damage, ischemia) are all situations associated with increased oxidative or nitrosative stress. Vitagenes, such as heme oxygenase, Hsp70, thioredoxin reductase and sirtuins represent an integrated system for cellular stress tolerance which can be activated by nutritional antioxidants including curcumin, resveratrol, acetyl-l-carnitine or carnosine. Acetyl-l-carnitine and polyphenols act through the activation of transcription factor Nrf2, which after binding to the ARE (antioxidant responsive element) in the HO-1 gene, up-regulates both HO-1 and TrxR, thus counteracting pro-oxidant conditions. In addition, carnosine has been shown to inhibit the induction of both HO-1 and Hsp70 following strong nitrosative conditions. In the same figure are indicated the redox cycling between bilirubin/ biliverdin as well as oxidized/reduced TrxR. OxS, oxidative stress; NitroS, nitrosative stress; HO-1, heme oxygenase-1; TrxR, thioredoxin reductase; Trx, Thioredoxin, SH: reduced form, S-S: oxidized form
Heme Oxygenase-1
The mechanisms responsible for neuronal death are not completely elucidated, even if many studies suggest that ROS are primarily involved in the genesis of neurodegenerative disorders [11, 12, 33–35]. Due to its strong antioxidant properties and wide distribution within the CNS, HO-1 has been proposed as a key enzyme in the prevention of brain damage [26–29, 36]. Panahian et al., using transgenic mice over-expressing HO-1 in neurons, demonstrated the neuroprotective effect of this enzyme in an experimental model of ischemic brain damage [37]. The neuroprotective effects of over-expressed HO-1 has been attributed to several factors such as: increased level of both cGMP and bcl-2 in neurons, inactivation of the pro-apoptotic transcription factor p53, increase in antioxidant sources in the iron sequestering protein, ferritin [37]. Particularly interesting is the role played by HO-1 in AD, a neurodegenerative disorder which involves a chronic inflammatory response associated with both oxidative brain injury and β-amyloid associated pathology. Significant increases in the levels of HO-1 have been observed in AD brains in association with neurofibrillary tangles and HO-1 mRNA was found to be increased in AD neocortex and cerebral vessels [38, 39]. HO-1 increase was not only associated with neurofibrillary tangles, but also co-localized with senile plaques and glial fibrillary acidic protein-positive astrocytes in AD brains [40]. Therefore, it is plausible to conclude that the dramatic increase in HO-1 in AD may be a direct response to an increase in free heme concentrations, associated with neurodegeneration, and can be considered as an attempt of brain cells to convert the highly toxic heme into the antioxidant BR. The protective role played by HO-1 and its products in AD prompted investigators to propose natural substances, which are able to increase HO-1 levels, as potential drugs for the treatment of AD. In this light, several in vitro studies have been focused on polyphenolic compounds contained in some herbs and spices, e.g. curcumin [41]. Curcumin is the active anti-oxidant principle in Curcuma longa, a colouring agent and food additive commonly used in Indian culinary preparations. In vitro, this polyphenolic substance has the potential to inhibit lipid peroxidation and to effectively intercept and neutralize ROS and RNS [42]. In addition, curcumin has been shown to significantly increase HO-1 in brain cells [43]. This latter effect on HO-1 can explain, at least in part, the anti-oxidant properties of curcumin, in particular keeping in mind that HO-1-derived BR has the ability to scavenge both ROS and RNS [29–32]. A single epidemiological study suggested that curcumin, as one of the most prevalent nutritional and medicinal compounds used by the Indian population, is responsible for the reduced (4.4-fold) prevalence of AD in India compared to United States [44]. Based on these findings, Lim and colleagues have provided evidence that dietary curcumin given to an Alzheimer transgenic APPSw mouse model (Tg2576) for 6 months resulted in a suppression of indices of inflammation and oxidative damage in the brain of these mice [45]. Furthermore, in a human neuroblastoma cell line curcumin inhibits NF-κB activation, efficiently preventing neuronal cell death [42].
Carbon monoxide (CO) is the gaseous products of HO and it has been found to play a role in several biological phenomena, including hippocampal long-term potentiation, non-adrenergic non-cholinergic gastrointestinal relaxation and vasodilatation, and is currently regarded as a neuromodulator in the peripheral and central nervous system (for extensive reviews on CO and its functions in the nervous system, see [28, 46] (Fig. 3). Evidence from in vitro and in vivo studies suggests that the HO-CO pathway is involved in the modulation of the neuroendocrine mechanism of stress. Thus, increased CO generation is clearly associated with the inhibition of K+ stimulated arginine vasopressin (AVP) and oxytocin release from rat hypothalamic explants, whereas the inhibition of HO activity significantly potentiates the LPS-induced increase in AVP circulating levels while reducing the hypothalamic content of this neuropeptide [47–49]. With regards to corticotropin-releasing hormone (CRH), the effects of CO on the release of this hormone are contradictory, since increases in CO generation induced by two HO substrates, hematin and hemin, were associated with reduced or enhanced CRH release respectively, in two different in vitro models [50, 51]. As far as the intracellular mechanism(s) by which CO exerts its biological functions, it is generally agreed that this gas activates the cytosolic form of guanylyl cyclase (sGC) which in turn increases intracellular cGMP levels [27]. However during the last 10 years many studies demonstrated that CO signals through the activation of alternative intracellular signal transduction pathways. Studies from our laboratory suggested that the activation of another hemoprotein, cyclooxygenase (COX), plays a significant role in CO signaling in the rat hypothalamus. In these studies we demonstrated that hemin, the precursor of CO via HO, dose-dependently increases PGE2 production from rat hypothalamus in vitro and this effect is specifically due to CO because it is counteracted by the HO inhibitor Sn-mesoporphyrin-IX and oxyhemoglobin, the latter being a well known scavenger for CO [52]. The direct evidence about the stimulatory role of CO on PGs production was obtained incubating hypothalami directly in CO saturated solutions and measuring significantly increased PGE2 levels with respect to control tissue [53]. Jaggar and colleagues demonstrated that exogenous or endogenously produced CO dilates cerebral arterioles by directly activating large-conductance Ca2+-activated K+ (KCa) channels primarily by increasing the coupling ratio and amplitude relationship between Ca2+ sparks and KCa channels [54]. Although CO is a potent and effective activator of KCa channels, the gas does not dilate arterioles in the absence of Ca2+ sparks. Therefore, CO appears to act by priming KCa channels for activation by Ca2+ sparks, and this ultimately leads to arteriole dilation via membrane hyperpolarization [54]. Finally, Otterbein and colleagues have shown that in organs and tissues different from brain, CO exerts anti-inflammatory and anti-apoptotic effects dependent on the modulation of the p38 MAPK-signaling pathway [55]. By virtue of these effects, CO confers protection in oxidative lung injury models, and likely plays a role in HO-1 mediated tissue protection [56].
Heat Shock Protein 70
The 70 kDa family of stress proteins is one of the most extensively studied. Included in this family are Hsc70 (heat shock cognate, the constitutive form), Hsp70 (the inducible form, also referred to as Hsp72) and GRP-75 (a constitutively expressed glucose-regulated protein found in the endoplasmic reticulum) [26, 57].
Only recently, the availability of transgenic animals and gene transfer allowed us to over-express the gene encoding for Hsp70, thus demonstrating that overproduction of this protein leads to protection in several different models of nervous system injury [58–60]. Following focal cerebral ischemia, Hsp70 mRNA is synthesized in most ischemic cells except in areas of very low blood flow, due to scarce ATP levels. Hsp70 proteins are produced mainly in endothelial cells, in the core of infarcts in the cells that are most resistant to ischemia, in glial cells at the edges of infarcts and in neurons outside the areas of infarction [61]. It has been suggested that this neuronal expression of Hsp70 outside an infarct can be used to define the ischemic penumbras, which means the zone of protein denaturation in the ischemic areas [61].
As mentioned above, Hsps are induced in many neurodegenerative disorders mainly in the view of its cytoprotective function. Hsp72 was overexpressed in post-mortem cortical tissue of AD patients and an increase in Hsp70 mRNA was found in cerebellum hippocampus and cortex of AD patients during the agonal phase of the disease [62–64]. Recently Kakimura et al. demonstrated that Hsp70 induces IL-6 and TNF-α in microglial cells and this event is associated with an increased phagocytosis and clearance of Aβ peptides [65] (Figs. 2, 3). The same authors hypothesize that Hsps could activate microglial cells through NF-κB and p-38 MAPK-dependent pathways [65].
A large body of evidence now suggests a correlation between mechanisms of nitrosative stress and Hsp induction. We have demonstrated in astroglial cell cultures that cytokine-induced nitrosative stress is associated with an increased synthesis of Hsp70 stress proteins. The molecular mechanisms regulating the NO-induced activation of heat-shock signal seems to involve cellular oxidant/antioxidant balance, mainly represented by the glutathione status and the antioxidant enzymes [66, 67].
Thioredoxin/Thioredoxin Reductase
The thioredoxin system, originally identified in Escherichia coli, in 1964, as a hydrogen donor for ribonucleotide reductase required for DNA synthesis, plays a key role in cell function by limiting oxidative stress directly via antioxidant effects and indirectly by protein–protein interactions [68]. It is well established that, in mammals, cellular redox regulation of many processes is provided by the cooperation between the Trx and glutathione systems [69]. In fact, Trx and GSH systems are involved in a variety of redox-dependent pathways such as supplying reducing equivalents for ribonucleotide reductase, and peptide methionine sulfoxide reductase, the latter being involved in antioxidant defence and regulation of the cellular redox state [70]. Therefore, Trx and GSH form a powerful system controlling redox regulation of gene expression, signal transduction, cell proliferation, protection against oxidative stress, anti-apoptotic functions, growth factor and co-cytokine effects, as well as regulation of the redox state of the extracellular environment [71]. The promoter of the Trx gene contains a series of stress-responsive elements, various transcription factor binding sites, such as SP1, AP-1, NF-κB, and the antioxidant-response element (ARE) [72–74] (Fig. 3). Importantly, induction of thioredoxin reductase and glutathione has been demonstrated to occur in parallel with other ARE-dependent phase 2 cytoprotective genes in several experimental systems, e.g., in cortical astrocytes, in human hepatoma cells and in human keratinocytes [75–77]. Similarly to induction of HO-1 gene expression, the ARE-mediated Trx-1 induction involves transcription factor Nrf2 [78] (Figs. 3, 4).

Mechanism of induction of cytoprotective phase 2 genes. Under basal conditions transcription factor Nrf2 is bound to the inducer sensor, the dimeric protein Keap1, and targeted for ubiquitination and proteasomal degradation via association with the Cullin 3-based E3 ubiquitin ligase complex. Inducers modify highly reactive cysteine residues of Keap1 which loses its ability to target Nrf2 for degradation. Consequently, Nrf2 is stabilized and migrates to the nucleus where it binds to the ARE (as a heterodimer with a small Maf transcription factor), and activated transcription of cytoprotective phase 2 genes
Importantly, it has been reported that Trx is constitutively present as a surface-associated sulfhydryl protein in plasma membrane of a wide range of cells [79]. Many physicochemical stimuli, such as UV irradiation and hydrogen peroxide, have been shown to induce Trx expression and secretion, as a redox-sensitive molecule with cytokine-like and chemokine-like activities to prevent cell injury against oxidative stress [70]. In addition to UV irradiation, treatment of cells in culture with phorbol esters, hydrogen peroxide, hypoxia, the cancer drug cisplatin and hemin has been reported to cause the translocation of Trx from the cytoplasm to the nucleus, where it regulates the redox-activation and DNA binding activity of critical transcription factors (Jun, Fos, p53, CREB, PEBP2/CBF, Myb), all involved in fundamental processes, such as gene expression, cell growth and apoptosis [79]. Thioredoxin plasma levels in normal individuals vary between 20 and 30 ng/ml [80, 81] and increase in certain human diseases including HIV infection and cancer [80, 82]. Noteworthy, several studies reported increased Trx-1 expression in many human primary cancers and tumor cell lines, including astrocytic brain tumors [83, 84]. Elevated Trx levels may contribute to increased cancer cell proliferation and resistance to chemotherapy by several mechanisms as the stimulation of DNA synthesis and the activation of redox-modulated transcription factors [79, 85]. Recent work suggests that Trx-1 is involved in nerve growth factor (NGF) signaling pathways [86]. NGF, a neurotrophic factor regulating development, maintenance and function of the CNS, has been shown to activate Trx-1 expression via cyclic AMP (cAMP)-response elements (CREs) present in the Trx-1 gene promoter, and also to induce nuclear translocation of Trx1 [87]. Recent data suggest that, beyond its ability to regulate the function of proteins through thiol-disulfide exchange reactions, Trx and its substrates may also have beneficial effects during oxidative stress by upregulating HO-1, with important cytoprotective pleiotropic effects deriving from heme degradation and bilirubin formation [88, 89]. Besides the role as a source of reducing equivalents, Trx per se acts as antioxidant or ROS scavenger. In fact, Trx eliminates singlet oxygen, hydroxyl radical and hydrogen peroxide [90]. It has also been reported that some of the neuroprotective effects of GSNO on beta-amyloid- or ferrous citrate-induced toxicity in rat cortical neurons or in rat substantia nigra can be due to the activation of multiple signalling pathways including thioredoxin [91, 92]. Interestingly, the interaction between Trx and GSNO seems to involve both the activation of sGC and the following cGMP generation and a direct S-nitrosylation reaction [92]. Finally, the NO-dependent expression of Trx has been shown to be involved in the neuroprotection against oxidative stress mediated by estrogens [93].
Sirtuins
The sirtuins are a group of proteins linked to aging, metabolism and stress tolerance in several organisms. In mammalian cells seven sirtuins have been identified. SIRT1, 2, 3, 6 and possibly 5 are NAD-dependent deacetylases (Fig. 5), SIRT4 and 6 are ADP-ribosyltransferases (Fig. 5), and the activity of SIRT7 has not been defined [94]. The sirtuin family of histone deacetylases (HDACs) was named after their homology to the Saccharomyces cerevisiae gene silent information regulator 2 (Sir2). In the yeast, Sir2 has been shown to mediate the effects of caloric restriction on the extension of life span, with high levels of Sir2 activity promoting longevity [95]. Like their yeast homologs, the mammalian sirtuins (SIRT1-7) are class III HDACs and require NAD+ as a cofactor to deacetylate substrates ranging from histones to transcriptional regulators. Through this activity, sirtuins are shown to regulate important biological processes, such as apoptosis, cell differentiation, energy transduction or glucose homeostasis (Fig. 6) [96]. In particular, the NAD+/NADH ratio can be considered as a “biochemical sensor” to evaluate the energetic status of the cell; in fact, among the several mechanisms through which dietary antioxidants may be useful for tissues, it is noteworthy to mention the improvement of metabolic conditions secondary to pro-inflammatory damage [96]. In this light, the interaction between NAD+/NADH and the members of the sirtuin family, puts in a single frame the cytoprotective activity of dietary antioxidants through the regulation of both cellular redox and metabolic state [96]. Since the Sir2 family of proteins exert their enzymatic activity not only on histones but also on numerous other proteins, including transcriptional factors, they are involved in many cellular processes, e.g., gene silencing, DNA repair, progression of the cell cycle, whereby controlling the mechanism of cellular ageing [96]. Sirtuin-mediated deacetylation and ADP-ribosylation are related in that both cleave NAD as the initial chemical step of the reaction cycle, as shown in Fig. 5. In deacetylation, the ADP-ribosyl transfer directly participates in the removal of the acetyl group from the protein substrate to generate 2,3-O-acetyl-ADP-ribose, whereas in ribosylation, the ADP ribosyl moiety is transferred to the protein substrate. Deacetylation of sirtuin substrates can inhibit or induce their activities, whereas ADP-ribosylation has only been shown to be inhibitory. [94]. The connection between the biochemical activation of Sir2 orthologs through NAD-dependent protein deacetylases and the involvement of NAD/NADH in several metabolic reactions within cells prompted the hypothesis of a biochemical relationship between diet, metabolism and aging processes (Fig. 7). In fact, many studies in Saccharomyces cerevisiae (yeast), Caenorhabditis elegans (worms), Drosophila melanogaster (fly) and rodents have shown that caloric restriction (CR) extends lifespan, and sirtuins are considered to mediate, at least partly, this effect [97, 98]. Most interestingly, overexpression of the gene encoding for Sir2 protein leads to a decrease in histone acetylation and, correspondingly, to an increase in life span in yeast, in the nematode C. elegans and in metazoans. Similarly, Sir2-activating compounds (STACs), such as resveratrol, promote longevity in yeast and other organisms such as worm, drosophila or mouse. In addition, both mutations of the Sir2 gene and pharmacological inhibition of Sir2 protein by nicotinamide induces an acceleration of ageing in yeast, and also SIRT1 knockout mice fail to display a prominent phenotype of CR (i.e. increased physical activity). Sirtuins apparently mediate their life-extending effects in different organisms by targeting different pathways. In mammals, SIRT1 deacetylates many key transcription factors and co-factors, such as p53, FOXO (forkhead) proteins, peroxisome proliferation activating receptor (PPAR)-gamma co-activator-1a (PGC-1a) and NF-κB, thereby affecting crucial cellular pathways involved in cellular stress resistance and metabolism (Figs. 5, 6), thus supporting the hypothesis that this class of conserved proteins are potential vitagenes [95] (Fig. 2). There are several factors supporting the action of sirtuins in mediating salutary physiological effects of CR in mammals. Oxidant/antioxidant balance perturbation and oxidative stress can induce sirtuin expression. It has been shown that hydrogen peroxide treatment increases SIRT2 expression. SIRT2 was found able to bind to FOXO3a and reduce its acetylation level, leading to an increase of FOXO DNA binding and to an elevation of the expression of FOXO target genes, such as manganese superoxide dismutase. Forkhead transcription factors of the FOXO subfamily are transactivators involved in growth control, differentiation and apoptosis [99]. SIRT1 targets FOXO3a and FOXO4 and represses their transcriptional activity in a deacetylation-dependent manner. Consistently, SIRT1 reduces FOXO4-triggered apoptosis, and the expression levels of FOXO-activated genes are higher in Sirt1−/− mice [100, 101]. Additional work shows that SIRT1-dependent FOXO3 deacetylation reduces the expression of proapoptotic genes and the ensuing cell death, whereas it favours cell cycle arrest and the expression of genes involved in stress resistance in eukaryotic cells. In keeping with this, deacetylation of FOXO1 by SIRT1 promotes the expression of p27kip1 and manganese superoxide dismutase (MnSOD). As a consequence, SIRT1 and SIRT2 upregulation is associated to a decrease in cellular levels of ROS [102]. Sirtuins have been shown to regulate important biological processes ranging from fat and glucose metabolism in mammals and cell survival [103]. Sirt1+/− mice did not mobilize as much fat to the blood as wild-type after overnight fasting showing a role of this sirtuin in fat mobilization [100, 101], as it has been demonstrated that the Sir2 homologue SIRT1, which modulates ageing in several species, controls the gluconeogenic/glycolytic pathways in liver in response to fasting signals through the transcriptional coactivator PGC-1alpha. They found that once SIRT1 is induced, it interacts with and deacetylates PGC-1alpha at specific lysine residues in an NAD(+)-dependent manner and induces gluconeogenic genes and hepatic glucose output, but does not regulate the effects of PGC-1alpha on mitochondrial genes [100, 101]. Several studies have determined a role for the human SIRT1 protein in cell survival. SIRT1 specifically associates with the p53 tumor suppressor protein and deacetylates it, resulting in negative regulation of p53-mediated transcriptional activation. Importantly, p53 deacetylation by SIRT1 also prevents cellular senescence and apoptosis induced by DNA damage and stress [104]. The identification of histones H3 and H4 as substrates for SIRT2 suggests a broader role in the cell, through transcription regulation. Additionally, Sir2 the yeast SIRT1 homologous, has been shown to mediate the effects of caloric restriction on the extension of life span, with high levels of Sir2 activity promoting longevity [96].

Sirtuin biochemistry. Sirtuin-mediated deacetylation and ADP-ribosylation are related in that both cleave NAD as the initial chemical step of the reaction cycle. In deacetylation, the ADP-ribosyl transfer directly participates in the removal of the acetyl group from the protein substrate to generate 2,3-O-acetyl-ADP-ribose, whereas in ribosylation, the ADP ribosyl moiety is transferred to the protein substrate. Free tryptophan is used as the precursor of NAD via the kynurenine pathway

Sirtuin activation and neuroprotection. Caloric restriction or nutritional interventions with SIRT activating compounds, such as polyphenols, induce up-regulation of SIRT1. Activated SIRT1 suppresses Nf-κB , UCP-2 (uncoupling protein 2) and PPARγ (peroxisome proliferator-activated receptor gamma), whereby modulating energy transduction processes, insulin secretion, gluconeogenesis and adipogenesis, with resulting neuroprotection and cardioprotection. SIRT inhibitors, nicotinamide, sirtinol and O-AA-ribose are also reported

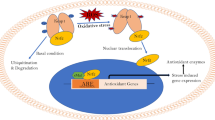
Sirtuin activation as function of NAD/NADH ratio. Interrelationship and overlap between sirtuin regulation, generation of altered proteins and mitochondrial biogenesis and activity, exerted by metabolic effects on NAD and NADH levels
SIRT1 regulates important aspects of mitochondrial biology, e.g. it deacetylates the essential cofactor PGC-1α (PPAR-γ coactivator-1α) in mitochondrial biogenesis. An up regulation of the mitochondrial activity might be of therapeutic benefit for various diseases related to aging such as metabolic disorders (e.g. diabetes type 2) or mitochondrial disorders [105–107]. In active tissues, such as the muscle, the metabolic rate increases, activates the glucose metabolism resulting in improved insulin sensitivity. It is also known that the number of functional mitochondria decreases with aging. Thus an increase of the mitochondrial biogenesis might have an anti-aging effect. More tangible evidence that SIRT1 activation might have benefit via mitochondrial function comes from studies of the polyphenol, resveratrol, in mice. Resveratrol and other polyphenolic compounds are made by plants in response to stress. Resveratrol was recently shown to affect the activity of SIRT1 in vitro although its effects seem to depend on the nature of the substrate for deacetylation [108] However, in vivo, resveratrol has been shown to exert effects dependent on sirtuin orthologs—extension of lifespan in yeast, C. elegans and Drosophila, and metabolic effects on mammalian cells [109]. Two recent studies show that deleterious effects of high fat, high caloric diets in mice were mitigated by resveratrol feeding. In one study, the shortening of lifespan by the high fat diet was reversed [110]. In a second study, resveratrol increased SIRT1 activation, PGC-1a deacetylation, and mitochondrial biogenesis in muscle [111]. These studies provide a powerful indication that SIRT1 activation offers a promising approach for treating metabolic disorders [108]. It has been suggested that metabolism of the redox couple NAD/NADH provides a link between sirtuin activity and the control of cell senescence and organism life-span: NAD-dependent protein deacetylation helps maintain the juvenile phenotype, whereas inhibition of deacetylation activity by NADH or nicotinamide, or by NAD unavailability, promote the onset of cellular aging and decrease organism lifespan [105]. Raising NAD levels, or lowering NADH levels by increasing its oxidation, also promote sirtuin activation, with concomitant beneficial effects on cell survival [106, 107]. There exists an interrelationship and overlap between sirtuin regulation, generation of altered proteins and mitochondrial activity, exerted by metabolic effects on NAD and NADH level, as reported in (Fig. 7). In the Wallerian degeneration slow (Wlds) mouse model, SIRT1 activation protects axons against neuronal injury [96]. This Wlds mouse bears in fact a dominant mutation producing an overexpressed chimeric Wlds protein composed of the ubiquitin assembly protein Ufd2a and the NAD+ salvage pathway enzyme NMNAT1. Decreasing SIRT1 activity reduces the axonal protection originally observed, whereas SIRT1 activation by resveratrol decreases the axonal degeneration after neuronal injury [112]. This suggests that the neuroprotection in the Wlds mouse model is achieved by increasing the neuronal NAD+ reserve and/or SIRT1 activity [96]. Furthermore it has been reported that the SIRT1 agonist resveratrol protects C. elegans neurons expressing a fragment of the Huntington disease-associated protein huntingtin and mammalian neurons from mutant polyglutamine cytotoxicity in an HdhQ111 knock-in mouse model of Huntington disease [96]. There have been also identified potent inhibitor of sirtuin 2 (SIRT2), such as nicotinamide (NAM), O-acetyl-ADP-ribose (O-AA-ribose) or AGK, with interesting data showing that inhibition of SIRT2 rescued a-synuclein toxicity and modified inclusion morphology in a cellular model of Parkinson’s disease [113]. Genetic inhibition of SIRT2 via small interfering RNA similarly rescued a-synuclein toxicity. Furthermore, the inhibitors protected against dopaminergic cell death both in vitro and in a Drosophila model of Parkinson’s disease. The results suggest a link between neurodegeneration and aging [113]. In addition, SIRT1 activation significantly decreases neuronal cell death induced by amyloid-beta (Aβ) peptides through inhibition of NF-κB signalling [96]. Specific brain hSIRT1 overexpression in transgenic animals induces a significant increase in the a-secretase activity, an enzyme that cleaves the amyloid precursor peptide (APP) within the Aβ peptide, favoring thereby the nonamyloidogenic pathway of the APP processing [96]. In addition, a recent study demonstrated the protective effect of CR against Alzheimer’s disease-type brain amyloidosis in monkeys [114]. Monkeys maintained on CR diet had significantly reduced contents of Aβ peptides in the temporal cortex that correlated with enhanced SIRT1 concentrations as compared to monkeys under normal diet. From these studies, it became clear that pharmacological strategies aiming at activating SIRT1 would impeded Aβ peptide deposition in the brain and prevent the development of Alzheimer’s disease [96, 114]. From the animal studies discussed above, it was suggested that SIRT1 could contribute to the pathogenesis of some complex diseases. In line with this hypothesis, genetic variants (SNPs) in the human SIRT1 gene have been shown to be tightly associated with energy expenditure [111]. Given their broad protective effects against oxidative stress, SIRT1 could hence be considered as a serious candidate target for therapeutic interventions in metabolic and neurodegenerative disorders.
Other Oxidative Stress-Related Neuroprotective Systems
Hsp90 chaperones are among the most abundant proteins in the cytosol of eukaryotic cells, and their abundance is increased further by condition of stress. Hsp90 chaperones not only have a role in the folding and assembly of cellular proteins, but they also regulate the activity of different signalling proteins, such as steroid hormone receptors and heme-dependent nitric oxide synthase [115].
Hsp60 family members are found in the mitochondria, where they interact with a cofactor of the HSP10 family and function in the folding and intracellular trafficking of many proteins [116].
Hsp40 proteins are important for protein translation, folding, unfolding, translocation, and degradation, primarily by stimulating the ATPase activity of chaperone proteins, Hsp70s [117].
Small heat shock proteins (sHSPs) belong to a family of 12- to 43-kDa proteins that are ubiquitous and are largely conserved in amino acid sequence among all organisms. The sHSPs (which include alpha crystallin) can form large multimeric structures and have a wide range of cellular functions, including endowing cells with thermotolerance in vivo and being able to act as molecular chaperones in vitro [118].
Natural Antioxidants, Vitagenes and Neurodegenerative Disorders
Curcumin
Curcumin (Figs. 2, 8a), the active principle of the turmeric Curcuma longa, has been proposed in the therapy of neurodegenerative disorders, but some problems may derive mainly due to its pharmacokinetic parameters. Although its stability at acidic pH, almost 40–80% of curcumin remains in the gastrointestinal tract upon ingestion [119]. Furthermore, studies in humans and rodents clearly demonstrated that a marked first-pass metabolism limits the systemic bioavailability of curcumin (~60%) [120–122]. Interestingly, in order to increase its bioavailability, the co-administration of curcumin with piperine or its complexation with phospholipids have been proposed [120, 123, 124]. Preclinical studies have shown that administration of 1 g/kg of curcumin to the rat allows the polyphenol to reach plasma concentrations around 0.5 µg/ml; on the other hand, patients affected by malignant or pre-malignant conditions of the bladder, skin, cervix, stomach or oral mucosa, treated with high dose curcumin (0.5–8 g/day for 3 months) had a plasma concentration of this compound of 1.75 ± 0.8 µM [120, 125]. In the rat, the volume of distribution of curcumin is ~190 l thus suggesting that this polyphenol may accumulate in many organs including colorectal tissue, and liver [120, 123, 126]. Evidence from rodents and humans have demonstrated that, after oral dosing, curcumin is transformed to curcumin glucuronide and curcumin sulfate as well as reduced into dihydrocurcumin (DHC), tetrahydrocurcumin (THC), hexahydrocurcumin, octahydrocurcumin and hexahydrocurcuminol [119, 127, 128]; curcumin, DHC and THC can be further converted in monoglucuronide conjugates [127, 129]. These metabolic changes do not occur only in the liver, the main organ deputed to biotransformation, but also in the intestinal tract [119, 128]. Interestingly, the metabolism of curcumin generates products such as THC endowed with anti-inflammatory activity comparable to that of the parental compound [119, 128]. In rodents and humans curcumin inhibits phase I and phase II enzymes such as cytochrome P450 enzymes, glutathione S-transferase and UDP-glucuronosyltransferases, therefore the ingestion of this spice may significantly alter the metabolism of drugs thus increasing their plasma concentrations and initiating potential toxic effects [130–133]. In the rat, curcumin is mainly excreted in the feces, only a small amount in the urine [121, 122] with a half-life of elimination of ~1.5 h [123]. The urinary elimination of curcumin and its metabolites seems to increase if curcumin is administered at large doses (for example 3.6 g/day for up to 4 months) [120, 124]. With regard to the toxicity profile of curcumin, studies in rodents and primates have shown that doses of up to 3.5 g/kg body weight administered for up to 3 months were well tolerated by the animals [120]. In humans, curcumin at doses ranging from 2.1 and 8 g/day for up to 3 months did not originate main toxic effects [125, 134]. However, patients affected by advanced colorectal cancer treated with curcumin (3.6 g/day) developed diarrhea whereas a dose of 0.9 g/day was associated with nausea, which resolved by suspending the treatment. In the same patients, blood test abnormalities related to curcumin administration were a rise in serum alkaline phosphatase and lactate dehydrogenase, but the possibility that they resulted from the progression of cancer rather than curcumin toxicity should be carefully evaluated [120, 135].

(a) Chemical structures of the polyphenols curcumin and resveratrol and (b) Chemical structures of the l-carnitine, acetyl-l-carnitine and l-carnosine
Early studies have shown that curcumin and related products such as THC, have antioxidant activity. In fact, these compounds reduced free radical- or copper-induced lipid peroxidation in several experimental systems [136–138]. Furthermore, structure–activity studies clearly demonstrated the importance of the beta-diketone moiety, especially, the hydroxyl group, for the antioxidant activity of curcumin and its analogues [136, 139]. Very recently, many papers demonstrated that curcumin and its metabolites interact with NF-κB, inducible nitric oxide synthase (iNOS), hypoxia-inducible factor-1 (HIF-1) and members of the vitagene family (see below).
Reyes-Gordillo et al., have shown that curcumin reduced the CCl4-induced liver toxicity in the rat; in particular, curcumin reduced the CCl4-related increase in pro-inflammatory cytokines and blocked the nuclear translocation of NF-κB [140]. Similarly, curcumin prevented the dinitrochlorobenzene-induced colitis in the rat by down-regulating both NF-κB and iNOS [141]. In lung epithelial cells, curcumin exerted anticarcinogenic activity and prevented the cigarette smoke-induced NF-κB activation through inhibition of IkBα kinase activation, IkBα phosphorylation and degradation [142]. The inhibition of the NF-κB activation was paralleled by the suppression of many NF-κB-related genes, including cyclin D1, cyclooxygenase-2 and matrix metalloproteinase-9 [142]. Comparable results have been found in a macrophage cell line (RAW 264.7) challenged with lipopolysaccharide. In these cells, curcumin and its reduced metabolites blocked the activation of NF-κB, and the downstream activation of iNOS, via inhibition of the IkB kinases 1 and 2, thus providing further evidence about the importance of the effects on NF-κB in the anti-inflammatory and anticarcinogenic activity of this phenolic compound [143]. Through interaction with NF-κB, curcumin exerts protective function also in the regulation of T-cell-mediated immunity. In fact, overexpression of NF-κB in T cells confers protection against tumor-induced apoptosis, whereas when NF-κB is inhibited, the cell becomes much more vulnerable and undergoes apoptosis [144]. By so doing, NF-κB plays an important role in the regulation of T cell apoptosis and the related thymic atrophy which occurs during carcinogenesis. In this experimental model, curcumin prevented the tumor-induced apoptosis and the following thymic atrophy by restoring the activity of NF-κB [144]. Another transcription factor involved in the anticarcinogenic effect of curcumin is HIF-1. Hypoxia-inducible factor-1 is composed of two proteins, HIF-1α and the aryl hydrocarbon receptor nuclear translocator (ARNT) and plays a major role in the development of hypoxic tumors [145]. Curcumin has been demonstrated to inactivate HIF-1 in several cell lines and this effect is related to its ability to promote ARNT degradation [145]. As a consequence of HIF-1 inactivation, several proteins downstream to HIF-1 were downregulated, such as erythropoietin and the vascular endothelial growth factor [145]. Particularly interesting is the interaction of curcumin with the vitagene system (Figs. 2–4). In particular, curcumin increased the expression of HO-1 in human cardiac myoblasts, hepatocytes, monocytes and endothelial cells, rat neurons and astrocytes as well as porcine endothelial cells [34, 146–150]. In rodents and human cells, curcumin-induced HO-1 overexpression was correlated with production of mitochondrial ROS, activation of transcription factors Nrf2 and NF-κB, induction of MAPK p38 and inhibition of phosphatase activity [149, 151, 152]. Moreover, curcumin up-regulated Hsp70 in human colorectal carcinoma cells, proximal tubule cells and rat glioma cells [153–157]. Quite different is the effect of curcumin on TrxR, as it has been shown that curcumin irreversibly inhibits TrxR activity. As a consequence, there was increased NADPH oxidase activity which, in turn, produced an abundance of ROS [158]. This latter paradoxical effect may explain, at least in part, the cancer chemopreventive activity of curcumin [158]. Having two Michael acceptor groups on its molecule, curcumin is an activator of nuclear factor-erythroid 2-related factor 2 (Nrf2), a 66-kDa transcription factor [159] that is responsible for both basal and inducible expression of many cytoprotective genes, classically known as phase 2 genes [160]. The family of phase 2 genes is very diverse and includes NAD(P)H:quinone oxidoreductase-1, glutathione transferases, thioredoxin reductase, heme oxygenase-1, and many others that have single or multiple copies of common upstream regulatory sequences known as antioxidant response elements (ARE) [161–163]. Nrf2 is a cap’-n’-collar (CNC) transcription factor and shares with the other members of this family of transcription factors a highly conserved basic region-leucine zipper (bZIP) domain [164, 165]. It forms heterodimers with members of the small Maf family, the resultant dimeric complex binds to the ARE and subsequently recruits the general transcriptional machinery to activate transcription of phase 2 genes [165–168]. Under normal conditions, Nrf2 has a very short half-life and is targeted for ubiquitination and proteasomal degradation via binding to a cytosolic repressor protein, Kelch-like ECH-associated protein 1 (Keap1) which in turn associates with Culin3 to form an E3 ubiquitin ligase complex [169]. Once in the cell, curcumin (and other Michael acceptor-containing molecules) modify specific highly reactive cysteine residues of Keap1 [170, 171]. As a result, Keap1 loses its ability to target Nrf2 for degradation, which then undergoes nuclear translocation. There is now overwhelming amount of experimental evidence that Nrf2 serves as a master regulator of the ARE-driven cellular defenses against various electrophiles and oxidants [172]. Indeed, nrf2-knockout mice exhibit enhanced sensitivity to many types of toxic chemicals, including carcinogens, allergens, and environmental pollutants [172–175]. Whereas inducers of Nrf2-dependent genes protect wild type mice against a variety of environmental challenges, they have diminished or no protective effects in nrf2-knockout mice. Figure 4 shows the current model for the mechanism of induction of cytoprotective phase 2 genes. In the absence of inducing stimuli Keap1 binds and targets transcription factor Nrf2 for ubiquitination and proteasomal degradation via association with the Culin 3 (Cul3)-based E3 ubiquitin ligase. Inducers (e.g., curcumin) react and chemically modify specific highly reactive cysteine residues of the sensor Keap1 rendering it unable to repress transcription factor Nrf2. Thus, Nrf2 is stabilized and undergoes nuclear translocation where it binds to the ARE (in heterodimeric combinations with a small Maf protein), and ultimately activated transcription of cytoprotective phase 2 genes.
Because both heme oxygenase 1 and the thioredoxin/ thioredoxin reductase system can be upregulated in an Nrf2/ARE-dependent manner, the questions arise whether: (i) the third member of the vitagene family, Hsp70, is also inducible by other phase 2 inducers, and (ii) there could be a common regulatory mechanism. Indeed, in addition to curcumin, several other inducers of Nrf2-dependent genes have been shown to increase the protein levels of Hsp70. Among them are the cyclopentenone prostaglandin 15-deoxy-∆12,14-prostaglandin J2 (15dPGJ2), and the vicinal dithiol reagent phenylarsine oxide [176, 177]. Importantly, all of these compounds react with sulfhydryl groups and the transcriptional activation of both Nrf2, the major transcription factor responsible for phase 2 gene expression, and heat shock factor 1 (HSF1), the major activator of Hsp70 gene expression, depend on cysteine modification either within the Nrf2 regulator Keap1 [167, 174], or within HSF1 itself [178].
As already mentioned above, neurodegenerative disorders, such as, such as AD and Parkinson’s disease (PD), belong to the family of the “protein conformational diseases” and affect a large portion of our aging population [179]. In general, conformational diseases are conditions that arise from the dysfunctional aggregation of proteins in non-native conformations. It is known that the beta conformation in proteins is particularly susceptible to perturbations in the quality control system and that ROS play an important role in the development and/or pathogenetic progression in aging and neurodegenerative diseases [180–182]. Chaperones can rescue misfolded proteins by breaking up aggregates and assisting in the refolding process [181, 183]. Proteins that cannot be rescued by refolding can be delivered to the proteasome by chaperones to be recycled [183]. If the cell is not able to eliminate misfolded proteins multiple metabolic derangements resulting in the excessive production of ROS and RNS occur [184]. The ability of a cell to deal with oxidative and nitrosative stress requires functional chaperones, antioxidant production, protein degradation and a cascade of intracellular events collectively known as the “unfolded protein response”, a form of cell stress response [185, 186]. As the cell’s quality control system becomes overwhelmed, conformational changes occur to amyloid polypeptide intermediates, generating stable oligomers with an anti-parallel crossed beta-pleated sheet structure that eventually accumulate as space-occupying lesions within neurons [182]. Although it is clear why mutant proteins form amyloid, it is harder to rationalize why a wild-type protein adopts a native conformation in most individuals, but it misfolds in a minority of others, in what should be a common extracellular environment. This discrepancy suggests that another event likely triggers misfolding in sporadic amyloid disease. One possibility is that an abnormal metabolite, generated only in some individuals, covalently modifies the protein or peptide and causes it to misfold. Candidate metabolites are suggested by the recently recognized links between AD and atherosclerosis, in which known chronic inflammatory metabolites, may play a critical pathogenic role. If this holds true, then new targets are disclosed for a prevention strategy brought about through nutritional antioxidants.
Alzheimer disease is characterized by a subtly impaired cognitive function or a disturbance of behaviour. With time there is a gradual memory loss and disorientation which eventually progress into dementia. Although, most cases are sporadic, 5–10% or more are familial. Gross examination of the brain in AD shows a variable degree of cortical atrophy with narrowed gyri and widened sulci most apparent in the frontal, parietal and temporal lobes. Microscopically, the features include neurofibrillary tangles, neurite (senile) plaques, the central core of which is amyloid-beta peptide, derived from the transmembrane amyloid precursor protein (APP), amyloid angiopathy, granulovacuolar degeneration and Hirano bodies. Importantly, all of these changes are present in the brains of nondemented older individuals but to a much lesser extent [187, 188]. Several lines of evidence now support a fundamental role for oxidative and nitrosative stress in the pathogenesis of this disease [20, 187, 189]. As said, the only evidence of a protective role of curcumin in the onset of AD was provided by Ganguli and colleagues demonstrated that Indian population, who have a curcumin-enriched diet, has a reduced prevalence of AD compared to United States [40]. Following this observation, many basic studies were conducted and the neuroprotective role of curcumin was corroborated. In vitro studies have shown that curcumin protects neuron-like PC12 cells from β-amyloid toxicity and, interestingly, the polyphenol displayed a neuroprotective effect greater than a well known antioxidant such as α-tocopherol [190]. By using an Alzheimer transgenic APPSw mouse model (Tg2576), Lim and colleagues have shown that dietary curcumin suppressed inflammation and oxidative damage in the brain of these mice [41]. More recently, Garcia-Alloza et al. in transgenic APPswe/PS1dE9 mice demonstrated that curcumin, given intravenously for 7 days, crosses the blood–brain barrier, binds to β-amyloid deposits in the brain and accelerates their rate of clearance [126]. These latter results are in good agreement with previous findings which demonstrated that curcumin disaggregates and inhibits β-amyloid aggregation [191, 192].
Parkinson’s disease, whose cardinal features include tremor, slowness of movement, stiffness and poor balance, is attributed to a profound deficit in dopamine that follows the loss of dopaminergic neurons in the substantia nigra pars compacta and dopaminergic nerve terminals in the striatum [20, 193]. Although the mechanisms leading to PD are still uncertain, a large amount of experimental evidence implicates oxidative and nitrosative stress as one of the crucial factors in the pathogenesis of PD [194, 195]. Considerable insights into the pathogenesis of PD, indeed, have been achieved by use of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is commonly used to induce an experiment model of PD [194, 196]. Excessive free radical formation or antioxidant deficiency and the resulting oxidative stress are all mechanisms involved in MPTP neurotoxicity [197]. Rajeswari has shown that curcumin protects rat brain from MPTP-induced neurotoxicity by virtue of its scavenger activity [198]. On the other hand, curcumin has been shown to protect PC12 cells from MPP+ (the active metabolite of MPTP) by inducing the antiapoptotic protein bcl-2, preventing the dissipation of mitochondrial membrane potential and reducing the intracellular iNOS levels [199]. The importance of mitochondria in the neuroprotective effect of curcumin has been also stressed by Mythri et al. [200] who demonstrated that curcumin prevents the formation of peroxynitrite which is responsible for the complex I damage which is a common feature in PD.
Friedreich’s ataxia (FRDA) is an autosomal recessive disease usually caused by a homozygous GAA expansion in the intron 1 of the frataxin encoding gene. This mutation results in a marked reduction in the amount of the FXN mRNA and subsequently of frataxin, a ubiquitously expressed mitochondrial protein [201–204]. Friedreich’s ataxia is the most common inherited ataxia. Its estimated prevalence in European populations is 1 in 50,000 [201]. Friedreich’s ataxia is characterized by onset before 20 years of age of ataxia of all four limbs, associated with cerebellar dysarthria, decreased/absent tendon reflexes, sensory loss and pyramidal signs. The progression of the disease is relentless. Skeletal deformities and cardiomyopathy are found in a majority of patients, who also have an increased frequency of impaired glucose tolerance and diabetes [201]. Generally within 15–20 years after the first appearance of symptoms, affected individuals are confined to a wheelchair. The neurological symptoms result from progressive degeneration of the dorsal root ganglia, posterior columns, corticospinal tracts and the dorsal spinocerebellar tracts of the spinal cord [205, 206]. How the GAA expansion leads to decreased FXN transcript levels remains not yet clarified [207, 208]. It could inhibit transcription initiation, block elongation, interfere with splicing, decrease mRNA stability or produce a combination of these effects. The absence of abnormally spliced frataxin mRNA species in FRDA cells suggests RNA splicing is not affected [204]. It has been suggested that the triplet repeats form a special structure, e.g. triplexes, which interferes with transcription elongation, however this was not confirmed in all studies [209, 210]. Transcription initiation could be inhibited by the formation of heterochromatin at the FXN locus in presence of the GAA expansion. Active promoters are associated with histone H3 and H4 acetylation (in particular H3/AcK9), with certain histone H3 methylations (in particular H3/(Me)3K4) and with the presence of the replacement histone H2A.Z; while inactive promoters are characterised by reduced histone acetylation, the presence of trimethylated H3/K27, K9 and K79 as well as trimethylated H4/K20. Highly repressed genes are also associated with DNA methylation [211]. Consistent with heterochromatin formation in presence of the GAA expansion, using a FRDA lymphoblast cell line decreased histones acetylation and increased trimethylated H3K9 levels were found in the FXN gene [212]. In an additional 3 FRDA lymphoblast lines the levels of H3/K9 methylation in FXN intron 1 and the methylation of 3 CpGs in intron 1 upstream of the GAA repeat were markedly increased [213]. In a recent study it was demonstrated that CpG sites in the region upstream the GAA repeat are consistently hypermethylated in FRDA patient brain, heart and cerebellum [214]. Furthermore, in the same study it was shown an overall decrease of histone H3K9 acetylation together with increase of H3K9 methylation in FRDA brain tissue. Moreover HDAC inhibitors had been shown to increase frataxin expression from a FXN reporter construct using butyrate and in FRDA lymphocytes using BML-210 and its analogs, however the analysis of the relationship between increased FXN transcripts and gene specific histone modification in these cells was limited and the influence of trans acting factors was not excluded [212, 215].
The role of frataxin is controversial. At least five different hypotheses for its function have been proposed: (1) mitochondrial iron transport, (2) mitochondrial iron storage, (3) Fe–S cluster biosynthesis, (4) Fe–S cluster protection and/or repair and (5) protection from oxidative stress. An involvement of frataxin in iron homeostasis has been proposed since iron accumulation has been detected in yfh1 knockout models, in FRDA patients heart and liver mitochondrial [216–219]. Iron accumulation could increase the formation of OH radicals, through Fenton reaction, leading to oxidative damage of Fe–S cluster containing proteins, whose activity is reduced in FRDA yeast model and FRDA patient tissues [216, 220]. Frataxin may interfere with the activity of the Fe–S cluster-containing subunits of mitochondrial respiratory complexes I, II and III, as a reduction in activity of these complexes was found in FRDA yeast models, in FRDA mouse models and patient tissues. The activity of aconitase, an Fe–S protein involved in iron homeostasis, was found to be deficient as well [216, 220, 221]. Bulteau et al. [222] demonstrated that frataxin protects the aconitase [4Fe–4S]2+ cluster from disassembly and promotes enzyme reactivation, suggesting a role of frataxin in Fe–S cluster protection and/or repair. An increased oxidative stress has been demonstrated in individuals with FRDA. Elevated concentrations of oxidative stress markers were evidenced in patients with FRDA [223–226]. Urinary concentrations of 8-hydroxy-2′-deoxyguanosine (8OH2′dG), a marker of oxidative DNA damage, were found to be increased in patients with FRDA as compared with controls [224]. Plasma malondialdehyde (MDA) levels, a product of lipid peroxidation, were found to be raised in Friedreich’s ataxia (FRDA) patients [225, 226]. These levels correlated with increasing age and disease duration, suggesting that lipid peroxidation increased with disease progression [226]. Piemonte et al. [223] evidenced a reduction of free glutathione levels in the blood of patients with Friedreich’s ataxia, suggesting a relevant role of free radical cytotoxicity in the pathophysiology of the disease. Moreover an increased ROS production in frataxin deficient cells has been described [227].
Since the GAA repeat expansion leads to a reduction in the FXN transcript and frataxin protein, a number of studies have been conducted to identify compounds that increase frataxin expression. Sturm et al. [228] showed that the recombinant human erythropoietin (rhuEPO) significantly increases frataxin expression in primary lymphocytes from FRDA patients by a still unknown mechanism. In a recent open-label clinical pilot study treatment of 12 FRDA patients with recombinant human erythropoietin led to a persistent and significant increase in frataxin levels after 8 weeks (P < 0.01) [229]. HDAC inhibitors have been shown to increase frataxin expression. Using a FXN reporter construct Sarsero et al. [215] showed that butyrate is able to increase frataxin expression. Herman et al. [212] described an increase in FXN transcript and frataxin protein in FRDA lymphoblasts and lymphocytes after the treatment with BML-210 and its analogs. Given the evidence of oxidative damage in FRDA patients the treatment as been focused on antioxidant protection [230, 231]. FRDA patients have been treated with a variety of antioxidants including idebenone and vitamin E [231]. Lodi et al. [232], using in vivo phosphorus magnetic resonance spectroscopy (31P-MRS), showed that after only 3 months of treatment with coenzyme Q10 and vitamin E (Coenzyme Q10 400 mg/day, vitamin E 2100 IU/day), the cardiac phosphocreatine to ATP ratio increased to 178% (P = 0.03) and the maximum rate of skeletal muscle mitochondrial ATP production increased to 139% (P = 0.01) of their respective baseline values in the FRDA patients. These improvements were sustained after 6 months of therapy. The treatment with these agents of ten FRDA patients for 47 months resulted in a significant and sustained improvement in cardiac and skeletal muscle bioenergetics. Moreover echocardiographic data revealed significantly improved cardiac function, defined by increased fractional shortening at the 35- and 47-month time points [233]. Clinical trials with idebenone in FRDA patients showed a reduction of the left ventricular mass, left ventricle posterior wall and interventricular septal thickness [234–237]. In some of these studies an improvement of cardiac function was also detected [235, 236].
In the previous studies, idebenone did not modify neurologic manifestations of the disease. However, a recent 6-month double-blind, placebo-controlled study conducted in 48 FRDA patients using three different doses of idebenone (approximately 5, 15, and 45 mg/kg) showed a significant improvement in ICARS and a dose-related response in ICARS, FARS, and ADL scores, when patients who required wheelchair assistance were excluded from the data analysis. The latter study suggests that higher doses may be necessary to have a beneficial effect on neurological function [238]. Since Friedreich Ataxia (FRDA) is characterized by an increased mitochondrial oxidative damage, antioxidants targeted to mitochondria should be particularly effective at slowing disease progression. Using FRDA fibroblasts, Jauslin et al. [239] showed that the mitochondria-targeted antioxidant MitoQ was several hundred fold more potent than the untargeted analog idebenone and that the mitochondria-targeted antioxidant MitoVit E was 350-fold more potent than the water soluble analog Trolox. Since induction of HSPs synthesis results in tolerance against oxidative stress-induced damage, molecules activating this defense mechanism are possible candidates for treatment of neurodegenerative diseases, such as Friedreich’s ataxia [240].
Transient forebrain ischemia is a common cause of stroke and occurs in people suffering from cardiovascular diseases [241]. As a consequence of ischemia and the following reperfusion, a cascade of events such as increased calcium release, the overexpression of COX-2 and iNOS both of which are important free-radical generators and trigger neuronal cell death in selected brain areas including the hippocampal cornu ammonis 1 (CA1) [1, 22, 52, 241]. Curcumin exerted a neuroprotective effect in rats who underwent ischemia/reperfusion injury and this effect has been related to the direct scavenger effect of curcumin as well as to a curcumin-induced interference with the apoptotic machinery, increase in antioxidant molecules (GSH) and enzymes (catalase, superoxide dismutase) [241–243].
l-Carnitine and Acetyl-l-Carnitine
l-Carnitine (LC) (Fig. 8b) is a natural compound and its biological role is to facilitate the transport of fatty acids to mitochondria. Dietary LC derives from the intake of red meats, but the endogenous synthesis of LC from the amino acid precursors lysine and methionine has been also documented [244]. The dietary intake of LC in humans ranges from 1 to 15 µmol/kg body weight/day, whereas the rate of biosynthesis is about 1–2 µmol/kg body weight/day [245]. Recently, exogenous LC, given by oral (p.o.) or intravenous (i.v.) routes, has been used for the treatment of cognitive disorders such as AD and dementia [244]. After oral ingestion, dietary LC is well absorbed by simple or carrier-mediated diffusion and its bioavailability is 54–86%; conversely, the bioavailability of exogenous LC is much lower, in the range 5–18% [244, 245]. This paradoxical effect can be explained considering that the absorption of LC decreases as the intake of LC increases, this to maintain the concentration of LC constant [244, 245]. The normal plasma concentration of LC in healthy adults with a mixed diet is 40–50 µM [244, 246]. When administered at doses 30–100 mg/kg p.o. in humans, LC peak plasma concentrations were 27–91 µM after 3 h, and returned to the baseline within 24 h [245, 247]. l-Carnitine undergoes acetylation in rodents and human intestine thus forming esterified compounds such as acetyl-l-carnitine (ALC) (Fig. 8b) which is endowed with biological activity per se [244, 245]. Interestingly, ALC diffuses across membranes much better than LC and its efflux in the systemic circulation has been calculated to be four times greater than that of LC [245, 248]. Data from AD patients have shown that after supplementation with pharmacological doses of ALC (2 g/day) for 55 days, its plasma concentrations increased from 7.2 to 10.3 µM [245]. In the plasma, neither LC nor ALC are bound to proteins [244]. The volume of distribution of LC differs considering the dietary or exogenous source being approximately 3000 l and 20–50 l, respectively [244]. This great difference in the volume of distribution between dietary and supplemental LC depends on the different degree of absorption, slow accumulation in tissues such as the muscle and rate of kidney elimination (see below), and therefore these numbers should be considered purely indicative [244]. It is interesting to underlie that ALC is able to cross blood–brain barrier; as shown by Parnetti et al. AD patients treated with ALC i.v. or p.o. for 10–60 days have an increased concentration of ALC in the cerebrospinal fluid up to 3.55 nmol/ml [249]. In human subjects treated with LC i.v. its elimination half-life ranged from 3 to 12 h [244]. However due to the long-lasting release of LC by the muscle, the total time of turnover from the body has been estimated to be 66 days [245]. l-Carnitine is metabolised by the intestine to γ-butyrobetaine and trimethylamine, the former excreted by the feces and the latter in urine [244, 245]. Accordingly, the renal clearance of LC which is about 1–3 ml/min suggesting an extensive rate of tubular reabsorption, significantly increases at values close to the creatinine clearance with the increase in LC plasma concentrations indicating that tubular reabsorption approaches full saturation [244]. This last finding is very important and contributes to explain how exogenous LC is almost completely excreted during the first 12 h after administration whereas dietary LC is reabsorbed [244]. Due to its elimination mainly through the kidney, LC and should be administered very carefully to patients affected by renal impairment [250].
Acetyl-l-carnitine has been proposed to have beneficial effects in preventing the loss of brain function which typically occurs during aging and neurodegenerative disorders. The main mechanism of action of ALC is the improvement of mitochondrial respiration which allows the neuron to produce ATP necessary to maintain the normal membrane potential [251]. However, ALC has been shown to be neuroprotective through a variety of other effects such as the increase in PKC activity [251]. Interestingly ALC counteracted the loss of NMDA receptors in neuronal membrane and increased the production of neurotrophins, two effects strictly related to synaptic plasticity [251]. Recent studies have shown that ALC reduces Aβ toxicity in primary cortical neuronal cultures by increasing both HO-1 and Hsp70 expression [252]. Studies in rats have shown that chronic ALC treatment increases life-span, improves cognitive behaviour in aged animals and guarantees long-term memory performance [251]. Furthermore, chronic ALC treatment has been shown to prevent age-related changes in mitochondrial respiration and decrease oxidative stress biomarkers through the up-regulation of HO-1, Hsp70 and superoxide dismutase-2 in senescent rat [253] (Fig. 3). Taken together, these pre-clinical studies suggested that ALC treatment could be beneficial for the treatment of age-related diseases and the potential use in humans has been encouraged. Patients affected by AD and treated with ALC at doses ranging from 1 to 2 g/day for 6–12 months, have shown an improved performance on several cognitive tests such as word recognition, name learning and world list recall with respect to placebo-treated patients, but none of these effects was significant [251, 254]. In two clinical studies, ALC 3 g/day for 1 year significantly reduced cognitive decline only in early-onset AD patients [255, 256], but this evidence was not confirmed in a later ad hoc designed study [257]. Consistently, we have demonstrated that ALC induces HO-1 in a dose and time dependent manner and that this effect was associated with up-regulation of other Hsps as well as high expression of the redox-sensitive transcription factor Nrf2. The results from this study show for the first time that acetylcarnitine induces heme oxygenase-1 and Hsp60 heat shock proteins, and that this effect may involve the transcription factor Nrf2, implying the conceivable possibility that acetylcarnitine, by promoting acetylation of DNA-binding proteins, can induce post-translational modifications of critical target proteins endowed with DNA competence and transactivating activity [258]. Very importantly, this new envisioned role of LAC as a molecule endowed with the capability of potentiating the cellular stress response pathways appear to be promising an alternative therapeutic approach for those pathophysiological conditions where stimulation of the HO pathway is warranted [258].
Carnosine
Carnosine is a natural dipeptide(β-alanyl-l-histidine) (Fig. 8b) present in long-lived mammalian tissues [259] and has numerous roles as proton buffer, metal chelator, antioxidant, antiglycating, immunostimulant, antitumoral and wound-healing agent [260, 261]. Since β-alanine is non-proteinogenic amino acid, it is obvious that carnosine is not product of protein catabolism: Instead it is synthesized enzymatically by carnosine synthetase, an enzyme present in brain and muscle that shows broad substrate specificity [262]. The hydrolysis of carnosine is catalyzed by two enzymes recently cloned and characterized [263]. Both enzymes belong to the M20 metalloprotease family. The enzyme named CN1 exhibits narrow specificity and the characteristics of the enzyme previously designated X-His dipeptidase or carnosinase [264]. The enzyme named CN2 displays broad substrate specificity and is ubiquitously expressed like the enzyme previously designated cytosol non-specific dipeptidase [265]. In the brain, carnosine has been found in glial cells and in some type of neurons [266]; uptake of carnosine has been found to be mediated by a high affinity, energy-dependent dipeptide transport system, identified as the peptide transporter PepT2 [267]. Carnosine has been shown to delay ageing in cultured human fibroblasts [268], male Drosophila [269] and senescence-accelerated mice [270]. Therapeutic potential has also been invoked in cataractogenesis [271] and diabetes [272–274]. The occurrence of carnosine and its analogue homocarnosine(γ-aminobutyryl-histidine) in brain, and homocarnosine in CSF, and their age-related alterations [275, 276] suggested a role for these peptides with respect to suppression of onset or progression of AD and other neurodegenerative diseases [277, 278]. In brain cells, carnosine has been shown to be neuroprotective because of its ability to counteract both oxidative and nitrosative stress related to several pathological conditions including ischemia [279–281], methamphetamine neurotoxicity [282] and neurodegenerative disorders [283]. Importantly, carnosine may indirectly influence neuronal excitability by modulating the effect of zinc and copper [284, 285]. Furthermore, carnosine has been shown prevent β-amyloid aggregation and toxicity [286] and this effect can be due to the known ability of this peptide to inhibit protein misfolding and avoid the formation of advanced-glycation end-products [261]. More recently, it has been reported that the peptide is able to protect against Aβ-induced neurotoxicity in differentiated rat PC12 cells by regulation of glutamate release and glutamate release and NMDA receptor trafficking [287, 288]. Interestingly, carnosine plasma levels have been found to be lower in AD patients than in age-matched controls [289]. Furthermore, carnosine has been shown to counteract peroxynitrite-dependent protein alterations such as tyrosine nitration [290] and to inhibit the No-dependent activation of guanylate cyclase [291]. Recent evidence demonstrated that carnosine prevents the up-regulation of iNos and the induction of both HO-1 and Hsp-70 following strong nitrosative conditions [292]. In addition, a correlation has been found between cell protection and NO free-radical scavenging activity of carnosine that showed direct NO-trapping ability in cell-free experiments [293]. Taken together, these findings allow us to propose the metabolic pathway leading to carnosine formation as a potential target for the prevention and/or treatment of neurodegenerative disorders. In addition, new carnosine, homocarnosine and anserine derivatives have been synthesized and characterized, showing chelating and antioxidant properties similar to those of the parent dipeptides [294–297]. In addition, these new compounds survive to attack by carnosinases [298] and should be explored for their ability to suppress age-related neurodegeneration where increased zinc and copper could play a role [299–301].
Resveratrol
Resveratrol (3,5,4′-trihydroxy-trans-stilbene) (Fig. 8a) is a phytoalexin found in grapes, cranberries and peanuts [302, 303]. Several in vitro studies have shown that resveratrol is a powerful molecule endowed with antioxidative, anticancer, anti-inflammatory and estrogenic activities [302, 304]. Studies in rodents and humans have shown that after oral ingestion resveratrol is readily absorbed (at least 50% in the rat) reaching peak plasma concentration after 10–60 min up to 2 µM total resveratrol (i.e. genuine resveratrol plus resveratrol derived from the hydrolysis of its conjugated products) whereas the amount of unchanged resveratrol was in the low nanomolar range [302–306]. In hepatic cells, resveratrol has been absorbed by passive diffusion and carrier mediated processes [307]. Interestingly, the amount of resveratrol adsorbed did not change in the presence of ethanol [302]. Resveratrol binds to plasma proteins such as albumin and lipoproteins [307]. The plasma half-life was estimated to be 12–15 min in the rat and 9–12 h in humans [303, 305]. Resveratrol undergoes massive metabolism both in gastrointestinal cells and liver. The main metabolites detected in humans are resveratrol monosulphate, resveratrol monoglucuronide (two isomeric forms), dihydroresveratrol monosulphate and monoglucuronide [303, 304]. The serum half-life of these metabolites is about 9.2 h, i.e. significantly higher than that of unmodified resveratrol [304, 305]; both in rodents and humans, enteric recirculation of resveratrol metabolites has been proposed and may account for the increased half-life of these metabolites with respect to the unchanged form. Resveratrol has been shown to inhibit CYP3A4 irreversibly and to be a reversible inhibitor of CYP2E1 [304, 308]. Furthermore, in rat cardiomyocytes resveratrol increased the activity of glutathione transferases, well known phase 2 enzymes involved in the detoxification of drugs [309]. Resveratrol is mainly excreted by the kidney and only a small portion is recovered in feces [302]. It is noteworthy to mention that in rodents and rabbits the tissue concentrations of resveratrol are always below 1 nmol/g fresh tissue [302, 310], whereas in humans the organs in which resveratrol shows greater accumulation are intestinal mucosa, stomach, liver, and kidney, but the exact concentrations have not been estimated [304].
The rationale to use resveratrol in the treatment of neurodegenerative disorders is based on the well-known antioxidant activity of this compound. Resveratrol has been shown to protect rat glioma cells from β-amyloid (Aβ) toxicity by reducing the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX-2), thus preventing the uncontrolled release of NO and prostaglandin E2 (PGE2) [311]. These effects, could be ascribed to the ability of resveratrol to prevent the Aβ-induced nuclear translocation of NF-κB [311]. Furthermore, in PC12 cells, resveratrol counteracted the Aβ25–35-induced toxicity by the down-regulation of pro-apoptotic factors such as Bax and c-Jun N-terminal kinase (JNK) proteins [312, 313]. In rat hippocampal neuronal cell cultures, resveratrol protected cells from Aβ toxic effects by inducing protein kinase-C (PKC) [312, 314]. The neuroprotective effect of resveratrol against Aβ toxic effects could be also mediated by promoting the intracellular degradation of Aβ through the ubiquitin proteasome system (UPS) [315]. Interestingly, resveratrol reduced sodium nitroprusside (SNP)-released NO toxicity in an experimental system of rat hippocampal mixed neuronal/glial cultures [316]. In the rat, the chronic administration of resveratrol (21 days) resulted in a marked inhibition of the cognitive impairment secondary to the intracerebroventricular administration of streptozotocin and this effect has been related to the increase of brain GSH levels [317]. Still under investigation is the neuroprotective effect of resveratrol through the overexpression of sirtuins [312, 318]. In particular, Sirt1–3 play a main role in protecting neurons in AD patients at least by 3 independent mechanisms: (i) the repression of the pro-apoptotic protein p53 and forkhead transcription factor3 (FOXO3) expression, (ii) the reduction of the hyperphosphorylation of tau protein and (iii) the reduction of ROS formation through the interaction with the uncoupling protein 4 (UCP4) [319]. Of note is the interaction of resveratrol with the vitagenes. Very recently, resveratrol has been demonstrated to increase the expression of HO-1 in PC12 cells and primary neuronal cultures presumably through the activation of NF-E2-related factor 2 (Nrf2) in PC12 cells [320, 321]. Although not directly related to AD, these data are in good agreement with previous papers which demonstrate how the overexpression of HO-1, and the related increase in antioxidant capacity, is neuroprotective in several models of AD. Indeed, Takahashi et al. found that cortical neurons cultured from mice expressing the Swedish mutation of AD had defects in BR production with subsequent increase of hydrogen peroxide toxicity [322]. Furthermore, in transfected neuroblastoma cells overexpressing HO-1, the activity of this enzyme was increased, and conversely, the level of tau protein was significantly decreased when compared with antisense HO-1 or vector transfected cells [38]. The suppression of tau protein expression was almost completely counteracted by zinc-deuteroporphyrin, a specific inhibitor of HO activity [38].
Conclusions and Perspectives
Protein conformational diseases, such as Alzheimer’s, Parkinson’s, Huntington’s and ALS affect a large portion of our aging population. In general, conformational diseases are conditions that arise from the dysfunctional aggregation of proteins in non-native conformations. Cells have evolved mechanisms for rescuing and recycling misfolded proteins, but these systems sometimes fail. However, chaperones can rescue misfolded proteins by breaking up aggregates and assisting in the refolding process. Proteins that cannot be rescued by refolding can be delivered to the proteasome by chaperones to be recycled. This often is associated with multiple metabolic derangements that result in the excessive production of ROS and oxidative stress. These ROS set in motion a host of redox reactions which can result in unstable nitrogen and thiol species that contribute to additional redox stress which trigger neuroinflammation [323, 324].
Aging is characterized by a progressive deterioration of physiological functions and metabolic processes. Healthy aging remains one of the ideals of modern society. In aging and in diseases associated with the elderly, such as Alzheimer’s or Parkinson’s, the loss of cells in vital structures or organs may be related to several factors, among which the production of ROS and or RNS by mitochondria is a common denominator, leading to DNA damage, apoptosis and cell death. Although a diet rich in antioxidants seems to offer hope in delaying the onset of unhealthy disorders that accompany aging, no clinical treatment as such has yet been developed and anti-aging drugs are still unavailable. It is well established that reducing food intake (caloric restriction) extends the life-span in a wide range of species. The protein implicated in this protective process is the silent information regulator 2 (SIR2, SIRT1 in mammals), a vitagene enzyme that belongs to a nicotinamide adenine dinucleotide (NAD)+-dependent protein deacetylases [318]. SIRTs regulate gene silencing, DNA repair, rDNA recombination, and ageing, apart from regulating programmed cell death. In this context, increasing SIRT1 has been found to protect cells against amyloid-beta-induced ROS production and DNA damage, thereby reducing apoptotic death in vitro. Moreover, it has been demonstrated that Alzheimer’s and Huntington’s disease neurons are rescued by the over-expression of SIRT1, induced by either caloric restriction or administration of resveratrol, a potential activator of this enzyme. The therapeutic use of polyphenols and other related compounds, which impact the vitagene network and, as such, acting as SIRT1 or Hsps pathway modulators, have been discussed in this review in the perspective of treating aging-related brain disorders. Although some beneficial phytochemicals might function solely as antioxidants, it is becoming clear that many of the beneficial chemicals in vegetables and fruits evolved as toxins (to dissuade insects and other predators) that, at subtoxic doses, activate adaptive cellular stress–response pathways, under control of genes defined by us as vitagenes in a variety of cells including neurons. Examples of such ‘preconditioning’ or ‘neurohormesis’ pathways include those involving cell-survival signaling kinases, the transcription factors NRF2 and histone deacetylases of the sirtuin family (Figs. 2, 3). In these ways, neurohormetic phytochemicals such as resveratrol, sulforaphanes and curcumin or other compounds such as acetyl-l-carnitine or carnosine might protect neurons against injury and disease by stimulating, through the vitagene system, the production of antioxidant enzymes, protein chaperones and other proteins that help cells to withstand stress.
The strong evidence that the vitagene network operates as a defense system in the brain during oxidative and nitrosative stress open new perspectives in the treatment of neurodegenerative disorders. Therefore, the nutritional manipulation of endogenous cellular defense mechanisms represents an innovative approach to therapeutic intervention in neurodegeneration, and propose potential novel therapeutic strategies relying upon the simultaneous activation of cytoprotective genes of the cell life program and down-regulation of proinflammatory and pro-oxidative genes involved in programmed cell death. Although the term vitagene was first proposed to indicate speculatively the existence of genes as opposed to gerontogenes, the first evidence based notion identifying vitagenes with stress responsive genes such as HO-1, Hsps, TrxR and sirtuins have been provided by our group (Fig. 3). Presented here is strong evidence that a functional interplay between stress response genes is important for cell stress tolerance, highlighting compelling reason for a renewed effort to understand the central role of this most extraordinary defense system in biology and medicine.
However, in the fight against oxidative and nitrosative stress, a major role is played by mitochondria which can be the target for cytoprotective effects of many dietary antioxidants. Acetyl-l-carnitine and lipoic acid (LA) has been shown to improve mitochondria bioenergetics and decrease oxidative stress associated with aging in many organs including rodent heart, liver and brain [325–329]. In particular, ALC ameliorates mitochondrial beta-oxidation whereas LA, due to its nature of thiol antioxidant as well as mitochondrial metabolite, increases the total antioxidant status and counteract free radical-induced cell damage [327, 329].
In this respect the concept of hormesis has been receiving greater interest in the biomedical and toxicological research communities over the past decade [330]. In the fields of biology and medicine hormesis is defined as an adaptive response of cells and organisms to a moderate (usually intermittent) stress [330, 331]. The findings suggest that hormetic effects represent evolutionary-based adaptive responses to environmentally induced disruptions in homeostasis. Such adaptive responses, which are incorporated into organismal integrative physiological systems and now clarified at the mechanistic level for more than two dozen receptor systems, provide a cogent basis for the application of hormetic mechanisms in the elucidation of fundamental evolutionary-based biological processes and in the development of novel clinical modalities. Examples include ischemic preconditioning, exercise, dietary energy restriction and exposures to low doses of certain phytochemicals. Recent findings have elucidated the cellular signaling pathways and molecular mechanisms that mediate hormetic responses which typically involve enzymes such as kinases and deacetylases, and transcription factors such as Nrf-2 and NF-κB. As a result, cells increase their production of cytoprotective and restorative proteins including growth factors, phase 2 and antioxidant enzymes, and protein chaperones [330, 331]. A better understanding of hormesis mechanisms at the cellular and molecular levels is leading to and to novel approaches for the prevention and treatment of many different diseases [332].
The several lines of evidence listed above supports also the notion that stimulation of various maintenance and repair pathways through exogenous intervention, such as mild stress or compounds targeting the heat shock signal pathway, such as polyphenols, l-acetyl-carnitine and carnosine may have biological significance as a novel approach to delay the onset of various age-associated alterations in cells, tissues and organisms. Hence, by maintaining or recovering the activity of vitagenes can be possible to delay the aging process and decrease the occurrence of age-related diseases with resulting prolongation of a healthy life span.
References
Halliwell B (2007) Oxidative stress and cancer: have we moved forward? Biochem J 401:1–11
Calabrese V, Scapagnini G, Colombrita C, Ravagna A, Pennisi G, Giuffrida Stella AM, Galli F, Butterfield DA (2003) Redox regulation of heat shock protein expression in aging and neurodegenerative disorders associated with oxidative stress: a nutritional approach. Amino Acids 25:437–444
Poon HF, Calabrese V, Scapagnini G, Butterfield DA (2004) Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontology 59:478–493
Forman HJ, Fukuto JM, Torres M (2004) Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol 287:246–256
Poon HF, Calabrese V, Scapagnini G, Butterfield DA (2004) Free radicals and brain aging. Clin Geriatr Med 20:329–359
Calabrese V, Scapagnini G, Ravagna A, Colombrita C, Spadaro F, Butterfield DA, Giuffrida Stella AM (2004) Increased expression of heat shock proteins in rat brain during aging: relationship with mitochondrial function and glutathione redox state. Mech Age Dev 125:325–335
Calabrese V, Giuffrida Stella AM, Butterfield DA, Scapagnini G (2004) Redox regulation in neurodegeneration and longevity: role of the heme oxygenase and HSP70 systems in brain stress tolerance. Antioxid Redox Signal 6:895–913
Halliwell B (2002) Hypothesis: proteasomal dysfunction: a primary event in neurogeneration that leads to nitrative and oxidative stress and subsequent cell death. Ann NY Acad Sci 962:182–194
Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192:1–15
Bergamini CM, Gambetti S, Dondi A, Cervellati C (2004) Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des 10:1611–1626
Pappolla MA, Chyan YJ, Omar RA, Hsiao K, Perry G, Smith MA, Bozner P (1998) Evidence of oxidative stress and in vivo neurotoxicity of beta-amyloid in a transgenic mouse model of Alzheimer’s disease: a chronic oxidative paradigm for testing antioxidant therapies in vivo. Am J Pathol 152:871–877
Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL, Tabaton M, Perry G (1998) Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 70:2212–2215
Butterfield DA, Drake J, Pocernich C, Castegna A (2001) Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med 7:548–554
Butterfield DA, Lauderback CM (2002) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic Biol Med 32:1050–1060
Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430:631–639
Drew B, Leeuwenburgh C (2002) Aging and the role of reactive nitrogen species. Ann NY Acad Sci 959:66–81
Kroncke KD (2003) Nitrosative stress and transcription. Biol Chem 384:1365–1377
Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA (2004) The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem 385:1–10
Calabrese V, Guagliano E, Sapienza M, Panebianco M, Calafato S, Puleo E, Pennisi G, Mancuso C, Butterfield DA, Stella AG (2007) Redox regulation of cellular stress response in aging and neurodegenerative disorders: role of vitagenes. Neurochem Res 32:757–773
Calabrese V, Boyd-Kimball D, Scapagnini G, Butterfield DA (2004) Nitric oxide and cellular stress response in brain aging and neurodegenerative disorders: the role of vitagenes. In Vivo 18:245–267
Mancuso C, Scapagnini G, Curro D, Giuffrida Stella AM, De Marco C, Butterfield DA, Calabrese V (2007) Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front Biosci 12:1107–1123
Vina J, Borras C, Gomez-Cabrera MC, Orr WC (2006) Part of the series: from dietary antioxidants to regulators in cellular signalling and gene expression. Role of reactive oxygen species and (phyto)oestrogens in the modulation of adaptive response to stress. Free Radic Res 40:111–119
McCord JM, Fridovich I (1988) Superoxide dismutase: the first twenty years (1968–1988). Free Radic Biol Med 5:363–369
Calabrese EJ, Staudenmayer JW, Stanek EJ (2006) Drug development and hormesis: changing conceptual understanding of the dose response creates new challenges and opportunities for more effective drugs. Curr Opin Drug Discov Devel 9:117–123
Zhang K, Kaufman RJ (2006) The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66:102–109
Calabrese V, Butterfield DA, Scapagnini G, Stella AM, Maines MD (2006) Redox regulation of heat shock protein expression by signaling involving nitric oxide and carbon monoxide: relevance to brain aging, neurodegenerative disorders, and longevity. Antioxid Redox Signal 8:444–477
Maines MD (1997) The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 37:517–554
Mancuso C (2004) Heme oxygenase and its products in the nervous system. Antioxid Redox Signal 6:878–887
Stocker R (2004) Antioxidant activities of bile pigments. Antioxid Redox Signal 6:841–849
Mancuso C, Pani G, Calabrese V (2006) Bilirubin: an endogenous scavenger of nitric oxide and reactive nitrogen species. Redox Rep 11:207–213
Mancuso C, Bonsignore A, Capone C, Di Stasio E, Pani G (2006) Albumin-bound bilirubin interacts with nitric oxide by a redox mechanism. Antioxid Redox Signal 8:487–494
Mancuso C, Bonsignore A, Di Stasio E, Mordente A, Motterlini R (2003) Bilirubin and S-nitrosothiols interaction: evidence for a possible role of bilirubin as a scavenger of nitric oxide. Biochem Pharmacol 66:2355–2363
Simonian NA, Coyle JT (1996) Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36:83–106
Sayre LM, Smith MA, Perry G (2001) Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem 8:721–738
Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10:18–25
Mancuso C, Perluigi M, Cini C, De Marco C, Giuffrida Stella AM, Calabrese V (2006) Heme oxygenase and cyclooxygenase in the central nervous system: a functional interplay. J Neurosci Res 84:1385–1391
Panahian N, Yoshiura M, Maines MD (1999) Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 72:1187–1203
Takeda A, Perry G, Abraham NG, Dwyer BE, Kutty RK, Laitinen JT, Petersen RB, Smith MA (2000) Overexpression of heme oxygenase in neuronal cells, the possible interaction with Tau. J Biol Chem 275:5395–5399
Premkumar DR, Smith MA, Richey PL, Petersen RB, Castellani R, Kutty RK, Wiggert B, Perry G, Kalaria RN (1995) Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer’s disease. J Neurochem 65:1399–1402
Schipper HM (2000) Heme oxygenase-1: role in brain aging and neurodegeneration. Exp Gerontol 35:821–830
Mancuso C, Bates TE, Butterfield DA, Calafato S, Cornelius C, De Lorenzo A, Dinkova Kostova AT, Calabrese V (2007) Natural antioxidants in Alzheimer’s disease. Expert Opin Investig Drugs 16:1921–1931
Butterfield D, Castegna A, Pocernich C, Drake J, Scapagnini G, Calabrese V (2002) Nutritional approaches to combat oxidative stress in Alzheimer’s disease. J Nutr Biochem 13:444–461
Scapagnini G, Colombrita C, Amadio M, D’Agata V, Arcelli E, Sapienza M, Quattrone A, Calabrese V (2006) Curcumin activates defensive genes and protects neurons against oxidative stress. Antioxid Redox Signal 8:395–403
Ganguli M, Chandra V, Kamboh MI, Johnston JM, Dodge HH, Thelma BK, Juyal RC, Pandav R, Belle SH, DeKosky ST (2000) Apolipoprotein E polymorphism and Alzheimer disease: The Indo-US Cross-National Dementia Study. Arch Neurol 57:824–830
Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM (2001) The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci 21:8370–8377
Wu L, Wang R (2005) Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev 57:585–630
Kostoglou-Athanassiou I, Forsling ML, Navarra P, Grossman AB (1996) Oxytocin release is inhibited by the generation of carbon monoxide from the rat hypothalamus—further evidence for carbon monoxide as a neuromodulator. Brain Res Mol Brain Res 42:301–306
Mancuso C, Kostoglou-Athanassiou I, Forsling ML, Grossman AB, Preziosi P, Navarra P, Minotti G (1997) Activation of heme oxygenase and consequent carbon monoxide formation inhibits the release of arginine vasopressin from rat hypothalamic explants, molecular linkage between heme catabolism and neuroendocrine function. Brain Res Mol Brain Res 50:267–276
Mancuso C, Ragazzoni E, Tringali G, Liberale I, Preziosi P, Grossman A, Navarra P (1999) Inhibition of heme oxygenase in the central nervous system potentiates endotoxin-induced vasopressin release in the rat. J Neuroimmunol 99:189–194
Parkes D, Kasckow J, Vale W (1994) Carbon monoxide modulates secretion of corticotropin-releasing factor from rat hypothalamic cell cultures. Brain Res 646:315–318
Pozzoli G, Mancuso C, Mirtella A, Preziosi P, Grossman AB, Navarra P (1994) Carbon monoxide as a novel neuroendocrine modulator: inhibition of stimulated corticotropin-releasing hormone release from acute rat hypothalamic explants. Endocrinology 135:2314–2317
Mancuso C, Pistritto G, Tringali G, Grossman AB, Preziosi P, Navarra P (1997) Evidence that carbon monoxide stimulates prostaglandin endoperoxide synthase activity in rat hypothalamic explants and in primary cultures of rat hypothalamic astrocytes. Brain Res Mol Brain Res 45:294–300
Mancuso C, Tringali G, Grossman A, Preziosi P, Navarra P (1998) The generation of nitric oxide and carbon monoxide produces opposite effects on the release of immunoreactive interleukin-1beta from the rat hypothalamus in vitro: evidence for the involvement of different signaling pathways. Endocrinology 139:1031–1037
Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, E S, Cheng X (2002) Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res 91:610–617
Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell R, Choi AM (2000) Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6:422–428
Ryter SW, Otterbein LE, Morse D, Choi AM (2002) Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem 234–235:249–263
Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK (1999) The neuroprotective potential of heat shock protein 70 (HSP70). Mol Med Today 5:525–531
Kelly S, Zhang ZJ, Zhao H, Xu L, Giffard RG, Sapolsky RM, Yenari MA, Steinberg GK (2002) Gene transfer of HSP72 protects cornu ammonis 1 region of the hippocampus neurons from global ischemia: influence of Bcl-2. Ann Neurol 52:160–167
Narasimhan P, Swanson RA, Sagar SM, Sharp FR (1996) Astrocyte survival and HSP70 heat shock protein induction following heatshock and acidosis. Glia 17:147–159
Fink SL, Chang LK, Ho DY, Sapolsky RM (1997) Defective herpes simplex virus vectors expressing the rat brain stress-inducible heat shock protein 72 protect cultured neurons from severe heat shock. J Neurochem 68:961–969
Hata R, Maeda K, Hermann D, Mies G, Hossmann KA (2000) Dynamics of regional brain metabolism and gene expression after middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 20:306–315
Perez N, Sugar J, Charya S, Johnson G, Merril C, Bierer L, Perl D, Haroutunian V, Wallace W (1991) Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer’s disease. Brain Res Mol Brain Res 11:249–254
Yoo BC, Seidl R, Cairns N, Lubec G (1999) Heat-shock protein 70 levels in brain of patients with Down syndrome and Alzheimer’s disease. J Neural Transm Suppl 57:315–322
Morrison-Bogorad M, Zimmerman AL, Pardue S (1995) Heat-shock 70 messenger RNA levels in human brain: correlation with agonal fever. J Neurochem 64:235–246
Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S (2002) Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J 16:601–603
Calabrese V, Testa G, Ravagna A, Bates TE, Stella AM (2000) HSP70 induction in the brain following ethanol administration in the rat: regulation by glutathione redox state. Biochem Biophys Res Commun 269:397–400
Calabrese V, Bates TE, Giuffrida Stella AM (2000) NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: the role of oxidant/antioxidant balance. Neurochem Res 65:1315–1341
Yamawaki H, Haendeler J, Berk BC (2003) Thioredoxin: a key regulator of cardiovascular homeostasis. Circ Res 93:1029–1033
Cho CG, Kim HJ, Chung SW, Jung KJ, Shim KH, Yu BP, Yodoi J, Chung HY (2003) Modulation of glutathione and thioredoxin systems by calorie restriction during the aging process. Exp Gerontol 38:539–548
Arner ES, Holmgren A (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 267:6102–6109
Sun QA, Kirnarsky L, Sherman S, Gladyshev VN (2001) Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc Natl Acad Sci USA 98:3673–3678
Bloomfield KL, Osborne SA, Kennedy DD, Clarke FM, Tonissen KF (2003) Thioredoxin-mediated redox control of the transcription factor Sp1 and regulation of the thioredoxin gene promoter. Gene 319:107–116
Kim YC, Yamaguchi Y, Kondo N, Masutani H, Yodoi J (2003) Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene 22:1860–1865
Tanito M, Masutani H, Kim YC, Nishikawa M, Ohira A, Yodoi J (2005) Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Invest Ophthalmol Vis Sci Mar 46:979–987
Eftekharpour E, Holmgren A, Juurlink BH (2000) Thioredoxin reductase and glutathione synthesis is upregulated by t-butylhydroquinone in cortical astrocytes but not in cortical neurons. Glia 31:241–248
Hintze KJ, Wald KA, Zeng H, Jeffery EH, Finley JW (2003) Thioredoxin reductase in human hepatoma cells is transcriptionally regulated by sulforaphane and other electrophiles via an antioxidant response element. J Nutr 133:2721–2727
Dinkova-Kostova AT, Cheah J, Samouilov A, Zweier JL, Bozak RE, Hicks RJ, Talalay P (2007) Phenolic Michael reaction acceptors: combined direct and indirect antioxidant defenses against electrophiles and oxidants. Med Chem 3:261–268
Sakurai A, Nishimoto M, Himeno S, Imura N, Tsujimoto M, Kunimoto M, Hara S (2005) Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J Cell Physiol 203:529–537
Hirota K, Nakamura H, Masutani H, Yodoi J (2002) Thioredoxin superfamily and thioredoxin-inducing agents. Ann NY Acad Sci 957:189–199
Baker AF, Dragovich T, Tate WR, Ramanathan RK, Roe D, Hsu CH, Kirkpatrick DL, Powis G (2006) The antitumor thioredoxin-1 inhibitor PX-12 (1-methylpropyl 2-imidazolyl disulfide) decreases thioredoxin-1 and VEGF levels in cancer patient plasma. J Lab Clin Med 147:83–90
Nakamura H, Bai J, Nishinaka Y, Ueda S, Sasada T, Ohshio G, Imamura M, Takabayashi A, Yamaoka Y, Yodoi J (2000) Expression of thioredoxin and glutaredoxin, redox-regulating proteins, in pancreatic cancer. Cancer Detect Prev 24:53–60
Nakamura H, Masutani H, Yodoi J (2002) Redox imbalance and its control in HIV infection. Antioxid Redox Signal 4:455–464
Haapasalo H, Kylaniemi M, Paunul N, Kinnula VL, Soini Y (2003) Expression of antioxidant enzymes in astrocytic brain tumors. Brain Pathol 13:155–164
Berggren MM, Powis G (2001) Alternative splicing is associated with decreased expression of the redox proto-oncogene thioredoxin-1 in human cancers. Arch Biochem Biophys 389:144–149
Biaglow JE, Miller RA (2005) The thioredoxin reductase/thioredoxin system: novel redox targets for cancer therapy. Cancer Biol Ther 4:6–13
Bai J, Nakamura H, Kwon YW, Hattori I, Yamaguchi Y, Kim YC, Kondo N, Oka S, Ueda S, Masutani H, Yodoi J (2003) Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. J Neurosci 23:503–509
Masutani H, Bai J, Kim YC, Yodoi J (2004) Thioredoxin as a neurotrophic cofactor and an important regulator of neuroprotection. Mol Neurobiol 29:229–242
Trigona WL, Mullarky IK, Cao Y, Sordillo LM (2006) Thioredoxin reductase regulates the induction of heme oxygenase-1 expression in aortic endothelial cells. Biochem J 394:207–216
Satoh T, Ishige K, Sagara Y (2004) Protective effects on neuronal cells of mouse afforded by ebselen against oxidative stress at multiple steps. Neurosci Lett 371:1–5
Das KC, Das CK (2000) Thioredoxin, a singlet oxygen quencher and hydroxyl radical scavenger: redox independent functions. Biochem Biophys Res Commun 277:443–447
Ju TC, Chen SD, Liu CC, Yang DI (2005) Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic Biol Med 38:938–949
Rauhala P, Andoh T, Yeh K, Chiueh CC (2002) Contradictory effects of sodium nitroprusside and S-nitroso-N-acetylpenicillamine on oxidative stress in brain dopamine neurons in vivo. Ann NY Acad Sci 962:60–72
Lee SY, Andoh T, Murphy DL, Chiueh CC (2003) 17beta-estradiol activates ICI 182, 780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. FASEB J 17:947–948
Westphal CH, Dipp MA, Guarente L (2007) A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci 32:555–560
Salminen A, Ojala J, Huuskonen J, Kauppinen A, Suuronen T, Kaarniranta K (2008) Interaction of aging-associated signaling cascades: inhibition of NF-kappaB signaling by longevity factors FoxOs and SIRT1. Cell Mol Life Sci 65:1049–1058
Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J (2007) Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med 39:335–345
Martin B, Mattson MP, Maudsley S (2006) Caloric restriction and intermittent fasting: two potential diets for successful brain aging. Ageing Res Rev 5:332–353
Mattson MP (2008) Hormesis defined. Ageing Res Rev 7:1–7
Giannakou ME, Goss M, Jacobson J, Vinti G, Leevers SJ, Partridge L (2007) Dynamics of the action of dFOXO on adult mortality in Drosophila. Aging Cell 6:429–438
Guarente L, Picard F (2005) Calorie restriction—SIR2 connection. Cell 120:473–482
Rodgers JT, Lerin C, Haas W, Cygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1and SIRT1. Nature 434:113–118
Wang F, Nguyen M, Qin FX, Tong Q (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6:505–514
Yang T, Sauve AA (2006) NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. AAPS J 8:632–643
Smith J (2002) Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol 12:404–406
Hipkiss AR (2008) Energy metabolism, altered proteins, sirtuins and ageing: converging mechanisms? Biogerontology 9:49–55
Hipkiss AR (2007) Could carnosine or related structures suppress Alzheimer’s disease? J Alzheimer’s Dis 11:229–240
Hipkiss AR (2007) On why decreasing protein synthesis can increase lifespan. Mech Ageing Dev 128:412–414
Baur JA, Sinclair DA (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506
Pallàs M, Verdaguer E, Tajes M, Gutierrez-Cuesta J, Camins A (2008) Modulation of sirtuins: new targets for antiageing. Recent Patents CNS Drug Discov 3:61–69
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450:712–716
Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122
Suzuki K, Koike T (2007) Resveratrol abolishes resistance to axonal degeneration in slow Wallerian degeneration (WldS) mice: activation of SIRT2, an NAD-dependent tubulin deacetylase. Biochem Biophys Res Commun 359:665–671
Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG (2007) Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317:461–462
Qin W, Chachich M, Lane M, Roth G, Bryant M, de Cabo R, Ottinger MA, Mattison J, Ingram D, Gandy S, Pasinetti GM (2006) Calorie restriction attenuates Alzheimer’s disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus). J Alzheimers Dis 10:417–422
Mayer MP, Bukau B (1999) Molecular chaperones: the busy life of Hsp90. Curr Biol 9:322–325
Deocaris CC, Kaul SC, Wadhwa R (2006) On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones 11:116–128
Qiu XB, Shao YM, Miao S, Wang L (2006) The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci 63:2560–2570
James M, Crabbe C, Hepburne-Scott HW (2001) Small heat shock proteins (sHSPs) as potential drug targets. Curr Pharm Biotechnol 2:77–111
Rahman I, Biswas SK, Kirkham PA (2006) Regulation of inflammation and redox signaling by dietary polyphenols. Biochem Pharmacol 72:1439–1452
Sharma RA, Gescher AJ, Steward WP (2005) Curcumin: the story so far. Eur J Cancer 41:1955–1968
Ravindranath V, Chandrasekhara N (1981–1982) Metabolism of curcumin—studies with [3H]curcumin. Toxicology 22:337–344
Ravindranath V, Chandrasekhara N (1980) Absorption and tissue distribution of curcumin in rats. Toxicology 16:259–265
Maiti K, Mukherjee K, Gantait A, Saha BP, Mukherjee PK (2007) Curcumin-phospholipid complex: preparation, therapeutic evaluation and pharmacokinetic study in rats. Int J Pharm 330:155–163
Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS (1998) Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med 64:353–356
Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, Ko JY, Lin JT, Lin BR, Ming-Shiang W, Yu HS, Jee SH, Chen GS, Chen TM, Chen CA, Lai MK, Pu YS, Pan MH, Wang YJ, Tsai CC, Hsieh CY (2001) Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 21:2895–2900
Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ (2007) Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem 102:1095–1104
Somparn P, Phisalaphong C, Nakornchai S, Unchern S, Morales NP (2007) Comparative antioxidant activities of curcumin and its demethoxy and hydrogenated derivatives. Biol Pharm Bull 30:74–78
Ireson CR, Jones DJ, Orr S, Coughtrie MW, Boocock DJ, Williams ML, Farmer PB, Steward WP, Gescher AJ (2002) Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol Biomarkers Prev 11:105–111
Lin JK, Pan MH, Lin-Shiau SY (2000) Recent studies on the biofunctions and biotransformations of curcumin. Biofactors 13:153–158
Hayeshi R, Mutingwende I, Mavengere W, Masiyanise V, Mukanganyama S (2007) The inhibition of human glutathione S-transferases activity by plant polyphenolic compounds ellagic acid and curcumin. Food Chem Toxicol 45:286–295
Thapliyal R, Maru GB (2001) Inhibition of cytochrome P450 isozymes by curcumins in vitro and in vivo. Food Chem Toxicol 39:541–547
Basu NK, Ciotti M, Hwang MS, Kole L, Mitra PS, Cho JW, Owens IS (2004) Differential and special properties of the major human UGT1-encoded gastrointestinal UDP-glucuronosyltransferases enhance potential to control chemical uptake. J Biol Chem 279:1429–1441
Oetari S, Sudibyo M, Commandeur JN, Samhoedi R, Vermeulen NP (1996) Effects of curcumin on cytochrome P450 and glutathione S-transferase activities in rat liver. Biochem Pharmacol 51:39–45
Deodhar SD, Sethi R, Srimal RC (1980) Preliminary study on antirheumatic activity of curcumin (diferuloyl methane). Indian J Med Res 71:632–634
Sharma RA, Euden SA, Platton SL, Cooke DN, Shafayat A, Hewitt HR, Marczylo TH, Morgan B, Hemingway D, Plummer SM, Pirmohamed M, Gescher AJ, Steward WP (2004) Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res 10:6847–6854
Sugiyama Y, Kawakishi S, Osawa T (1996) Involvement of the beta-diketone moiety in the antioxidative mechanism of tetrahydrocurcumin. Biochem Pharmacol 52:519–525
Osawa T, Sugiyama Y, Inayoshi M, Kawakishi S (1995) Antioxidative activity of tetrahydrocurcuminoids. Biosci Biotechnol Biochem 59:1609–1612
Chen WF, Deng SL, Zhou B, Yang L, Liu ZL (2006) Curcumin and its analogues as potent inhibitors of low density lipoprotein oxidation: H-atom abstraction from the phenolic groups and possible involvement of the 4-hydroxy-3-methoxyphenyl groups. Free Radic Biol Med 40:526–535
Priyadarsini KI, Maity DK, Naik GH, Kumar MS, Unnikrishnan MK, Satav JG, Mohan H (2003) Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic Biol Med 35:475–484
Reyes-Gordillo K, Segovia J, Shibayama M, Vergara P, Moreno MG, Muriel P (2007) Curcumin protects against acute liver damage in the rat by inhibiting NF-kappaB, proinflammatory cytokines production and oxidative stress. Biochim Biophys Acta 1770:989–996
Venkataranganna MV, Rafiq M, Gopumadhavan S, Peer G, Babu UV, Mitra SK (2007) NCB-02 (standardized Curcumin preparation) protects dinitrochlorobenzene- induced colitis through down-regulation of NFkappa-B and iNOS. World J Gastroenterol 13:1103–1107
Shishodia S, Potdar P, Gairola CG, Aggarwal BB (2003) Curcumin (diferuloylmethane) down-regulates cigarette smoke-induced NF-kappaB activation through inhibition of IkappaBalpha kinase in human lung epithelial cells: correlation with suppression of COX-2, MMP-9 and cyclin D1. Carcinogenesis 24:1269–1279
Pan MH, Lin-Shiau SY, Lin JK (2000) Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappaB activation in macrophages. Biochem Pharmacol 60:1665–1676
Bhattacharyya S, Mandal D, Sen GS, Pal S, Banerjee S, Lahiry L, Finke JH, Tannenbaum CS, Das T, Sa G (2007) Tumor-induced oxidative stress perturbs nuclear factor-kappaB activity-augmenting tumor necrosis factor-alpha-mediated T-cell death: protection by curcumin. Cancer Res 67:362–370
Choi H, Chun YS, Kim SW, Kim MS, Park JW (2006) Curcumin inhibits hypoxia-inducible factor-1 by degrading aryl hydrocarbon receptor nuclear translocator: a mechanism of tumor growth inhibition. Mol Pharmacol 70:1664–1671
Abuarqoub H, Green CJ, Foresti R, Motterlini R (2007) Curcumin reduces cold storage-induced damage in human cardiac myoblasts. Exp Mol Med 39:139–148
Jeong GS, Oh GS, Pae HO, Jeong SO, Kim YC, Shin MK, Seo BY, Han SY, Lee HS, Jeong JG, Koh JS, Chung HT (2006) Comparative effects of curcuminoids on endothelial heme oxygenase-1 expression: ortho-methoxy groups are essential to enhance heme oxygenase activity and protection. Exp Mol Med 38:393–400
McNally SJ, Harrison EM, Ross JA, Garden OJ, Wigmore SJ (2006) Curcumin induces heme oxygenase-1 in hepatocytes and is protective in simulated cold preservation and warm reperfusion injury. Transplantation 81:623–626
Rushworth SA, Ogborne RM, Charalambos CA, O’Connell MA (2006) Role of protein kinase C delta in curcumin-induced antioxidant response element-mediated gene expression in human monocytes. Biochem Biophys Res Commun 341:1007–1016
Balogun E, Foresti R, Green CJ, Motterlini R (2003) Changes in temperature modulate heme oxygenase-1 induction by curcumin in renal epithelial cells. Biochem Biophys Res Commun 308:950–955
McNally SJ, Harrison EM, Ross JA, Garden OJ, Wigmore SJ (2007) Curcumin induces heme oxygenase 1 through generation of reactive oxygen species, p38 activation and phosphatase inhibition. Int J Mol Med 19:165–172
Andreadi CK, Howells LM, Atherfold PA, Manson MM (2006) Involvement of Nrf2, p38, B-Raf, and nuclear factor-kappaB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol Pharmacol 69:1033–1040
Rashmi R, Santhosh Kumar TR, Karunagaran D (2003) Human colon cancer cells differ in their sensitivity to curcumin-induced apoptosis and heat shock protects them by inhibiting the release of apoptosis-inducing factor and caspases. FEBS Lett 538:19–24
Sood A, Mathew R, Trachtman H (2001) Cytoprotective effect of curcumin in human proximal tubule epithelial cells exposed to shiga toxin. Biochem Biophys Res Commun 283:36–41
Chen YC, Tsai SH, Shen SC, Lin JK, Lee WR (2001) Alternative activation of extracellular signal-regulated protein kinases in curcumin and arsenite-induced HSP70 gene expression in human colorectal carcinoma cells. Eur J Cell Biol 80:213–221
Chen YC, Kuo TC, Lin-Shiau SY, Lin JK (1996) Induction of HSP70 gene expression by modulation of Ca(+2) ion and cellular p53 protein by curcumin in colorectal carcinoma cells. Mol Carcinog 17:224–234
Kato K, Ito H, Kamei K, Iwamoto I (1998) Stimulation of the stress-induced expression of stress proteins by curcumin in cultured cells and in rat tissues in vivo. Cell Stress Chaperones 3:152–160
Fang J, Lu J, Holmgren A (2005) Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem 280:25284–25290
Moi P, Chan K, Asunis I, Cao A, Kan YW (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA 91:9926–9930
Talalay P (2000) Chemoprotection against cancer by induction of phase 2 enzymes. Biofactors 12:5–11
Nguyen T, Scherratt PJ, Pickett CB (2003) Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol 43:233–260
Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD (2003) Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J 374:337–348
Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM (1995) Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE). Mol Med 1:827–837
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86
Motohashi H, Yamamoto M (2004) Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 10:549–557
Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M (2005) Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol 25:8044–8051
Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA (2005) Nrf2, a multi-organ protector? FASEB J 19:1061–1066
Kensler TW, Wakabayashi N, Biswal S (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47:89–116
McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD (2006) Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem 281:24756–24768
Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA 99:11908–11913
Dinkova-Kostova AT, Holtzclaw WD, Kensler TW (2005) The role of Keap1 in cellular protective responses. Chem Res Toxicol 18:1779–1791
Kobayashi M, Yamamoto M (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul 46:113–140
Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S (2004) Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 114:1248–1259
Rangasamy T, Guo G, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S (2005) Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202:47–59
Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S (2006) Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 116:984–995
Zhang X, Lu L, Dixon C, Wilmer W, Song H, Chen X, Rovin BH (2004) Stress protein activation by the cyclopentenone prostaglandin 15-deoxy-delta12, 14-prostaglandin J2 in human mesangial cells. Kidney Int 65:798–810
Rokutan K, Miyoshi M, Teshima S, Kawai T, Kawahara T, Kishi K (2000) Phenylarsine oxide inhibits heat shock protein 70 induction in cultured guinea pig gastric mucosal cells. Am J Physiol Cell Physiol 279:1506–1515
Liu H, Lightfoot R, Stevens JL (1996) Activation of heat shock factor by alkylating agents is triggered by glutathione depletion and oxidation of protein thiols. J Biol Chem 271:4805–4812
Tabner BJ, Turnbull S, El-Agnaf O, Allsop D (2001) Production of reactive oxygen species from aggregating proteins implicated in Alzheimer’s disease, Parkinson’s disease and other neurodegenerative diseases. Curr Top Med Chem 1:507–517
Barnham KJ, Cappai R, Beyreuther K, Masters CL, Hill AF (2006) Delineating common molecular mechanisms in Alzheimer’s and prion diseases. Trends Biochem Sci 31:465–472
Hinault M, Ben-Zvi A, Goloubinoff P (2006) Chaperones and proteases: cellular fold-controlling factors of proteins in neurodegenerative diseases and aging. J Mol Neurosci 30:249–265
Butterfield DA (2002) Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic Res 36:1307–1313
Zhang K, Kaufman RJ (2006) The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66:102–109
Nakamura T, Lipton SA (2007) Molecular mechanisms of nitrosative stress-mediated protein misfolding in neurodegenerative diseases. Cell Mol Life Sci 64:1609–1620
Schroder M (2006) The unfolded protein response. Mol Biotechnol 34:279–290
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Calabrese V, Sultana R, Scapagnini G, Guagliano E, Sapienza M, Bella R, Kanski J, Pennisi G, Mancuso C, Stella AM, Butterfield DA (2006) Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal 8:1975–1986
Katzman R, Saitoh T (1991) Advances in Alzheimer’s disease. FASEB J 5:278–286
Guix FX, Uribesalgo I, Coma M, Muñoz FJ (2005) The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol 76:126–152
Kim DS, Park SY, Kim JK (2001) Curcuminoids from Curcuma longa L. (Zingiberaceae) that protect PC12 rat pheochromocytoma and normal human umbilical vein endothelial cells from betaA(1–42) insult. Neurosci Lett 303:57–61
Ono K, Hasegawa K, Naiki H, Yamada M (2004) Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J Neurosci Res 75:742–750
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM (2005) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901
Voelker R (2006) Parkinson disease guidelines aid diagnosis, management. JAMA 295:2126–2128
Przedborski S, Ischiropoulos H (2005) Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal 7:685–693
Hald A, Lotharius J (2005) Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp Neurol 193:279–290
Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ (1983) A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Proc Natl Acad Sci USA 80:4546–4550
Chiueh CC, Andoh T, Lai AR, Lai E, Krishna G (2000) Neuroprotective strategies in Parkinson’s disease: protection against progressive nigral damage induced by free radicals. Neurotox Res 2:293–310
Rajeswari A (2006) Curcumin protects mouse brain from oxidative stress caused by 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Eur Rev Med Pharmacol Sci 10:157–161
Chen J, Tang XP, Zhi JL, Cui Y, Yu HM, Tang EH, Sun SN, Feng JQ, Chen PX (2006) Curcumin protects PC12 cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by bcl-2-mitochondria-ROS-iNOS pathway. Apoptosis 11:943–953
Mythri RB, Jagatha B, Pradhan N, Andersen J, Bharath MM (2007) Mitochondrial complex I inhibition in Parkinson’s disease: how can curcumin protect mitochondria? Antioxid Redox Signal 9:399–408
Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (1996) Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 335:1169–1175
Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427
Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Dürr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M (1997) Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet 6:1771–1780
Bidichandani SI, Ashizawa T, Patel PI (1998) The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet 62:111–1121
Lamarche JB, Lemieux B, Lieu HB (1984) The neuropathology of “typical” Friedreich’s ataxia in Quebec. Can J Neurol Sci 11:592–600
Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, Storey E, MacMillan JC, Collins V, Williamson R, Forrest SM (1999) Clinical and genetic study of Friedreich ataxia in an Australian population. Am J Med Genet 87:168–174
Montermini L, Richter A, Morgan K, Justice CM, Julien D, Castellotti B, Mercier J, Poirier J, Capozzoli F, Bouchard JP, Lemieux B, Mathieu J, Vanasse M, Seni MH, Graham G, Andermann F, Andermann E, Melançon SB, Keats BJ, Di Donato S, Pandolfo M (1997) Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol 41:675–678
Filla A, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Campanella G, Cocozza S (1996) The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet 59:554–560
Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD (2001) Sticky DNA, a selfassociated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. J Biol Chem 276:27171–27177
Krasilnikova MM, Mirkin SM (2004) Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol Cell Biol 24:2286–2295
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM (2006) Histonedeacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol 2:551–558
Greene E, Mahishi L, Entezam A, Kumari D, Usdin K (2007) Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res 35:3383–3390
Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, Trabzuni D, Pook M (2008) The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet 17:735–746
Sarsero JP, Li L, Wardan H, Sitte K, Williamson R, Ioannou PA (2003) Upregulation of expression from the FRDA genomic locus for the therapy of Friedreich ataxia. J Gene Med 5:72–81
Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH (2000) Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum Mol Genet 9:275–282
Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M (1997) Kaplan regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276:1709–1712
Foury F, Cazzalini O (1997) Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett 411:373–377
Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (1997) Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat Genet 16:345–351
Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (1997) Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet 17:215–217
Foury F (1999) Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Lett 456:281–284
Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI (2004) Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 305:242–245
Piemonte F, Pastore A, Tozzi G, Tagliacozzi D, Santorelli FM, Carrozzo R, Casali C, Damiano M, Federici G, Bertini E (2001) Glutathione in blood of patients with Friedreich’s ataxia. Eur J Clin Invest 31:1007–1011
Schulz JB, Dehmer T, Schöls L, Mende H, Hardt C, Vorgerd M, Bürk K, Matson W, Dichgans J, Beal MF, Bogdanov MB (2000) Oxidative stress in patients with Friedreich ataxia. Neurology 55:1719–1721
Emond M, Lepage G, Vanasse M, Pandolfo M (2000) Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 55:1752–1753
Bradley JL, Homayoun S, Hart PE, Schapira AH, Cooper JM (2004) Role of oxidative damage in Friedreich’s ataxia. Neurochem Res 29:561–567
Santos MM, Ohshima K, Pandolfo M (2001) Frataxin deficiency enhances apoptosis in cells differentiating into neuroectoderm. Hum Mol Genet 10:1935–1944
Sturm B, Stupphann D, Kaun C, Boesch S, Schranzhofer M, Wojta J, Goldenberg H, Scheiber-Mojdehkar B (2005) Recombinant human erythropoietin: effects on frataxin expression in vitro. Eur J Clin Invest 35:711–717
Boesch S, Sturm B, Hering S, Goldenberg H, Poewe W, Scheiber-Mojdehkar B (2007) Friedreich’s ataxia: clinical pilot trial with recombinant human erythropoietin. Ann Neurol 62:521–524
Cooper JM, Schapira AH (2003) Friedreich’s ataxia: disease mechanisms, antioxidant and coenzyme Q10 therapy. Biofactors 18:163–171
Cooper JM, Schapira AH (2007) Friedreich’s ataxia: coenzyme Q10 and vitamin E therapy. Mitochondrion 7:127–135
Lodi R, Hart PE, Rajagopalan B, Taylor DJ, Crilley JG, Bradley JL, Blamire AM, Manners D, Styles P, Schapira AH, Cooper JM (2001) Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann Neurol 49:590–596
Hart PE, Lodi R, Rajagopalan B, Bradley JL, Crilley JG, Turner C, Blamire AM, Manners D, Styles P, Schapira AH, Cooper JM (2005) Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol 62:621–626
Rustin P, von Kleist-Retzow JC, Chantrel-Groussard K, Sidi D, Munnich A, Rötig A (1999) Effect of idebenone on cardiomyopathy in Friedreich’s ataxia: a preliminary study. Lancet 354:477–479
Hausse AO, Aggoun Y, Bonnet D, Sidi D, Munnich A, Rötig A, Rustin P (2002) Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart 87:346–349
Buyse G, Mertens L, Di Salvo G, Matthijs I, Weidemann F, Eyskens B, Goossens W, Goemans N, Sutherland GR, Van Hove JL (2003) Idebenone treatment in Friedreich’s ataxia: neurological, cardiac, and biochemical monitoring. Neurology 60:1679–1681
Mariotti C, Solari A, Torta D, Marano L, Fiorentini C, Di Donato S (2003) Idebenone treatment in Friedreich patients: one-year-long randomized placebo-controlled trial. Neurology 60:1676–1679
Di Prospero NA, Baker A, Jeffries N, Fischbeck KH (2007) Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial. Lancet Neurol 6:878–886
Jauslin ML, Meier T, Smith RA, Murphy MP (2003) Mitochondria-targeted antioxidants protect Friedreich ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J 17:1972–1974
Calabrese V, Scapagnini G, Ravagna A, Giuffrida Stella AM, Butterfield DA (2002) Molecular chaperones and their roles in neural cell differentiation. Dev Neurosci 24:1–13
Al-Omar FA, Nagi MN, Abdulgadir MM, Al Joni KS, Al-Majed AA (2006) Immediate and delayed treatments with curcumin prevents forebrain ischemia-induced neuronal damage and oxidative insult in the rat hippocampus. Neurochem Res 31:611–618
Wang Q, Sun AY, Simonyi A, Jensen MD, Shelat PB, Rottinghaus GE, MacDonald RS, Miller DK, Lubahn DE, Weisman GA, Sun GY (2005) Neuroprotective mechanisms of curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. J Neurosci Res 82:138–148
Ghoneim AI, Abdel-Naim AB, Khalifa AE, El-Denshary ES (2002) Protective effects of curcumin against ischaemia/reperfusion insult in rat forebrain. Pharmacol Res 46:273–279
Evans AM, Fornasini G (2003) Pharmacokinetics of L-carnitine. Clin Pharmacokinet 42:941–967
Rebouche CJ (2004) Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann NY Acad Sci 1033:30–41
Lombard KA, Olson AL, Nelson SE, Rebouche CJ (1989) Carnitine status of lactoovovegetarians and strict vegetarian adults and children. Am J Clin Nutr 50:301–306
Brass EP, Hoppel CL, Hiatt WR (1994) Effect of intravenous L-carnitine on carnitine homeostasis and fuel metabolism during exercise in humans. Clin Pharmacol Ther 55:681–692
Gross CJ, Savaiano DA (1993) Effect of development and nutritional state on the uptake, metabolism and release of free and acetyl-L-carnitine by the rodent small intestine. Biochim Biophys Acta 1170:265–274
Parnetti L, Gaiti A, Mecocci P, Cadini D, Senin U (1992) Pharmacokinetics of IV and oral acetyl-L-carnitine in a multiple dose regimen in patients with senile dementia of Alzheimer type. Eur J Clin Pharmacol 42:89–93
Kelly JG, Hunt S, Doyle GD, Laher MS, Carmody M, Marzo A, Arrigoni Martelli E (1990) Pharmacokinetics of oral acetyl-L-carnitine in renal impairment. Eur J Clin Pharmacol 38:309–312
McDaniel MA, Maier SF, Einstein GO (2003) “Brain-specific” nutrients: a memory cure? Nutrition 19:957–975
Abdul HM, Calabrese V, Calvani M, Butterfield DA (2006) Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1–42-mediated oxidative stress and neurotoxicity: implications for Alzheimer’s disease. J Neurosci Res 84:398–408
Calabrese V, Colombrita C, Sultana R, Scapagnini G, Calvani M, Butterfield DA, Stella AM (2006) Redox modulation of heat shock protein expression by acetylcarnitine in aging brain: relationship to antioxidant status and mitochondrial function. Antioxid Redox Signal 8:404–416
Rai G, Wright G, Scott L, Beston B, Rest J, Exton-Smith AN (1990) Double-blind, placebo controlled study of acetyl-l-carnitine in patients with Alzheimer’s dementia. Curr Med Res Opin 11:638–647
Spagnoli A, Lucca U, Menasce G, Bandera L, Cizza G, Forloni G, Tettamanti M, Frattura L, Tiraboschi P, Comelli M (1991) Long-term acetyl-L-carnitine treatment in Alzheimer’s disease. Neurology 41:1726–1732
Thal LJ, Carta A, Clarke WR, Ferris SH, Friedland RP, Petersen RC, Pettegrew JW, Pfeiffer E, Raskind MA, Sano M, Tuszynski MH, Woolson RF (1996) A 1-year multicenter placebo-controlled study of acetyl-L-carnitine in patients with Alzheimer’s disease. Neurology 47:705–711
Thal LJ, Calvani M, Amato A, Carta A (2000) A 1-year controlled trial of acetyl-l-carnitine in early-onset AD. Neurology 55:805–810
Calabrese V, Ravagna A, Colombrita C, Guagliano E, Scapagnini G, Calvani M, Butterfield DA, Giuffrida Stella AM (2005) Acetylcarnitine induces heme oxygenase in rat astrocytes and protects against oxidative stress: involvement of the transcription factor Nrf2. J Neurosci Res 79:509–521
Snyder SH (1980) Brain peptides as neurotransmitters. Science 209:976–983
Abe H (2000) Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochemistry (Mosc) 65:757–765
Hipkiss AR, Preston JE, Himsworth DT, Worthington VC, Keown M, Michaelis J, Lawrence J, Mateen A, Allende L, Eagles PA, Abbott NJ (1998) Pluripotent protective effects of carnosine, a naturally occurring dipeptide. Ann NY Acad Sci 854:37–53
Bakardjiev A (1997) Biosynthesis of carnosine in primary cultures of rat olfactory bulb. Neurosci Lett 227:115–118
Teufel M, Saudek V, Ledig JP, Bernhardt A, Boularand S, Carreau A, Cairns NJ, Carter C, Cowley DJ, Duverger D, Ganzhorn AJ, Guenet C, Heintzelmann B, Laucher V, Sauvage C, Smirnova T (2003) Sequence identification and characterization of human carnosinase and a closely related non-specific dipeptidase. J Biol Chem 278:6521–6531
Bauer K (2004) X-His dipeptidase. In: Barret AJ, Rawlings ND, Woessner JF (eds) Handbook of proteolytic enzymes, 2nd edn. Elsevier, Amsterdam, pp 1023–1024
Bauer K (2004) Cytosol non-specific dipeptidase. In: Barret AJ, Rawlings ND, Woessner JF (eds) Handbook of Proteolytic Enzymes, 2nd edn. Amsterdam, Elsevier, pp 1020–1022
De Marchis S, Modena C, Peretto P, Giffard C, Fasolo A (2000) Carnosine-like immunoreactivity in the central nervous system of rats during postnatal development. J Comp Neurol 426:378–390
Bauer K (2005) Carnosine and homocarnosine, the forgotten, enigmatic peptides of the brain. Neurochem Res 30:1339–1345
McFarland GA, Holliday R (1994) Retardation of senescence in cultured human diploid fibroblasts by carnosine. Exp Cell Res 212:167–175
Yuneva AO, Kramarenko GG, Vetreshchak TV, Gallant S, Boldyrev AA (2002) Effect of carnosine on Drosophila melanogaster lifespan. Bull Exp Biol Med 133:559–561
Yuneva AO, Bulygina ER, Gallant S, Kramarenko GG, Stvolinsky SL, Semyonova ML, Boldyrev AA (1999) Effect of carnosine on age-induced changes in senescence-accelerated mice. J Anti-Aging Med 2:337–342
Babizhayev MA (2006) Biological activities of the natural imidazole-containing peptidomimetics n-acetylcarnosine, carcinine and l-carnosine in ophthalmic and skin care products. Life Sci 78:2343–2357
Lee Y, Hsu C, Lin M, Liu K, Yin M (2005) Histidine and carnosine delay diabetic deterioration in mice and protect human low density lipoprotein against oxidation and glycation. Eur J Pharmacol 512:145–150
Rashid I, van Reyk DM, Davies MJ (2007) Carnosine and its constituents inhibit glycation of low-density lipoproteins that promotes foam cell formation in vitro. FEBS Letts 581:1067–1070
Sauerhofer S, Yuan G, Braun GS, Deizer M, Neumaier M, Gretz N, Floege J, Kriz W, van der Woude F, Moeller MJ (2007) L-Carnosine, a substrate of carnosinase-1, influences glucose metabolism. Diabetes 56:2425–2432
Hipkiss AR (2005) Glycation, aging and carnosine: are carnivorous diets beneficial? Mech Ageing Dev 126:1034–1039
Huang Y, Duan J, Chen H, Chen M, Chen G (2005) Separation and determination of carnosine-related peptides using capillary electrophoresis with laser-induced fluorescence detection. Electrophoresis 26:593–599
Hipkiss AR (2005) Could carnosine suppress zinc-mediated proteasome inhibition and neurodegeneration? Therapeutic potential of a non-toxic but non-patentable dipeptide. Biogerontology 6:147–149
Hipkiss AR (2007) Could carnosine or related structures suppress Alzheimer’s disease? J Alzheimer’s Dis 11:229–240
Dobrota D, Fedorova T, Stvolinsky S, Babusikova E, Likavcanova K, Drgova A, Strapkova A, Boldyrev A (2005) Carnosine protects the brain of rats and Mongolian gerbils against ischemic injury: after-stroke-effect. Neurochem Res 30:1283–1288
Stvolinsky S, Kukley M, Dobrota D, Mezesova V, Boldyrev A (2000) Carnosine protects rats under global ischemia. Brain Res Bull 53:445–448
Tang S, Arumugam TV, Cutler RG, Jo D, Magnus T, Chan SL, Mughal MR, Telljohann RS, Nassar M, Ouyang X, Calderan A, Ruzza P, Guiotto A, Mattson MP (2007) J Neurochem 101:729–736
Pubill D, Verdaguer E, Sureda FX, Camins A, Pallas M, Camarasa J, Escubedo E (2002) Carnosine prevents methamphetamine-induced gliosis but not dopamine terminal loss in rats. Eur J Pharmacol 448:165–168
Dukic-Stefanovic S, Schinzel R, Riederer P, Munch G (2001) AGES in brain ageing: AGE-inhibitors as neuroprotective and anti-dementia drugs? Biogerontology 2:19–34
Trombley PQ, Horning MS, Blakemore LJ (1998) Carnosine modulates zinc and copper effects on amino acid receptors and synaptic transmission. Neuroreport 9:3503–3507
Horning MS, Blakemore LJ, Trombley PQ (2000) Endogenous mechanisms of neuroprotection: role of zinc, copper, and carnosine. Brain Res 852:56–61
Preston JE, Hipkiss AR, Himsworth DT, Romero IA, Abbott JN (1998) Toxic effects of beta-amyloid(25–35) on immortalised rat brain endothelial cell: protection by carnosine, homocarnosine and beta-alanine. Neurosci Lett 242:105–108
Fu Q, Dai H, Hu W, Fan Y, Zhang Y, Chen Z (2008) Carnosine protects against Aβ-42-induced neurotoxicity in differentiated rat PC12 cells. Cell Mol Neurobiol 28:307–316
Shen Y, Hu W, Dai H, Fu Q, Wei E, Luo J, Chen Z (2007) Carnosine protects against NMDA-induced neurotoxicity in differentiated rat PC12 cells through carnosine-histidine-histamine pathway and H1/H3 receptors. Biochem Pharmacol 73:709–717
Fonteh AN, Harrington RJ, Tsai A, Liao P, Harrington MG (2007) Free amino acid and dipeptide changes in the body fluids from Alzheimer’s disease subjects. Amino Acids 32:213–224
Fontana M, Pinnen F, Lucente G, Pecci L (2002) Prevention of peroxynitrite-dependent damage by carnosine and related sulphonamido pseudodipeptides. Cell Mol Life Sci 59:546–551
Severina IS, Bussygina OG, Pyatakova NV (2000) Carnosine as a regulator of soluble guanylate cyclase. Biochemistry (Mosc) 65:783–788
Calabrese V, Colombrita C, Guagliano E, Sapienza M, Ravagna A, Cardile V, Scapagnini G, Santoro AM, Mangiameli A, Butterfield DA, Giuffrida Stella AM, Rizzarelli E (2005) Protective effect of carnosine during nitrosative stress in astroglial cell cultures. Neurochem Res 30:797–807
Nicoletti VG, Santoro AM, Grasso G, Vagliasindi LI, Giuffrida ML, Cuppari C, Spina Purrello V, Stella Giuffrida AM, Rizzarelli E (2007) Carnosine interaction with nitric oxide and astroglial protection. J Neurosci Res 85:2239–2245
La Mendola D, Sortino S, Vecchio G, Rizzarelli E (2002) Synthesis of new carnosine derivatives of β-cyclodextrin and their hydroxyl scavenger ability. Helv Chim Acta 85:1633–1643
Bonomo RP, Bruno V, Conte E, De Guidi G, La Mendola D, Maccarrone G, Nicoletti F, Rizzarelli E, Sortino S, Vecchio G (2003) Potentiometric, spectroscopic and antioxidant activity studies of SOD mimics containing carnosine. J Chem Soc Dalton Trans 4406–4415
Mineo P, Vitalini D, La Mendola D, Rizzarelli E, Scamporrino E, Vecchio G (2004) ESI-MS and spectroscopic investigations on 6A, 6D-di-(β-alanyl-L-histidine)-6A, 6D-dideoxy-β-cyclodextrin and 6A, 6C-di-(β-alanyl-L-histidine)-6A, 6C-dideoxy-β-cyclodextrin and their copper(II) complexes. J Inorg Biochem 98:254–265
Bellia F, La Mendola D, Maccarrone G, Mineo P, Vitalizi D, Scamporrino E, Sortino S, Vecchio G, Rizzarelli E (2007) Copper(II) complexes with β-cyclodextrin-homocarnosine conjugates and their antioxidant activity. Inorg Chim Acta 360:949–954
Amorini AM, Bellia F, Di Pietro V, Giardina B, La Mendola D, Lazzarino G, Sortino S, Tavazzi B, Rizzarelli E, Vecchio G (2007) Synthesis and antioxidant activity of new homocarnosine β-cyclodextrin conjugates. Eur J Med Chem 42:910–920
Bellia F, Amorini AM, La Mendola D, Vecchio G, Gavazzi B, Giardina B, Di Pietro V, Lazzarino G, Rizzarelli E (2008) New glycosidic derivatives of histidine-containing dipeptides with antioxidant properties and resistant to carnosinase activity. Eur J Med Chem 43:373–380
Adlard PA, Bush AI (2006) Metals and Alzheimer’s disease. J Alzheimer’s Dis 10:145–163
Chrouch PJ, White AR, Bush AI (2007) The modulation of metal bio-availability as a therapeutic strategy for the treatment of Alzheimer’s disease. FEBS J 274:375–3783
Wenzel E, Somoza V (2005) Metabolism and bioavailability of trans-resveratrol. Mol Nutr Food Res 49:472–481
Gescher AJ, Steward WP (2003) Relationship between mechanisms, bioavailability, and preclinical chemopreventive efficacy of resveratrol: a conundrum. Cancer Epidemiol Biomarkers Prev 12:953–957
Baur JA, Sinclair DA (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506
Walle T, Hsieh F, DeLegge MH, Oatis JE Jr, Walle UK (2004) High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos 32:1377–1382
Soleas GJ, Angelini M, Grass L, Diamandis EP, Goldberg DM (2001) Absorption of trans-resveratrol in rats. Methods Enzymol 335:145–154
Jannin B, Menzel M, Berlot JP, Delmas D, Lançon A, Latruffe N (2004) Transport of resveratrol, a cancer chemopreventive agent, to cellular targets: plasmatic protein binding and cell uptake. Biochem Pharmacol 68:1113–1118
Zhou S, Koh HL, Gao Y, Gong ZY, Lee EJ (2004) Herbal bioactivation: the good, the bad and the ugly. Life Sci 74:935–968
Cao Z, Li Y (2004) Potent induction of cellular antioxidants and phase 2 enzymes by resveratrol in cardiomyocytes: protection against oxidative and electrophilic injury. Eur J Pharmacol 489:39–48
Asensi M, Medina I, Ortega A, Carretero J, Baño MC, Obrador E, Estrela JM (2002) Inhibition of cancer growth by resveratrol is related to its low bioavailability. Free Radic Biol Med 33:387–398
Kim YA, Kim GY, Park KY, Choi YH (2007) Resveratrol inhibits nitric oxide and prostaglandin E2 production by lipopolysaccharide-activated C6 microglia. J Med Food 10:218–224
Anekonda TS (2006) Resveratrol—a boon for treating Alzheimer’s disease? Brain Res Rev 52:316–326
Jang JH, Surh YJ (2003) Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic Biol Med 34:1100–1110
Bastianetto S, Brouillette J, Quirion R (2007) Neuroprotective effects of natural products: interaction with intracellular kinases, amyloid peptides and a possible role for transthyretin. Neurochem Res 32:1720–1725
Marambaud P, Zhao H, Davies P (2005) Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J Biol Chem 280:37377–37382
Bastianetto S, Zheng WH, Quirion R (2000) Neuroprotective abilities of resveratrol and other red wine constituents against nitric oxide-related toxicity in cultured hippocampal neurons. Br J Pharmacol 131:711–720
Sharma M, Gupta YK (2002) Chronic treatment with trans resveratrol prevents intracerebroventricular streptozotocin induced cognitive impairment and oxidative stress in rats. Life Sci 71:2489–2498
Pallàs M, Verdaguer E, Tajes M, Gutierrez-Cuesta J, Camins A (2008) Modulation of sirtuins: new targets for antiageing. Recent Patents CNS Drug Discov 3:61–69
Anekonda TS, Reddy PH (2006) Neuronal protection by sirtuins in Alzheimer’s disease. J Neurochem 96:305–313
Chen CY, Jang JH, Li MH, Surh YJ (2005) Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem Biophys Res Commun 331:993–1000
Zhuang H, Kim YS, Koehler RC, Doré S (2003) Potential mechanism by which resveratrol, a red wine constituent, protects neurons. Ann NY Acad Sci 993:276–286
Takahashi M, Doré S, Ferris CD, Tomita T, Sawa A, Wolosker H, Borchelt DR, Iwatsubo T, Kim SH, Thinakaran G, Sisodia SS, Snyder SH (2000) Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 28:461–473
Halliwell B (2007) Biochemistry of oxidative stress. Biochem Soc Trans 35:1147–1150
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Giuffrida Stella AM (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8:766–775
Hagen TM, Liu J, Lykkesfeldt J, Wehr CM, Ingersoll RT, Vinarsky V, Bartholomew JC, Ames BN (2002) Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci USA 99:1870–1875
Hagen TM, Moreau R, Suh JH, Visioli F (2002) Mitochondrial decay in the aging rat heart: evidence for improvement by dietary supplementation with acetyl-L-carnitine and/or lipoic acid. Ann NY Acad Sci 959:491–507
Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN (2002) Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci USA 99:2356–2361
Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, Ames BN (2008) Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J 22:703–712
Milgram NW, Araujo JA, Hagen TM, Treadwell BV, Ames BN (2007) Acetyl-L-carnitine and alpha-lipoic acid supplementation of aged beagle dogs improves learning in two landmark discrimination tests. FASEB J 21:3756–3762
Mattson MP (2008) Hormesis defined. Ageing Res Rev 7:1–7
Calabrese E (2008) Hormesis: why it is important to toxicology and toxicologists. Environ Toxicol Chem 14:1
Calabrese EJ (2008) Another california milestone: the first application of hormesis in litigation and regulation. Int J Toxicol 27:31–33
Acknowledgments
This work was supported by grants of MIUR, FIRB RBNE03PX83, and FIRB RBRN07BMCT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue article in honor of Dr. Anna Maria Giuffrida-Stella.
Rights and permissions
About this article
Cite this article
Calabrese, V., Cornelius, C., Mancuso, C. et al. Cellular Stress Response: A Novel Target for Chemoprevention and Nutritional Neuroprotection in Aging, Neurodegenerative Disorders and Longevity. Neurochem Res 33, 2444–2471 (2008). https://doi.org/10.1007/s11064-008-9775-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-008-9775-9