
Abstract
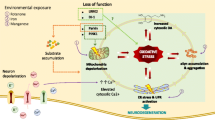
Parkinson’s disease (PD) is the second most prevalent age-related neurodegenerative disease with physiological manifestations including tremors, bradykinesia, abnormal postural reflexes, rigidity and akinesia and pathological landmarks showing losses of dopaminergic neurons in the substantia nigra. Although the etiology of PD has been intensively pursued for several decades, biochemical mechanisms and genetic and epigenetic factors leading to initiation and progression of the disease remain elusive. Environmental toxins including (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) MPTP, paraquat and rotenone have been shown to increase the risk of PD in humans. Oxidative stress remains the leading theory for explaining progression of PD. Studies with cell and animal models reveal oxidative and inflammatory properties of these toxins and their ability to activate glial cells which subsequently destroy neighboring dopaminergic neurons. This review describes pathological effects of neurotoxins on cells and signaling pathways for production of reactive oxygen species (ROS) that underline the pathophysiology of PD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Oxidative Stress and Neurodegenerative Diseases
Many cellular reactions utilize molecular oxygen for catalysis and energy production. These reactions in turn produce reactive oxygen species (ROS) such as superoxide anions, hydrogen peroxide, hydroxyl radicals, peroxy radicals and in the presence of nitric oxide, reactive nitrogen species (RNS) such as peroxynitrite and nitro-tyrosyl radicals are also produced. While these reactive species are important for execution of physiological functions, excessive production is detrimental to cell membranes and can cause cell death [1]. To cope with the many oxidative reactions, antioxidants such as glutathione and vitamin E as well as antioxidant enzymes such as superoxide dismutase, catalase and gluthathione peroxidase are present in cells to balance the oxidative mechanisms. Oxidative stress in cells due either to excessive production of ROS or insufficient antioxidant defense (particularly in the elderly) can damage cellular proteins, lipids and DNA and activate apoptotic pathways [1–4].
The brain is only 2–3% of the total body mass, but it consumes 20% of body oxygen [3, 5]. Cells in the brain are particularly susceptible to oxidative damage due to high levels of polyunsaturated fatty acids in their membranes and relatively low activity of endogenous antioxidant enzymes [3]. Aging is associated with increased oxidative stress and accumulation of oxidatively damaged biomolecules which gradually weakens cognition [3, 6]. More oxidized proteins, including carboxyls and nitro-protein adducts are found in brains as they age [3]. High levels of 8-hydroxy-2-deoxy-guanosine, a marker of oxidized DNA, were detected in both nuclear and mitochondrial DNA in elderly brain [3]. The increase in oxidative stress together with the decline in endogenous antioxidants are important underlying risk factors for older people to develop neurodegenerative diseases including Alzheimer’s and Parkinson’s disease [3, 6].
Parkinson’s Disease
Over one million people in the United States are affected by PD [7]. PD, a movement disorder, is the second most common neurodegenerative disease in the elderly, generally affecting people over 55 years of age [8, 9]. This disease is characterized by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta and a gradual accumulation of Lewy bodies which are deposits of specific cytoplasmic proteins such as ubiquitin, α-synuclein, and oxidized neurofilaments [10–14]. Dopaminergic neurons are necessary for motor function and thus loss of these neurons can cause resting tremors, bradykinesia, abnormal postural reflexes, rigidity and akinesia [11, 15]. By the time symptoms appear, 50–80% of the dopaminergic neurons have been lost [8, 15]. Normally there are around 400,000 pigmented nerve cells in the substantia nigra but with normal aging, approximately 2,500 of these nerve cells are lost every year [15]. Mechanism(s) that account for the accelerated loss of dopaminergic neurons in PD patients remains elusive [16].
Post mortem studies demonstrated increased oxidation of proteins, lipids, and DNA in neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, stroke, progressive supranuclear palsy, Huntington’s disease, Creutzfedt-Jakob disease, and amyotrophic lateral sclerosis [3, 6, 15, 17–20]. Dopaminergic neurons oxidize dopamine by monoamine oxidase, a reaction known to cause production of superoxide and hydrogen peroxide [3, 6, 21, 22]. Consequently, dopaminergic neurons are in a perpetual state of oxidative stress, and this imbalance may lead to reduced levels of endogenous antioxidants [6, 18, 19, 21].
Herbicide Use and PD
Epidemiological studies have identified several risk factors for PD including using well water, living on a farm, occupational use of herbicides, family history of PD, and head trauma [23, 24]. Among these, the greatest risk for developing PD is age, followed by family history, head injuries and herbicide use [24]. Mutations in α-synuclein and parkin have been shown in a small fraction of early onset familial Parkinson’s disease [7]. It is unclear whether the familial component of PD reflects genetic reasons or exposure to the same environmental toxins, but most likely it is the combination of both [24]. The majority of patients do not report a positive family history of PD, suggesting that nongenetic factors are more important risk factors [24]. Non-familial PD occurs after the age of 50 and is over 90% of cases [7, 9, 14, 25]. Twin studies have shown head trauma to be a significant risk factor [24]. A twin that had suffered a head injury was twice as likely to develop PD as their healthy twin [24]. An increased risk for Parkinson’s disease is correlated with exposure to environmental factors including herbicides containing paraquat, but there was no increased risk with occupational exposures to chemicals in general, heavy metals, and minerals [26–28]. However, despite a three-fold increase in PD diagnosis, only 10% of patients reported using any herbicide [24].
Neurotoxins Producing PD-like Symptoms
6-Hydroxydopamine
6-Hydroxydopamine (6-OHDA) was the first dopaminergic neurotoxin discovered and has been used experimentally in models of PD for over 30 years [15, 29]. This analog of dopamine and norepinephrine can be taken up by catecholaminergic neurons through the dopamine and norepinephrine transporters [8, 29]. 6-OHDA can damage catecholaminergic neurons throughout the body; it can even deplete norepinephrine in sympathetic nerves in the heart [8, 29]. This compound not only is toxic to neurons but also can induce activation of glial cells [29]. Like dopamine, 6-OHDA does not readily cross the blood-brain barrier, but it accumulates (or remains trapped) in the brain if injected directly into the ventricles [8, 29]. Interestingly, accumulation of endogenous 6-OHDA has been shown in PD patients [8]. Upon transport to the neurons, 6-OHDA is oxidized like dopamine to generate free radicals and quinines [8, 29]. 6-OHDA inhibits mitochondrial complex I and produces superoxide and hydroxyl radicals through participation in the Fenton reaction [8]. Although administration of 6-OHDA produces many symptoms resembling those in PD, it is worth noting that treatment with this compound does not cause the formation of Lewy bodies [15, 29].
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
In 1982, several drug abusers in Northern California developed severe Parkinson-like symptoms from a contaminant in synthetic heroin, MPTP [8, 29–31]. This discovery gave the first indication that exogenous toxins may be involved in the pathophysiology of PD [30]. MPTP-intoxicated individuals exhibit clinical neurological features of PD including tremor, rigidity, slowness of movement, postural instability, and freezing [8, 29, 32]. As in PD, patients also show, upon autopsy, damage to the dopaminergic system in the substantia nigra, but unlike PD, Lewy bodies were not found [8, 29, 31].
MPTP is a lipophilic molecule that readily crosses the blood-brain barrier. This parent compound is not harmful to dopaminergic neurons, but is readily metabolized into a toxic metabolite [8, 15]. In astrocytes, monoamine oxidase type B first converts MPTP into MPDP and then oxidizes it to a toxic pyridinium ion (MPP+) [8, 22, 30]. This polar metabolite is released from astrocytes and taken up by dopaminergic neurons [8]. MPP+ binds to dopaminergic transporters with high affinity, and mice lacking these transporters fail to exhibit toxicity to MPTP [8, 15, 22]. Normal dopaminergic neurons accumulates MPP+ to high levels in mitochondria where it inhibits electron transport chain complex I and in turn produces ROS [8, 15, 21, 22, 29, 33, 34]. MPP+ is also transported into synaptic vesicles through vesicular monoamine transporters that store dopamine [8, 21]. This uptake system serves as a detoxification mechanism. Mice with significant depletion of vesicular monoamine transporters are more sensitive to MPP+ toxicity because MPP+ is preferentially taken up by mitochondria [8]. In dopaminergic cell lines, MPP+ produces ROS in mitochondria which then triggers the classic apoptosis cascade including caspase activation and DNA fragmentation [34]. MPTP toxicity shares many similarities to PD except there are no Lewy bodies [8, 29, 31]. The accidental intoxication of drug abusers with MPTP in the early 1980’s demonstrated that even limited exposure to this compound can result in neurodegeneration that is both self perpetuating and long lasting [31].
Paraquat
Paraquat (PQ), 1,1′-dimethyl-4,4-bipyridinium, is a widely used herbicide in the 20th and early 21st centuries [26, 27, 35]. PQ is a cationic non-selective herbicide used to control weeds and grasses in agriculture fields such as cotton [13, 25, 36]. Unfortunately, the waste greenery from cotton is fed to beef cattle [13]. PQ, regulated in the United States, is considered safe by industry and worldwide standards, but some of the long-term effects of paraquat include Parkinson’s disease, lung damage, and skin cancer [26, 27, 35]. There is significant overlap between geographic areas where paraquat is used and areas where PD is prevalent [37]. People exposed to paraquat develop Parkinson-like symptoms. Paraquat is structurally similar to MPP+, the active metabolite for MPTP [7, 12, 13, 26, 27, 37]. Epidemiological data have shown that exposure to MPP+ or paraquat are risk factors for Parkinson’s disease [7]. Similar to MPTP, paraquat also depletes dopaminergic neurons in substantia nigra and cause the symptoms of Parkinsonism [13, 27, 35, 38, 39]. In fact, a study by Kang et al. [40] showed that paraquat is more cytotoxic than MPP+.
Despite being a polar molecule, paraquat has been shown to accumulate in the brain [7]. It crosses the blood-brain barrier, mainly through the neutral amino acid transporter [7, 13, 26, 27]. l-valine can competitively inhibit transport of paraquat across the blood brain barrier by neutral amino acid transporters [25, 41]. Once in the brain, paraquat is selectively taken up into terminals of dopaminergic melanin-containing neurons in substantia nigra through the dopamine transporter [7, 13, 25–27, 36]. Therefore, inhibition of dopamine transporter by cocaine and other compounds is protective against paraquat toxicity to dopaminergic neurons [25, 36]. Paraquat injected intraperitoneally concentrates in the substantia nigra and seemingly causes the loss of dopaminergic neurons [27, 38]. Interestingly, this toxicity is selective since GABAergic cells in the same region were not affected [27, 38].
Within neurons, paraquat can produce intracellular ROS leading to production of malondialdehyde, protein carbonyls, and DNA fragmentation [12, 36, 42]. Paraquat is reduced by NADPH in redox cycling to give rise to paraquat radicals and eventually to hydroxyl free radicals and superoxide anions [13, 27, 39, 40]. Paraquat inhibits mitochondrial complex I and perturbs the mitochondrial respiration chain causing impaired energy metabolism, proteasomal dysfunction and intracellular ROS production [25, 36, 43].
Rotenone
One of the most recent approaches to develop cell models to study PD results from use of the pesticide rotenone, which was previously used by Indians to poison fish [15, 29]. Rotenone is the most potent member of a family of cytotoxic compounds called rotenoids [29]. Around the world, rotenone is used commonly as an insecticide and pesticide [15, 29, 34]. Rotenone is used in vegetable gardens to kill insects and in lakes to kill fish that are invasive and overpopulated [15, 34]. To date, soil and water treatment with rotenone in the USA is restricted to times when crops are not growing and people are not swimming [29]. Rotenone is a naturally occurring complex ketone extracted from the roots of Lonchocarpus species [15, 34]. Rotenone is easily degraded by exposure to sunlight, soil and water. Therefore, this clearance makes the likelihood of environmental exposure to rotenone very low, which make it advantageous as a pesticide [15, 29].
Ingestion of rotenone is not likely to have adverse effects in healthy people. It is slowly and incompletely absorbed in the stomach and intestines and then efficiently degraded by the liver’s first pass effect [29]. Thus, one would have to either ingest a large amount of this compound or have a liver disease in order for rotenone to exceed the liver’s ability to detoxify it and only then would it enter the general circulation [29]. Once rotenone enters the body, like MPTP, it is very lipophilic and crosses the blood-brain barrier to distribute evenly throughout the brain [15, 29].
Since the 1950s, it is believed that rotenone acts as an inhibitor for mitochondria respiratory chain [34]. Because it is very lipophilic, rotenone not only distributes throughout the body and brain, it can enter organelles including double-membrane encased mitochondria where it inhibits with high affinity complex I of the mitochondrial electron transport chain [15, 22, 29, 34]. Fortunately, most cells are able to detoxify the oxidative damage caused by rotenone through their antioxidant protective enzyme pathways. Unfortunately, chronic inhibition of this mitochondrial complex can cause selective degeneration of dopaminergic neurons despite being uniformly distributed throughout the brain [15]. This raises the questions why dopaminergic cells are targeted for destruction and whether rotenone in the environment contributes to the etiology of PD in certain individuals. Due to its inhibition of complex I, rotenone is known to cause ROS generation, ATP depletion, and cell death in neurons [15]. In agreement with the involvement of ROS in rotenone toxicity, antioxidants are capable of attenuating rotenone-mediated cell death [15]. Interestingly, rotenone also inhibits the formation of microtubules [29]. Similar to PD, treatment with rotenone leads to the aggregation of α-synuclein and formation of Lewy bodies [29]. Another similarity to PD is that rotenone depletes glutathione, a primary cell antioxidant [15]. Thus, rotenone toxicity mimics many pathological hallmarks of PD, including neurodegeneration of dopaminergic neurons in substantia nigra and formation of Lewy bodies which is presumably due to oxidative damage, depletion of glutathione, and disruption of axonal transport [15, 29].
Microglia Involvement in Oxidative Stress
All known dopaminergic neurotoxins exhibit a common characteristic. They inhibit either mitochondrial complex I or complex III in neurons [8]. Additionally, oxidative stress in the CNS comes not only from mitochondrial-generated ROS in neurons but also from activated microglia [7]. Reactive microglia are known to play a role in several neurodegenerative disorders including PD [44–48]. Indeed, Parkinson’s patients can have more than six times the number of reactive microglia as compared to controls [49–51]. However, it is unknown whether these microglia initiate or aggravate neurodegeneration [48]. Stimulation of microglial and astrocyte activity is known to correlate with neuronal injury induced by paraquat [7, 38]. However, a study by Gao et al. [10] showed greater contribution to MPTP-induced dopaminergic neurodegeneration by microglial than by astrocytes.
Microglia behave as macrophages in the brain and are one of four major cell types in the central nervous system (CNS) [52, 53]. Similar to macrophages, a major function of microglia is to fight infection and remove debris in the brain [53]. Microglia become activated after brain damage or exposure to immune mediators such as IL-1beta or TNF-alpha [52]. Besides producing cytokines, proteases, and prostanoids, activated microglia also produce superoxide and nitric oxide [53]. In order to survive in the environment of these oxidative generators, it is not surprising that microglia also contain high levels of endogenous antioxidants [53].
Activated microglia in the substantia nigra are found in several models for PD, including exposure to MPTP, rotenone, substance P, and methamphetamine [10, 44, 54–58] (see Table 1). In many instances, activation of microglia and generation of ROS coincide with neurochemical changes such as the decrease in dopamine synthesis [56, 58]. While some neurochemical changes happen quickly and can be detected before evidence of any lesions, others are delayed and continue to be manifested long after neurotoxin exposure [59]. These results suggest that a brief exposure to an insult can initiate a process of continuous neurodegeneration [60]. Microglia may play a role in initiation and progression of PD and enhance neurotoxicity elicited by neurotoxins [55, 61]. Inhibition of microglial activation by minocycline, an antibiotic, could attenuate the neurotoxicity of 6-OHDA, MPTP and rotenone [61, 62].
Contribution of Oxidative Stress by NADPH Oxidase
NADPH oxidase is an enzyme that metabolizes molecular oxygen and generates superoxide as a product [58]. There is evidence that NADPH oxidase is up-regulated in the substantia nigra in PD patients. NADPH oxidase is comprised of three cytosolic components (p47phox, p67phox, and p40phox) and two membrane bound subunits, gp91phox and p22phox [64]. Besides translocation of the cytosolic subunits to the membrane subunits, the enzyme complex also requires a small GTPase Rac for full function [64]. Translocation of the cytosolic subunits is initiated by phosphorylation by protein kinases. In the p47phox subunit, phosphorylation results in conformational change and the SH3 domain then binds with the p22phox membrane subunit [64]. In recent studies, activation of NADPH oxidase is regarded a major source of superoxide in a number of neurodegenerative diseases including PD [46]. Mice defective in the NADPH oxidase show less neurotoxic response to LPS [65], MPTP [58], and rotenone [61]. NADPH oxidase appears to be ubiquitously expressed in all brain regions and cell types including neurons and glial cells [66]. Similar to other immune cells, high levels of this enzyme are found in microglia. Recent studies show that paraquat can induce microglia to produce ROS through NADPH oxidase [63, 67] and confer neurotoxicity by damaging neighboring neurons [68]. Microglia lacking a functional NADPH oxidase in coculture with neurons failed to produce neurotoxicity to paraquat as compared with microglia having a functional NADPH oxidase [68]. Table 2 describes involvement of NADPH oxidase in PD and PD models.
Protein Kinase C
Several protein kinases including protein kinase C (PKC) and MAPK have been implicated in activation of NADPH oxidase and therefore contribute to oxidative stress. PKC isoforms are ubiquitously expressed in brain tissue and are involved in regulation of ion channels and receptors, neurotransmitter release, and synaptic development [72–76]. Abnormal PKC has been shown in a number of neurodegenerative diseases including PD [72, 75, 77].
There are more than 10 PKC isoforms in mammalian cells generally grouped as conventional, novel and atypical types based on their structure and function. Both the conventional and novel isoforms contain a conserved C1 domain that binds diacylglycerol or phorbol esters [77]. The conventional PKCs also contain a C2 domain for binding calcium [77]. The C3 and C4 domains are conserved throughout all the isoforms and contain the ATP binding and the catalyst domains, respectively [77]. All PKC isoforms contain a pseudosubstrate domain and under normal conditions, the PKC kinase region is autoinhibited by the pseudosubstrate domain [76]. The conventional and novel PKC are translocated to the membrane and are activated at the membrane by diacylglycerol. The presence of several different isoforms of PKC allows substrate specificity and differential regulation [73].
Protein kinase C delta (PKCδ) is a novel isoform of PKC that is calcium-independent. This PKC isoform is expressed in many cell types and is involved in the mitochondrial-dependent pathway of apoptosis [78, 79]. Cells derived from PKCδ-null transgenic mice lack activation of the mitochondrial-dependent apoptosis pathway [79, 80] and over-expression of PKCδ in certain cell types can exacerbate apoptosis [79, 81]. PKCδ is a substrate for caspase; cleavage by caspase-3 permanently activates PKCδ [82, 83]. Rottlerin, a selective PKCδ inhibitor, can inhibit caspase-3 activation and thereby decrease PKCδ cleavage. In turn, the latter can also activate caspase 3 thus form a positive feedback between caspase-3 and PKCδ [82, 83]. In pheochromocytoma cells, generation of ROS resulted in proteolytic cleavage of PKCδ, release of cytochrome C from the mitochondria and activation of caspase 9 and caspase 3 [83]. PKCδ inhibition by prolonged treatment with PMA or pretreatment with rottlerin blocked H2O2-induced JNK activation and apoptosis [80].
Ample evidence indicates a role for PKCδ in phosphorylation of cytosolic subunits and activation of NADPH oxidase. ROS production is inhibited by rottlerin or dominant negative mutants of PKC [84–88]. For example, cells deficient in PKCδ failed to generate ROS and overexpression of PKCδ increased the generation of superoxide anion [87, 89]. Phosphorylation of p67phox and p47phox subunits could be inhibited by rottlerin or antisense oligodeoxyribonucleotides specific for PKCδ [89, 90]. PKCδ phosphorylates p67phox and p47phox [84, 91]. Rottlerin or PKCδ antisense oligodeoxyribonucleotides also inhibited translocation of the subunits to the membrane [63, 84].
Mitogen Activated Protein Kinases
MAPKs are highly conserved and ubiquitously expressed in all tissue and activate many enzymatic pathways contributing to oxidative stress [92, 93]. They are activated by phosphorylation of neighboring threonines and tyrosines, and they phosphorylate serines or threonines that are adjacent to prolines on other proteins [93, 94]. There are five MAPK families including extracellular-regulated kinase (ERK)1/2; JNK1/2/3; p38α,β,γ,δ; ERK5; and ERK7 [92, 94–96]. Each MAPK is activated by a MEK (also called MAPK kinase), which is activated by a MEKK (also called MAPK kinase kinase) [93, 96, 97]. The GTPase Ras phosphorylates and activates MEKK [94, 98]. In total there are at least 11 MAPKs, 7 MEKs and 20 MEKKs [96]. MAPKs activate a wide range of proteins including transcription factors, other kinases and enzymes [93, 95]. MAPKs can translocate from the cytosol to the nucleus and affect gene expression [92, 95, 98]. ERK1/2 are the most studied MAPKs. ERK1/2 are activated by growth factors, serum, and phorbol esters and are involved in proliferation, differentiation and migration [92–94, 98]. ERK1/2 can also be activated by environmental stresses, oxidative stress, LPS, and cytokines [92, 94, 97]. Environmental stresses, oxidative stress, UV radiation, hypoxia, ischemia, LPS, and inflammatory cytokines activate the p38 isoforms [94, 95, 97]. P38 MAPK is involved in inflammation, cell growth, cell differentiation, and cell death [93, 95]. JNK is activated by similar stimuli as p38 MAPK including cytokines, UV irradiation, LPS, and osmotic stress [93–95]. The MAPKs are involved in the expression of inducible nitric oxide synthase in macrophages and ultimately, inflammation [94]. The MAPK are involved in a wide variety of cellular processes including activation of NADPH oxidase [99]. The ERK1/2 inhibitor, PD98059, was able to inhibit the phosphorylation of p47phox [99]. Paraquat induces apoptotic cell death through activation of the JNK signaling pathway in cultured neuronal cell line [12].
Nitric Oxide Synthase
Another possible source of oxidative stress is nitric oxide (NO) which is produced by nitric oxide synthase (NOS) by converting of l-agrinine to l-citrulline utilizing NADPH and O2 as cofactors [100–104]. Three isoforms of NOS have been identified and they are classified as neuronal or type 1 NOS, immunologic or type 2 NOS, and endothelial or type 3 NOS [100, 101]. NOS1 and NOS3 are constitutive whereas NOS2 is inducible [100, 105]. The constitutive NOS isoforms require calcium and calmodulin for activity and the inducible NOS requires de novo synthesis of the enzyme upon activation of transcriptional pathway by cytokines or LPS [101, 102, 106]. NOS2 plays a role in host defense and can produce a 1000-fold higher amounts of NO in the micromolar concentrations compared to the constitutive isoforms that only produce nanomolar NO concentrations [104, 107]. NOS2 was first discovered in macrophages where it is involved in host defense, but NOS2 is also present in many other cells including certain types of neurons, astrocytes, and microglia [105, 108].
NO has diverse roles in the body depending on the concentration, the type of cell and the isoform expressed. It is regarded as a signaling molecule involved in a wide array of physiological events including smooth muscle relaxation, inflammation, host defense, cell death, blood pressure regulation, inhibition of platelet aggregation, atherosclerosis, and septic shock [100, 102, 104–106, 109]. In the brain, NO is involved in neurotransmitter release and reuptake, neurodevelopment, synaptic plasticity, and gene expression [100–102, 104, 110]. Because NO is a vasodilator, increased production may benefit the brain after injury [111]. On the other hand, excess production of NO can cause neurodegeneration manifesting as stroke, PD and HIV dementia [101, 112]. NO can form adducts with mitochondrial enzymes and dysregulate mitochondrial function. There is a positive correlation between NO levels and cellular oxidation from the mitochondrial electron transport chain [101, 104, 107]. Other possible mechanisms associating NO with neurodegeneration are inhibition of cytochrome oxidase, ribonucleotide reductase, superoxide dismutase, and glyceraldehydes-3-phosphate; impairment of iron metabolism; and generation of hydroxyl radicals and peroxynitrite [101, 104, 107].
Under many conditions, activated glia can simultaneously produce NO, ROS and cytokines and together, form the basis of many neuroinflammatory responses [106, 113] (see Table 3). Increased proinflammatory cytokines such as interleukin-1β (IL-1β) and interferon gamma (IFNγ) are observed in PD brains, and they can induce iNOS in activated microglia. Among the proinflammatory cytokines, IFNγ is produced by immune effector cells in response to intracellular pathogens [70, 114–117]. It is a pro-inflammatory cytokine that rapidly alerts immune cells of an infection [116]. Macrophages have a more rapid and heightened response to lipopolysacharide (LPS) on the surface of gram-negative bacteria when first exposed to IFNγ [116]. The activated morphology of macrophages including expulsion of cytoplasmic granules and extension of pseudopodia is achieved after exposure to IFNγ and LPS [116]. In the murine immortalized microglial cells (BV-2), INFγ can induce iNOS and production of NO without other pathogens [118, 119].
Lipopolysacharides
Endotoxins such as LPS from bacteria are potent inflammatory molecules known to activate microglia and damage dopaminergic neurons [49, 120]. LPS is a component of the outer membrane of gram-negative bacteria [115, 121, 122] and is a highly toxic and a strong stimulus for inflammatory responses in CNS [115, 122]. LPS is more neurotoxic to dopaminergic neurons than neurons lacking tyrosine hydroxylase such as the gamma-aminobutyric acidergic and serotonergic neurons [49, 120]. LPS can induce loss of dopaminergic neurons in substantia nigra [70] and activate astrocytes and microglia to produce NO. However, in the absence of glial cells, LPS is not neurotoxic to neurons, suggesting the important role of glial cells in mediating neurotoxicity [114]. Microglia have been shown to be major mediators of the neurotoxicity of dopaminergic neurons by LPS [49]. LPS failed to induce neurotoxicity in dopaminergic neurons when activation of microglial cells was inhibited [46]. NOS2 inhibitors can also attenuate the loss of dopaminergic neurons induced by LPS, suggesting that NO is an important factor in mediating toxicity [114]. Pregnant rats treated with LPS gave birth to pups that had fewer dopaminergic neurons than normal controls [123]. The NOS2 gene has regulatory regions that have consensus elements for interferons and LPS [102].
Toxins that target dopaminergic neurons combine with LPS to synergistically enhance toxicity; toxicity was increased with MPTP and LPS treatment together or in tandem [70]. LPS alone did not induce dopaminergic neurotoxicity but enhanced the toxicity of MPTP [115]. MPTP and LPS induced additive generation of superoxide and NO, and neurotoxicity was found only in the presence of microglia [70]. Apparently, MPTP and LPS induce ROS production through NADPH oxidase and activate inflammatory cytokine pathways for NO production [101, 115]. Inhibitors of NADPH oxidase and NOS2 can attenuate dopaminergic neurotoxicity due to MPTP and LPS [70]. These reactions require close proximity of neuron-glia; mixed cultures lacking functional NADPH oxidase were spared toxicity of MPTP and LPS [70]. Similarly, a combination of rotenone and LPS at nontoxic levels induced significant loss of dopaminergic neurons [71]. Prenatal exposure to LPS lead to increased levels of oxidized proteins, and formation of Lewy bodies-like inclusions containing alpha-synuclein [123]. Rotenone together with LPS further increased the number of activated microglia and synergistic loss of dopaminergic neurons [123]. Taken together, these results suggest that combinations of environmental toxins or drugs, even at nontoxic levels, may increase neuroinflammation and risk for development of PD [115, 123].
Besides MPTP and rotenone, NO enhances the toxicity of paraquat. NO donors exacerbate the toxicity of paraquat despite being minimally toxic by themselves [111, 112]. Paraquat generates superoxide intracellularly in glial cells by redox cycling and increases generation of NO by glial cultures [106, 111]. Pro-inflammatory cytokines may increase paraquat’s toxicity through combined production of superoxide and NO [112]. In the presence of superoxide, NO can form peroxynitrite, a compound more toxic than either of the two agents [111]. However, nitrotyrosine, an indicator of peroxynitrite, was not detected when an NO donor and paraquat were combined, suggesting that other factors are involved in the increased toxicity [111].
Toxins such as paraquat may cause toxicity by interacting directly with NOS; the underlying mechanism remains to be determined. Our results indicate that paraquat decreases NO production induced by IFNγ and LPS. However, paraquat combined with cytokines significantly increased ROS generation and disrupted membrane integrity more than paraquat alone (Miller et al., unpublished data). Similar to cytochrome P-450, NOS can be uncoupled by shunting electrons away from the heme in the reductase domain to other molecules [103]. It is possible that in the presence of paraquat, electrons are shunted away from the heme to paraquat which thus decreases NO production and increases superoxide production [67, 103]. When paraquat undergoes a one electron reduction, it produces a radical that can transfer its electron to oxygen to produce superoxide [67].
Conclusion
A number of models are available for PD but each has its strengths and weaknesses in mimicking the pathophysiological features of the disease. There are models that mimic mitochondrial dysfunction and raise oxidative stress; both features have been shown in postmortem studies of PD patients. There is also evidence that glia cells, especially microglia, play an active role in generation of ROS in PD. NADPH oxidase and NOS are known to play a role in inflammation and generation of ROS, and inhibition of these enzymes offers protection to dopaminergic neurons. Activation of NADPH oxidase involves phosphorylation of the cytosolic subunits by protein kinases such as PKC and MAPK. These models suggest that besides the genetic component, environmental factors also play a role in the progression of PD. Furthermore, exposure of neurons in substantia nigra to neurotoxins can cause dopaminergic cell loss similar to PD. Future investigations to develop therapeutics for PD and its prevention should focus on anti-oxidant and anti-inflammatory compounds that may inhibit ROS formation and suppress microglial activation.
References
Loh KP, Huang SH, De Silva R, Tan BK, Zhu YZ (2006) Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res 3:327–337
Betarbet R, Sherer TB, Greenamyre JT (2005) Ubiquitin-proteasome system and Parkinson’s diseases. Exp Neurol 191:S17–S27
Mariani E, Polidori MC, Cherubini A, Mecocci P (2005) Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B: Analyt Technol Biomed Life Sci 827:65–75
Mattson MP (2006) Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid Redox Signal 8:1997–2006
Moreira PI, Siedlak SL, Aliev G, Zhu X, Cash AD, Smith MA, Perry G (2005) Oxidative stress mechanisms and potential therapeutics in Alzheimer disease. J Neural Transm 112:921–932
Cardoso SM, Moreira PI, Agostinho P, Pereira C, Oliveira CR (2005) Neurodegenerative pathways in Parkinson’s disease: therapeutic strategies. Curr Drug Targets CNS Neurolog Disord 4:405–419
Chun HS, Gibson GE, DeGiorgio LA, Zhang H, Kidd VJ, Son JH (2001) Dopaminergic cell death induced by MPP+, oxidant and specific neurotoxicants shares the common molecular mechanism. J Neurochem 76:1010–1021
Schober A (2004) Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res 318:215–224
Siderowf A, Stern M (2003) Update on Parkinson’s disease. Ann Intern Med 138:651–658
Gao HM, Liu B, Zhang W, Hong JS (2003) Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. FASEB J 17:1954–1956
Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Dev Disease 131:2183–2194
Peng J, Stevenson FF, Doctrow SR, Andersen JK (2005) Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat-mediated dopaminergic neuron death in the substantial nigra. J Biol Chem 280:29194–29198
Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA (2000) The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson’s disease. J Neurosci Res 20:9207–9214
Gelinas S, Martinoli M-G (2002) Neuroprotective effect of estradiol and phytoestrogens on MPP+-induced cytotoxicity in neuronal PC12 cells. J Neurosci Res 70:90–96
Uversky VN (2004) Neurotoxicant-induced animal models of Parkinson’s disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissure Res 318:225–241
Tieu K, Ischiropoulos H, Przedborski S (2003) Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 55:329–335
Ahlskog JE (2005) Challenging conventional wisdom: the etiologic role of dopamine oxidative stress in Parkinson’s disease. Movement Disord 20:271–282
Beal MF (2004) Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J Bioenerg Biomembr 36:381–386
Gandhi S, Wood NW (2005) Molecular pathogenesis of Parkinson’s disease. Human Mol Genet 14:2749–2755
Hald A, Lotharius J (2005) Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp Neurol 193:279–290
Greenamyre JT, Sherer TB, Betarbet R, Panov AV (2001) Complex I and Parkinson’s disease. IUBMB Life 52:135–141
Maguire-Zeiss KA, Short DW, Federoff HJ (2005) Synuclein, dopamine and oxidative stress: co-conspirators in Parkinson’s disease? Brain Research. Mol Brain Res 134:18–23
Firestone JA, Smith-Weller T, Franklin G, Swanson P, Lonstreth WT, Checkoway H (2005) Pesticides and risk of Parkinson disease. Arch Neurol 62:91–95
Semchuk KM, Love EJ, Lee RG (1993) Parkinson’s disease: a test of the multifactorial etiologic hypothesis. Neurology 43:1173–1180
Shimizu K, Matsubara K, Ohtaki K, Fujimaru S, Saito O, Shiono H (2003) Paraquat induces long-lasting dopamine overflow through the excitotoxic pathway in the striatum of freely moving rats. Brain Res 976:243–252
Yang W-L, Sun AY (1998) Paraquat-induced free radical reaction in mouse brain microsomes. Neurochem Res 23:47–53
Brooks AI, Chadwick CA, Gelbard HA, Cory-Slechta DA, Federoff HJ (1999) Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res 823:1–10
Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC (1997) Environmental risk factors and Parkinson’s disease: a case-control study in Taiwan. Neurology 48:1583–1588
Bove J, Prou D, Perier C, Przedborski S (2005) Toxin-induced models of Parkinson’s disease. NeuroRx 2:484–494
Wersinger C, Sidhu A (2006) An inflammatory pathomechanism for Parkinson’s disease? Curr Med Chem 13:591–602
Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R, Droller D (2005) Early environmental origins of neurodegenerative disease in later life. Environ Health Persp 113:1230–1233
Brown TP, Rumsby PC, Capleton AC, Rushton L, Levy LS (2006) Pesticides and Parkinson’s disease-is there a link? Environ Health Persp 114:156–164
Schmidt WJ, Alam M (2006) Controversies on new animal models of Parkinson’s disease pro and con: the rotenone model of Parkinson’s disease (PD). J Neural Transm Suppl 273–276
Kotake Y, Ohta S (2003) MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr Med Chem 10:2507–2516
Wesseling C, van Wendel de Joode B, Ruepert C, Leon C, Monge P, Hermosillo H, Partanen TJ (2001) Paraquat in developing countries. Int J Occup Environ Health 7:275–286
Yang W, Tiffany-Castiglioni E (2005) The bipyridyl herbicide paraquat produces oxidative stress-mediated toxicity in human neuroblastoma SH-SY5Y cells: relevance to the dopaminergic pathogenesis. J Toxicol Environ Health Part A 68:1939–1961
Shimizu K, Matsubara K, Ohtaki K, Shiono H (2003) Paraquat leads to dopaminergic neural vulnerability in organotypic midbrain culture. Neurosci Res 46:523–532
McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA (2002) Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis 10:119–127
Yang W-L, Sun AY (1998) Paraquat-induced cell death in PC12 cells. Neurochem Res 23:1387–1394
Kang D, Miyako K, Kuribayashi F, Hasegawa E (1997) Changes in energy metabolism induced by 1-methyl-4-phenylpyridinium (MPP+)-related compounds in rat pheochromocytoma PC12 cells. Arch Biochem Biophys 337:75–80
McCormack A, Di Monte D (2003) Effect of l-dopa and other amino acids against paraquat-induced nigrostriatal degeneration. J Neurochem 85:82–86
Thiruchelvam M, Prokopenko O, Cory-Slechta D, Richfield E, Buckley B, Mirochnitchenko O (2005) Overexpression of superoxide dismutase or glutathione peroxidase protects against the paraquat + maneb-induced Parkinson disease phenotype. J Biol Chem 280:22530–22539
Bretaud S, Lee S, Guo S (2004) Sensitivity of zebrafish to environmental toxins implicated in Parkinson’s disease. Neurotoxicol Teratol 26:857–864
Block ML, Li G, Qin L, Wu X, Pei Z, Wang T, Wilson B, Yang J, Hong JS (2006) Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: substance P vs. dynorphin. FASEB J 20:251–258
Li G, Cui G, Tzeng NS, Wei SJ, Wang T, Block ML, Hong JS (2005) Femtomolar concentrations of dextromethorphan protect mesencephalic dopaminergic neurons from inflammatory damage. FASEB J 19:489–496
Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA (2005) Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. PNAS 102:9936–9941
Qin L, Liu Y, Qian X, Hong JS, Block ML (2005) Microglial NADPH oxidase mediates leucine enkephalin dopaminergic neuroprotection. Ann NY Acad Sci 1053:107–120
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J (2005) Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19:533–542
Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y, Mochizuki H (2004) Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1beta, and expression of caspase-11 in mice. J Biol Chem 279:51647–51653
McGeer PL, Itagaki S, Akiyama H, McGeer EG (1988) Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol 24:574–576
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291
Mander PK, Jekabsone A, Brown GC (2006) Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol 176:1046–1052
Dringen R (2005) Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal 7:1223–1233
Delgado M, Ganea D (2003) Neuroprotective effect of vasoactive intestinal peptide (VIP) in a mouse model of Parkinson’s disease by blocking microglial activation. FASEB J 17:944–946
Gao HM, Hong JS, Zhang W, Liu B (2002) Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22:782–790
Scheller C, Sopper S, Jenuwein M, Neuen-Jacob E, Tatschner T, Grunblatt E, ter Meulen V, Riederer P, Koutsilieri E (2005) Early impairment in dopaminergic neurotransmission in brains of SIV-infected rhesus monkeys due to microglia activation. J Neurochem 95:377–387
Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM (2004) Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther 311:1–7
Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. PNAS 100:6145–6150
Barcia C, Bahillo AS, Fernandez-Villalba E, Bautista V, Poza MP, Fernandez-Barreiro A, Hirsch EC, Herrero M-T (2004) Evidence of the active microglia in substantia nigra pars compacta of Parkinsonian monkeys 1 year after MPTP exposure. GLIA 46:402–409
Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D (1999) Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol 46:598–605
Casarejos MJ, Menendez J, Solano RM, Rodriguez-Navarro JA, Garcia de Yebenes J, Mena MA (2006) Susceptibility of rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J Neurochem 97:934–946
Croisier E, Moran LB, Dexter DT, Pearce RKB, Graeber MB (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation 2:14–22
Miller R, Sun G, Sun A (2007) Cytotoxicity of paraquat in microglial cells: involvement of the PKC delta- and ERK 1/2-dependent NADPH oxidase. Brain Res 1167:129–139
Sumimoto H, Ueno N, Yamasaki T, Taura M, Takeya R (2004) Molecular mechanism underlying activation of superoxide-producing NADPH oxidases: roles for their regulatory proteins. Jpn J Infect Dis 57:S24–S25
Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS (2004) NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279:1415–1421
Infanger DW, Sharma RV, Davisson RL (2006) NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal 8:1583–1596
Bonneh-Barkay D, Reaney SH, Langston WJ, Di Monte DA (2005) Redox cycling of the herbicide paraquat in microglial cultures Brain Res Mol Brain Res 134:52–56
Wu XF, Block ML, Zhang W, Qin L, Wilson B, Zhang WQ, Veronesi B, Hong JS (2005) The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxid Redox Signal 7:654–661
Gao HM, Liu B, Hong JS (2003) Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopamingeric neurons. J Neurosci 23:6181–6187
Gao HM, Liu B, Zhang W, Hong JS (2003) Synergistic dopaminergic neurotoxicity of MPTP and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson’s disease. FASEB J 17:1957–1959
Gao H-M, Hong J-S, Zhang W, Liu B (2003) Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson’s disease. J Neurosci 23:1228–1236
Wagey R, Hu J, Pelech SL, Raymond LA, Krieger C (2001) Modulation of NMDA-mediated excitotoxicity by protein kinase C. J Neurochem 78:715–726
Ran X, Miao H-H, Sheu F-S, Yan D (2003) Structural and dynamic characterization of a neuron-specific protein kinase C substrate, neurogranin. Biochemistry 42:5143–5150
Catarsi S, Drapeau P (1997) Requirement for tyrosine phosphatase during serotonergic neuromodulation by protein kinase C J Neurosci Res 17:5792–5797
Ahlemeyer B, Kolker S, Zhu Y, Hoffmann GF, Krieglstein J (2002) Increase in glutamate-induced neurotoxicity by activated astrocytes involves stimulation of protein kinase C. J Neurochem 82:504–515
Battaini F (2001) Protein kinase C isoforms as therapeutic targets in nervous system disease states. Pharmacol Res 44:353–361
Kozikowski AP, Nowak I, Petukhov PA, Etcheberrigaray R, Mohamed A, Tan M, Lewin N, Hennings H, Pearce LL, Blumber PM (2003) New amide-bearing benzolactam-based protein kinase C modulators induce enhanced secretion of the amyloid precursor protein metabolite sAPPα. J Med Chem 46:364–373
Gallagher HC, Murphy KJ, Foley AG, Regan CM (2001) Protein kinase C delta regulates neural cell adhesion molecule polysialylation state in the rat brain. J Neurochem 77:425–434
DeVries TA, Neville MC, Reyland ME (2002) Nuclear import of PKCδ is required for apoptosis: identification of a novel nuclear import sequence. EMBO J 21:6050–6060
Shibukawa Y, Takahashi M, Laffont I, Honke K, Taniguchi N (2003) Down-regulation of hydrogen peroxide-induced PKCd activation in N-acetylglucosaminyltransferase III-transfected HeLaS3 cells. J Biol Chem 278:3197–3203
Cataisson C, Joseloff E, Murillas R, Wang A, Atwell C, Torgerson S, Gerdes M, Subleski J, Gao J-L, Murphy PM, Wiltrout RH, Vinson C, Yuspa SH (2003) Activation of cutaneous protein kinase Ca induces keratinocyte apoptosis and intraepidermal signaling pathways. J Immunol 171:2603–2713
Leverrier S, Vallentin A, Joubert D (2002) Positive feedback of protein kinase C proteolytic activation during apoptosis. Biochem J 368:905–913
Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG (2002) Caspase-3-dependent proteolytic cleavage of protein kinase Cδ is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J Neurosci Res 22:1738–1751
Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, Subbulakshmi V, Feldman GM, Wientjes FB, Cathcart MK (2004) Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol 173:5730–5738
He R, Nanamor M, Sang H, Yin H, Dinauer MC, Ye RD (2004) Reconstitution of chemotactic peptide-induced nicotinamide adenine dinucleotide phosphate (reduced)oxidase activation in transgenic COS-phox cells. J Immunol 173:7462–7470
Shao MX, Nadel JA (2005) Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. PNAS 102:767–772
Talior I, Tennebaum T, Kuroki T, Eldar-Finkelman H (2005) PKC-delta-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice: role for NADPH oxidase. Am J Physiol-Endocrinol Metab 288:E405–E411
Waki K, Inanami O, Yamamori T, Nagahata H, Kuwabara M (2006) Involvement of protein kinase Cdelta in the activation of NADPH oxidase and the phagocytosis of neutrophils. Free Radic Res 40:359–367
Zhang X, Dong F, Ren J, Driscoll MJ, Culver B (2005) High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol 191:318–325
Serezani CH, Aronoff DM, Jancar S, Mancuso P, Peters-Golden M (2005) Leukotrienes enhance the bactericidal activity of alveolare macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood 106:1067–1075
Zhao X, Xu B, Bhattacharjee A, Oldfield CM, Wientjes FB, Feldman GM, Cathcart MK (2005) Protein kinase Cdelta regulates p67phox phosphorylation in human monocytes. J Leukocyte Biol 77:414–420
Nishimoto S, Nishida E (2006) MAPK signalling: ERK5 versus ERK1/2. EMBO Rep 7:782–786
Bardwell L (2006) Mechanisms of MAPK signalling specificity. Biochem Soc Trans 34:837–841
Cuschieri J, Maier RV (2005) Mitogen-activated protein kinase (MAPK). Crit Care Med 33:S417–S419
Kaminska B (2005) MAPK signalling pathways as molecular targets for anti-inflammatory therapy-from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta 1754:253–262
Uhlik MT, Abell AN, Cuevas BD, Nakamura K, Johnson GL (2004) Wiring diagrams of MAPK regulation by MEKK1, 2, and 3. Biochem Cell Biol 82:658–663
Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344
Murphy LO, Blenis J (2006) MAPK signal specificity: the right place at the right time. Trend Biochem Sci 31:268–275
Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J (2000) The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J Immunol 165:5238–5244
Day BJ, Patel M, Calavetta L, Chang L-Y, Stamlet JS (1999) A mechanism of paraquat toxicity involving nitric oxide synthase. PNAS 96:12760–12765
Ebadi M, Sharma SK (2003) Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Antioxid Redox Signal 5:319–335
Duval DL, Miller DR, Collier J, Billings RE (1996) Characterization of hepatic nitric oxide synthase: identification as the cytokine-inducible form primarily regulated by oxidants. Mol Pharmacol 50:277–284
Margolis AS, Porasumphatana S, Rosen GM (2000) Role of paraquat in the uncoupling of nitric oxide synthase. Biochim Biophys Acta 1524:253–257
Guo Q, Tirosh O, Packer L (2001) Inhibitory effect of α-lipoic acid and its positively charged amide analogue on nitric oxide production in RAW 264.7 macrophages. Biochem Pharmacol 61:547–554
Shih C-L, Chi S-I, Chiu TH, Sun GY, Lin T-N (2001) Ethanol effects on nitric oxide production in cerebral pial cultures. Alcohol: Clin Exp Res 25:612–618
Noack H, Possel H, Rethfeldt C, Keilhoff G, Wolf G (1999) Peroxynitrite mediated damage and lowered superoxide tolerance in primary cortical glial cultures after induction of the inducible isoform of NOS. GLIA 28:13–24
Yamamoto F, Ohgari Y, Yamaki N, Kitajima S, Shimokawa O, Matsui H, Taketani S (2007) The role of nitric oxide in δ-aminolevulinic acid (ALA)-induced photosensitivity of cancerous cells. Biochem Biophys Res Commun 353:541–546
Connelly L, Madhani M, Hobbs AJ (2005) Resistance to endotoxic shock in endothelial nitric-oxide synthase (eNOS) knock-out mice. J Biol Chem 280:10040–10046
Mitsumoto A, Nakagawa Y (2001) DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic Res 35:885–893
Xia J, Simonyi AS, Sun GY (1999) Chronic ethanol and iron administration of iron content, neuronal nitric oxide synthase, and superoxide dismutase in rat cerebellum. Alcohol: Clin Exp Res 23:702–707
Gobbel GT, Chan TY-Y, Chan PH (1997) Nitric oxide- and superoxide-mediated toxicity in cerebral endothelial cells. J Pharmacol Exp Ther 282:1600–1607
Tomita M, Okuyama T, Ishikawa T, Hidaka K, Nohno T (2001) The role of nitric oxide in paraquat-induced cytotoxicity in the A549 lung carcinoma cell line. Free Radic Res 34:193–202
Shin CY, Choi J-W, Jang ES, Ju C, Kim W-K, Kim H-C, Choi C-R, Ko KH (2001) Dehydroepiandrosterone inhibits the death of immunostimulated rat C6 glioma cells deprived of glucose. Brain Res 922:267–275
Iravani MM, Leung CCM, Sadeghian M, Haddon CO, Rose S, Jenner P (2005) The acute and the long-term effects of nigral lipopolysaccharide administration on dopaminergic dysfunction and glial cell activation. Eur J Neurosci 22:317–330
Goralski KB, Renton KW (2004) Brain inflammation enhances 1-methyl-4-phenylpyridinium-evoked neurotoxicity in rats. Toxicol Appl Pharmacol 196:381–389
Schroder K, Sweet MJ, Hume DA (2006) Signal integration between IFNγ and TLR signaling pathways in macrophages. Immunobiology 211:511–524
Whitehead GS, Grasman KA, Kimmel EC (2003) Lung function and airway inflammation in rats following exposure to combustion products of carbon-graphite/epoxy composite material: comparison to a rodent model of acute lung injury. Toxicology 183:175–197
Shen S, Yu S, Binek J, Chalimoniuk M, Zhang X, Lo SC, Hannink M, Wu J, Fritsche K, Donato R, Sun GY (2005) Distinct signaling pathways for induction of type II NOS by IFNgamma and LPS in BV-2 microglial cells. Neurochem Int 47:298–307
Li W, Xia J, Sun GY (1999) Cytokine induction of iNOS and sPLA2 in immortalized astrocytes (DITNC): response to genistein and pyrrolidine dithiocarbamate. J Interf Cytok Res 19:121–127
Herrera AJ, Tomas-Camardiel M, Venero JL, Cano J, Machado A (2005) Inflammatory process as a determinant factor for the degeneration of substantia nigra dopaminergic neurons. J Neural Transm 112:111–119
Chandel NS, Trzyna WC, McClintock DS, Schumacker PT (2000) Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J Immunol 165:1013–1021
West NP, Jungnitz H, Fitter JT, McArthur JD, Guzman CA, Walker MJ (2000) Role of phosphoglucomutase of Bordetella bronchiseptica in lipopolysaccharide biosynthesis and virulence. Infect Immun 68:4673–4680
Ling Z, Chang QA, Tong CW, Leurgans SE, Lipton JW, Carvey PM (2004) Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Exp Neurol 190:373–383
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue article in honor of Dr. George DeVries.
Rights and permissions
About this article
Cite this article
Miller, R.L., James-Kracke, M., Sun, G.Y. et al. Oxidative and Inflammatory Pathways in Parkinson’s Disease. Neurochem Res 34, 55–65 (2009). https://doi.org/10.1007/s11064-008-9656-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-008-9656-2