
Abstract
Environmental toxins, genetic predisposition and old age are major risk factors for Parkinson’s disease (PD). Although the mechanism(s) underlying selective dopaminergic (DA) neurodegeneration remain unclear, molecular studies in both toxin based models and genetic based models of the disease suggest a major etiologic role for mitochondrial dysfunction in the pathogenesis of PD. Further, recent studies have presented clear evidence for a high burden of mtDNA deletions within the substantia nigra neurons in individuals with PD. Ultimately, an understanding of the molecular events which precipitate DA neurodegeneration in idiopathic PD will enable the development of targeted and effective therapeutic strategies. We review recent advances and current understanding of the genetic factors, molecular mechanisms and animal models of PD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive movement disorder characterized pathologically by the relatively selective degeneration of midbrain dopamine (DA) neurons and the presence of prominent cytoplasmic inclusions, termed Lewy bodies [33]. The percentage of affected individuals within a population rises from ∼1% at 65 years to ∼5% at 85 years, making age the main risk factor for PD. The majority of cases are thought to be idiopathic. However, in ∼5–10% of cases, PD is thought to have a genetic component, showing both recessive and dominant modes of inheritance [129]. In these cases, the causative genes that have been identified are: α-synuclein, parkin, nuclear receptor-related 1 (Nurr1), ubiquitin carboxy-terminal hydrolase L1 (UCHL1), PTEN-induced putative kinase 1(PINK1), leucine-rich repeat kinase 2 (LRRK2) and DJ-1 [14, 108].
Although the mechanisms responsible for neurodegeneration in PD are largely unknown, they result in damage and subsequent loss of DA neurons [82]. Both environmental and genetic factors have been implicated in the pathogenesis of PD [103].
While mitochondrial dysfunction has been indirectly linked to idiopathic PD [33, 110], studies of families with rare inherited forms of PD have identified genes involved in regulating mitochondrial function [27, 94, 102]. Recently, clear evidence has been presented for a high burden of mtDNA deletions within the substantia nigra neurons in individuals with PD [9, 67]. Additional support comes from experimental studies with toxins that inhibit Complex I of the mitochondrial respiratory chain and cause selective death of midbrain DA neurons [109]. However, the interpretation of results from experiments with neurotoxins is complicated by the fact that they may have pleiotropic pharmacological effects in DA neurons, effects on non-DA cell types, or both [109]. Although the roles of mitochondrial dysfunction and mtDNA mutations in the pathogenesis of PD remain controversial, parkinsonism has been associated with several mtDNA mutations, including large-scale rearrangements [15, 20, 106] and point mutations or microdeletions [34, 110, 120].
The most compelling evidence yet comes from MitoPark mice. These are mice possessing conditional knockout of mitochondrial transcription factor A (Tfam) in DA neurons. These mice have reduced mtDNA expression with subsequent respiratory chain deficiency in midbrain DA neurons. This in turn leads to a parkinsonism phenotype characterized by adult onset of a progressive impairment of motor function accompanied by formation of intraneuronal inclusions and DA nerve cell death [37]. Further support for mitochondrial involvement in the pathogenesis of PD is supported by post-mortem biochemical studies which show a disease specific and drug independent defect of Complex I in substantia nigra and other tissues in persons with idiopathic PD [95, 99].
Somatic mtDNA mutations and Parkinson’s disease
Recent research has shown that the substantia nigra neurons from individuals with PD have a high level of deleted mtDNA compared to controls [110]. Kraytsberg et al. [67] using a novel single-molecule PCR approach, have quantified the total burden of mtDNA deletions in aged human substantia nigra neurons compared to neurons from younger individuals as controls. They found high levels of mtDNA deletions in the aged neurons. They also found that affected substantia nigra neurons also lost cytochrome c oxidase (COX) expression. In a parallel study, Bender et al. [9] using long range PCR in single cells confirmed the presence of somatic and clonally expanded mtDNA deletions that were associated with respiratory chain deficiency in individuals with PD. These studies suggest direct involvement of mtDNA deletions in the development of respiratory chain deficiency in substantia nigra neurons in individuals with PD. Another study, however, has shown that somatic mtDNA mutations are capable of inducing aging phenotypes without affecting reactive oxygen species (ROS) production suggesting that oxidative stress is not a key modulator of neurodegeneration in aging [121].
Environmental factors and mitochondrial dysfunction in PD
The discovery in 1983 of persons developing typical signs of PD after intravenous injection of drugs contaminated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and the subsequent finding that MPTP selectively damages DA cells in the substantia nigra [72], led to the hypothesis that exposure to environmental toxins might be related to the risk of PD. Complex I is inhibited in dopamine-containing neurons by systemic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which can induce parkinsonism in animal models and in humans [7]. The defect appears to be restricted both to Complex I and the substantia nigra, with other brain areas showing normal activity of the ETC [29]. Studies on platelets of PD patients show a consistent Complex I defect either alone or in combination with a mild defect in other Complexes [95]. The question then arises as to the origin of the Complex I-deficiency in PD. It could result from an environmental toxin or an acquired or inherited mtDNA mutation(s). To define the origin of oxidative phosphorylation deficit in PD, cybrid PD models have been developed. PD platelet mitochondrial genes expressed in cybrids produce reduced Complex I activity [48, 116], suggesting that the Complex I deficiency was determined by the mtDNA derived from PD patient platelets. Since then, many epidemiological studies have been done to examine the association between exposure to pesticides and herbicides, as well as hypothesized surrogate measures, such as farming, living in rural areas, and drinking of well water, and the risk of PD. Evidence fairly consistently points towards a positive association between pesticide exposure and PD risk, although results were statistically significant in only half of the studies [35]. Environmental neurotoxins such as rotenone, paraquart, maneb, and dieldrin are also inhibitors of Complex I and able to induce dopaminergic loss [98].
Genetic factors and mitochondrial dysfunction in PD
Many of the genes associated with familial forms of PD also implicate mitochondria in disease pathogenesis. At least nine nuclear genes have been identified as causing PD or affecting PD risk. Of the nuclear genes, α-synuclein, parkin, DJ-1, tensin homologue (PTEN)-induced kinase 1 (PINK1), leucine-rich-repeat kinase 2 (LRRK2) and high temperature requirement A2 (HTRA2) directly or indirectly impact mitochondrial function [8, 75]. In transgenic mice, α-synuclein overexpression impairs mitochondrial function, increases oxidative stress and enhances the toxicity of MPTP [111]. Parkin is an ubiquitin E3 ligase that can associate with the outer mitochondrial membrane and protects against cytochrome c release and caspase activation [32]; it may also associate with Tfam and enhance mitochondrial biogenesis [71]. When oxidized, DJ-1 translocates to mitochondria (intermembrane space and matrix), downregulates the PTEN-tumour suppressor protein, and protects the cell from oxidative-stress-induced cell death [64]. PINK-1 is a nuclear-encoded kinase localized to the mitochondrial matrix [107] that seems to protect against apoptosis, an effect that is reduced by PD-related mutations or kinase inactivation [97]. About 10% of the kinase LRRK2 is localized to mitochondria, and PD-related mutations augment its kinase activity [128]. Mutations in other nuclear-encoded genes such as HTRA2, UCH-L1, synphilin-1, glucocerebrosidase, POLG, NR4A2 and tau, have been identified in individual patients with idiopathic PD [1, 87]. HTRA2, a serine protease localized to the mitochondrial intermembrane space, degrades denatured proteins within mitochondria and, if released into the cytosol, promotes programmed cell death by binding inhibitor of apoptosis proteins [113, 115]. Table 1 provides a general overview of how the majority of these gene products directly or indirectly implicate mitochondria in the disease pathogenesis. This suggests that mitochondrial dysfunction may be central to the molecular pathogenesis of both idiopathic and familial forms of PD.
Oxidative stress and PD
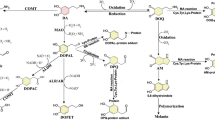
Oxidative stress has been demonstrated in PD [51, 126] and evidence also clearly supports the involvement of impaired mitochondrial function in PD [63, 100]. In particular, increased iron, oxidation of proteins, and lipid peroxidation in the SN appear to be common [2, 36, 132]. Reduced levels of glutathione (GSH) in the midbrain may be indicative of increased free radical levels [105]. Dopamine metabolism can also be a source of ROS in nigral neurons. Cytosolic dopamine produces electrophilic semiquinones and quinones which themselves act as oxidants by supporting ROS formation [114]. The possible involvement of oxidative stress as an etiological factor of PD is further supported by studies with specific neurotoxins that are potent inducers of Parkinsonism in humans and animals. MPTP treatment in mice generates hydroxyl radicals in the striatum which in turn leads to oxidant damage and could cause initiation of apoptosis [23]. Activated glial cells were observed at sites of neurodegeneration in PD, and might participate in the mechanism of nerve cell death by producing more ROS and reactive nitrogen species (RNS). Antioxidants have been proposed as a means to accomplish neuroprotection in PD [17]. It has also been recognized that oxidative stress leading to caspase activation and consequent apoptosis are clearly evident in PD [42].
Animal models
Several animal models of PD, such as the rotenone [11, 43] Drosophila DJ-1 mutants, [84, 85], and 6-hydroxydopamine (6-OHDA) [26] and MPTP [31], have been shown to have multiple mitochondrial dysfunctions including increased ROS generation and striking sensitivity to stressors. Of these, the mammalian models have activated migroglia.
Already substantial and still accumulating evidence shows that lipopolysaccharide (LPS)-induced microglial activation causes DA neurodegeneration in vitro and in vivo [4, 45, 57, 79]. Also, inducible nitric oxide synthase (iNOS) is generally found to be upregulated in experimental PD models [60, 74] and inhibition of iNOS reduces the toxicity of LPS or LPS and interferon γ (IFN- γ-activated microglia on DA neurons in vitro by approximately 75%) [56, 73]. Evidence now clearly indicates that inflammatory cytokines, such as Tumor necrosis factor a (TNF-α), interleukin-1 and 6 (IL-1 and -6)and the signaling molecule NO are toxic to neurons [3, 41, 45, 77, 81, 112]. Inactivation of the genes involved in the synthesis of proinflammatory molecules such as COX-2 [40], nicotinamide adenine dinuleotide phosphate (NADPH) oxidase [130] and both TNF-α receptors [112] were shown to protect DA neurons against MPTP-induced neurotoxicity, thus indicating that inflammation plays an important role in MPTP-mediated as well as other types of nigrostriatal neurodegeneration. Interestingly, it has been observed that MPTP and LPS act synergistically to mediate nigral DA neurotoxicity, probably by stimulating release of the superoxide radical [44].
Cell models of PD
It is difficulty to assess the rate of neuronal loss in PD since it is very slow. In vitro studies, in spite of their limitations, are therefore a better alternative to explore degenerative pathways involved in PD. Most in vitro studies have been conducted in SHSY5Y, MN9D, NB41, and PC12 cell lines, in primary mesencephalic cultures or in cerebellar granule cells.
Cybrids bearing mitochondrial DNA from patients with idiopathic PD produce Lewy body inclusions [122]. They have decreased Complex I activity that is associated with increased apoptosis via the p38 and JNK pathways as compared to age and sex matched control cybrids [93]. They are also more susceptible to oxidative stress [92]. This suggests that mtDNA, if not causative, modulates susceptibility to PD [123].
MPP+ was found to cause apoptotic cell death in PC12 and SH-SY5Y cells and primary midbrain (mesencephalic) cell cultures through the activation of caspase-3 [39, 53], treatment of SH-SY5Y cells with MPP+ induced ROS production, lactate release, inhibition of ETC, p53 expression, cleavage of caspase-3 and PARP, and apoptotic cell death with DNA fragmentation. bcl-2 over expression protected these cells against MPP+ toxicity whereas decreased bcl-2 levels enhanced MPP+-induced cell death [39, 66]. It has now been shown that upon in vivo inhibition of Complex I, p53 mediates Bax transcription and translocation to mitochondria [96].
6-OHDA induces an early increase in p53 cellular content in PC12 cultures [13]. Activation of caspase-3 and caspase-9 was also demonstrated in 6-OHDA induced apoptosis in SHSY5Y cells [28], which was inhibited by caspase inhibitor [117].
Chronic low-grade Complex I inhibition by rotenone exposure induces accumulation and aggregation of α-synuclein and ubiquitin, as well as progressive oxidative stress and caspase-dependent apoptotic death in human neuroblastoma cells [104]. In PC 12 cells inhibition of Complex I by rotenone caused apoptotic cell death [52]. They found both rotenone and MPTP induced apoptosis at low concentrations and necrosis at high concentrations.
Dopamine induced apoptosis in SH-SY5Y neuroblastoma cells through activation of caspase-9 and caspase-3 and cleavage of PARP. In this model nuclear condensation was mediated by the activation of p38 mitogen activated protein (MAP) kinase and mitochondrial cytochrome-c release [62]. These studies suggest a role for apoptosis in PD neurodegeneration although the signaling events involved are yet to be clearly elucidated. Unfortunately, the strategy of inhibiting apoptosis by preventing caspase activation has failed clinically [12].
Mitogen activated signaling cascades and mitochondria in PD
Kinase signaling pathways impact major mitochondrial functions including oxidative phosphorylation, antioxidant protein expression, mitochondrial fission, and execution of survival-death decisions. Extracellular signal regulated kinase (ERK) has also been implicated in promoting oxidative neuronal injury [24] in PD [47, 68, 69]. Phospho-ERK was found at high labeling densities within a subset of mitochondria in degenerating neurons from patients with Parkinson’s disease and Lewy body dementia [25, 134], corresponding to a distinct granular cytoplasmic pattern of staining not observed in age-matched control patients [135]. It is interesting to note that mitochondrial ROS and the permeability transition have both been implicated upstream of JNK activation under both physiologic and pathologic conditions [18, 89]. In a similar manner, p38 MAPK is activated by mitochondrially derived ROS [70, 92]. Thus, these kinases, like ERK, may signal to influence mitochondrial functions as well as to communicate mitochondrial signals to the rest of the cell.
The proteasome system and PD
A significant feature of PD pathology is the presence of Lewy bodies, which contain a variety of proteins including α-synuclein, ubiquitin, proteasome subunits, chaperone proteins, and neurofilament proteins [125]. This is indicative of incomplete clearance of the target proteins by the ubiquitin-proteasome system (UPS). This may arise from increased protein oxidation products overwhelming the proteolytic capacity of the proteasome [38, 91]. Furthermore, mitochondrial inhibition by neurotoxins can deplete intracellular ATP levels, thereby adversely affecting ATP-dependent proteasomal degradation. Oligomerization of α-synuclein following exposure to toxins, DA, DA metabolites, and upregulation of α-synuclein expression inhibit proteasomal function [22, 46, 76, 91]. Nitrosylation, altered solubility or covalent modification of Parkin diminishes its ubiquitin ligase activity, and Uch-L1 oxidation reduces its hydrolase activity [49, 91]. These findings suggest that interactions between the UPS and mitochondrial function promote the degenerative processes in DA neurons.
Inflammation and PD
A large cohort study of patients has shown that the risk of developing PD in regular non steroidal anti inflammatory drugs (NSAID) users (for cardiovascular protection) was decreased by up to 45% compared with those who take NSAIDs on a non-regular basis [21]. Thus, it is suggested that the use of NSAIDs may lead to neuroprotection in PD [50]. However, the neuroprotective effect of COX-2 inhibitors against MPTP in vivo may not be due to the reduced microglial activation, but rather has been linked to inhibition of COX-induced DA oxidation [118, 119].
Inflammation also has been proposed to contribute to PD pathogenesis, [83] in part through upregulation of inflammatory cytokines such as TNF-α [58, 88]. Various PD models demonstrate inflammation in neurodegeneration, and anti-inflammatory drugs attenuate toxin-induced PD [43, 55, 124]. The well known association between encephalitis and parkinsonism [10, 16] and a report of parkinsonism induced by accidental exposure to lipopolysaccharide (LPS) from Salmonella minnesota [90] have supported the role of inflammation in the etiology of PD. Intranigral and intrapallidal LPS induce microglial activation and DA neuronal death [4, 5, 19, 60, 78, 80, 133]. Microglial activation initiates or perpetuates neuronal loss by increasing cytotoxic molecules like superoxide, NO, various pro-inflammatory cytokines, and prostaglandins [6, 65, 86]. Recently, LPS-induced mitochondrial dysfunction has been demonstrated in vitro [131], where mitochondrial dysfunction precedes cell death [30, 127]. In these studies, LPS toxicity was associated with respiratory chain dysfunction. In mitochondria isolated from the striatum, there was evidence of oxidative damage to mitochondrial components, which suggests that mitochondria may be a target of free-radical stress initiated by activated microglia. The fact that both celecoxib and pioglitazone can reduce mitochondrial dysfunction suggests that mitochondrial impairment may be secondary to inflammation [59].
Future directions
Mitochondrial dysfunction and oxidative stress are thought to play an important role in the pathogenesis of idiopathic PD [54, 61, 101]. Although the mechanisms by which this leads to neurodegeneration in DA neurons are still unknown, recent advances in recessive PD implicate parkin, PINK1, LRRK2 and DJ-1 in mitochondrial function. Further studies in both in both toxin based models and genetic based models of the disease will help elucidate the molecular relationships between the two models and hopefully lead to development of therapeutic interventions that prevent, reduce or ameliorate PD associated mitochondrial dysfunction and oxidative stress.
References
Abou-Sleiman P, Muqit M, Wood N (2006) Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci 7:207–219
Alam Z, Daniel S, Lees A, Marsden D, Jenner P, Halliwell B (1997) A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J Neurochem 69:1326–1329
Allan S, Rothwell N (2001) Cytokines and acute neurodegeneration. Nat Rev Neurosci 2:734–744
Arimoto T, Bing G (2003) Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol Dis 12:35–45
Arimoto T, Choi D, Lu X, Liu M, Nguyen X, Zheng N, Stewart C, Kim H, Bing G (2006) Interleukin-10 protects against inflammation-mediated degeneration of dopaminergic neurons in substantia nigra. Neurobiol Aging 28:894–906
Banati R, Gehrmann J, Schubert P, Kreutzberg GW (1993) Cytotoxicity of microglia. Glia 7:111–118
Beal M (2003) Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann Neurol 53:S39–S47
Beal M (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58:495–505
Bender A, Krishnan K, Morris C, Taylor G, Reeve A, Perry R, Jaros E, Hersheson J, Betts J, Klopstock T, Taylor R, Turnbull D (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517
Berger J, Glitza I (2003) Von’s Economo’s Encephalitis. In: Nath A Jr. (ed) Clinical neurovirology. Marcel Dekker, New York, p 523–542
Betarbet R, Sherer T, MacKenzie G, Garcia-Osuna M, Panov A, Greenamyre J (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature Neurosci 3:1301–1306
Biglan K, Ravina B (2007) Neuroprotection in Parkinson’s disease: an elusive goal. Semin Neurol 27:106–112
Blum D, Wu Y, Nissou M, Arnaud S, Benabid AL, Verna JM (1997) P53 and bax activation in 6-hydroxydopamine-induced apoptosis in PC12 cells. Brain Res 751:139–142
Bossy Wetzel E, Schwarzenbacher R, Lipton S (2004) Molecular pathways to neurodegeneration. Nat Med 10:S2–S9
Casali C, Bonifati V, Santorelli F, Casari G, Fortini D, Patrignani A, Fabbrini G, Carrozzo R, D’Amati G, Locuratolo N, Vanacore N, Damiano M, Pierallini A, Pierelli F, Amabile G, Meco G (2001) Mitochondrial myopathy, parkinsonism, and multiple mtDNA deletions in a Sephardic Jewish family. Neurology 56:802–805
Casals J, Elizan T, Yahr M (1998) Postencephalitic parkinsonism—a review. J Neural Transm 105:645–676
Casetta I, Govoni V, Granieri E (2005) Oxidative stress, antioxidants and neurodegenerative diseases. Curr Pharm Des 11:2033–2052
Cassarino DS, Halvorsen EM, Swerdlow RH, Abramova NN, Parker WD Jr, Sturgill TW, Bennett JP Jr (2000) Interaction among mitochondria, mitogen-activated protein kinases, and nuclear factor-kappaB in cellular models of Parkinson's disease. J Neurochem 74(4):1384–1392
Castano A, Herrera A, Cano J, Machado A (1998) Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem 70:1584–1592
Chalmers R, Brockington M, Howard R, Lecky B, Morgan-Hughes J, Harding A (1996) Mitochondrial encephalopathy with multiple mitochondrial DNA deletions: a report of two families and two sporadic cases with unusual clinical and neuropathological features. J Neurol Sci 143:41–45
Chen H, Zhang S, Hernan M, Schwartzschild M, Willett W, Colditz G, Speizer F, Ascherio A (2003) Nonsteriodal anti-inflammatory drugs and the risk of Parkinson's disease. Arch Neurol 60:1059–1064
Chen L, Thiruchelvam M, Madura K, Richfield E (2006) Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol Dis 23:120–126
Chiueh C, Haung S, Murphy D (1992) Enhanced hydroxyl radical generation by 2′-methyl analog of MPTP: suppression by clorgyline and deprenyl. Synapse 11:346–348
Chu C, Levinthal D, Kulich S, Chalovich E, DeFranco D (2004) Oxidative neuronal injury: the dark side of ERK1/2. Eur J Biochem 271:2060–2066
Chu C, Zhu J-H (2003) Subcellular compartmentalization of P-ERKs in the Lewy body disease substantia nigra. Ann NY Acad Sci 991:288–290
Ciccetti F, Brownell A, Williams K, Chen Y, Livni E, Isacson O (2002) Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci 15:991–998
Clark I, Dodson M, Jiang C, Cao J, Huh J, Seol J, Yoo S, Hay Ba, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166
Coelln R, Kugler S, Bahr M, Weller M, Dichgans J, Schulz J (2001) Rescue from death but not from functional impairment: caspase inhibition protects dopaminergic cells against 6-hydroxydopamine induced apoptosis but not against the loss of their terminals. J Neurochem 77:263–273
Cooper J, Daniel S, Marsden C, Schapira A (1995) L-dihydroxyphenylalanine and complex I deficiency in Parkinson’s disease brain. Mov Disord 10:295–297
Crouser E, Julian M, Blaho D, Pfeiffer D (2002) Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med 30:276–284
Czlonkowska A, Kohuknika M, Kurkowska-Jatrzebska I, Czlonkowski A (1996) Microglial reaction in MPTP (1-methyl-4phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration 5:137–143
Darios F, Corti O, Lucking C, Hampe C, Muriel M, Abbas N, Gu W, Hirsch E, Rooney T, Ruberg M, Brice A (2003) Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet 12:517–526
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909
De Coo I, Renier W, Ruitenbeek W, Ter Laak H, Bakker M, Schagger H, Van Oost B, Smeets H (1999) A 4-base pair deletion in the mitochondrial cytochrome b gene associated with Parkinsonism/MELAS overlap syndrome. Ann Neurol 45:130–133
de Lau L, Breteler M (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5:525–535
Dexter D, Wells F, Agid F, Agid Y, Lees AJ, Jenner P, Marsden CD (1987) Increased nigral iron content in postmortem parkinsonian brain. Lancet 8569:1219–1220
Ekstrand M, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson F, Trifunovic A, Hoffer B, Cullheim S, Mohammed A, Olson L, Larsson N (2007) Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci USA 104:1325–1330
Elkon H, Melamed E, Offen D (2004) Oxidative stress, induced by 6-hydroxydopamine, reduces proteasome activities in PC12 cells: implications for the pathogenesis of Parkinson’s disease. J Mol Neurosci 24:387–400
Fall CP, Bennett JP Jr (1999) Visualization of cyclosporin A and Ca2+-sensitive cyclical mitochondrial depolarizations in cell culture. Biochim Biophys Acta 1410(1):77–84
Feng Z, Wang T, Li D, Fung P, Wilson B, Liu B, Ali S, Langenbach R, Hong J (2002) Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl-1,2,3,6-tetradydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci Lett 329:354–358
Fisher J, Mizrahi T, Schori H, Yoles E, Levkovitch-Verbin H, Haggaig S, Revel M, Schwartz M (2001) Increased post-traumatic survival of neurons in IL-6-knockout mice on a background of EAE susceptibility. J Neuroimmunol 119:1–9
Friedlander R (2003) Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 348:1365–1375
Gao H, Hong J, Zhang W, Liu B (2002) Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22:782–790
Gao H, Liu B, Zhang W, Hong J (2003) Synergistic dopaminergic neurotoxicity of MPTP and inflammogen lipopolysaccharide: relevance to the etioloy of Parkinson's disease. FASEB J 17:1957–1971
Gayle D, Ling Z, Tong C, Landers T, Lipton J, Carvey P (2002) Lipopolysacchride (LPS)-induced dopamine cell loss in culture: roles of tumor necrosis factor-alpha, interleukin-1beta, and nitric oxide. Brain Res Dev 133:27–35
Ghee M, Fournier A, Mallet J (2000) Rat alpha-synuclein interacts with Tat binding protein 1, a component of the 26S proteasomal complex. J Neurochem 75:2221–2224
Gomez-Santos C, Ferrer I, Reiriz J, Vinals F, Barrachina M, Ambrosio S (2002) MPP+ increases α-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res 935:32–39
Gu M, Cooper J, Taanman J, Schapira A (1998) Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann Neurol 44:177–186
Gu Z, Nakamura T, Yao D, Shi Z, Lipton S (2005) S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Cell Death Differ 12:1202–1204
Hald A, Lotharius J (2005) Oxidative stress and inflammation in Parkinson’s disease: is there a casual link? Exp Neurol 193:279–190
Halliwell B, Gutteridge J (1999) Oxidative stress in PD. In: Halliwell B, Gutteridge J (eds) Free radicals in biology and medicine. Oxford University Press, New York pp 744–758
Hartley A, Stone J, Heron C, Cooper J, Schapira A (1994) Complex I inhibitors induce dose-dependent apoptosis in PC12 cells: relevance to Parkinson’s disease. J Neurochem 63:1987–1990
Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC (2000) Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc Natl Acad Sci USA 97(6):2875–2880
Hattori N, Tanaka M, Ozawa T, Mizuno Y (1991) Immunohistochemical studies on complexes I, II, III, IV of mitochondria in Parkinson’s disease. Ann Neurol 30:563–571
He Y, Appel S, Le W (2001) Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res 909:187–193
Hemmer K, Fransen I, Vanderstichele H, VanmeChelen E, Heuschling P (2001) An in vitro model for the study of microglial-induced neurodegeneration: involvement of nitric oxide and tumour necrosis factor-alpha. Neurochem Int 38:557–565
Hererra A, Castano A, Venero J, Cano J, Machado A (2000) The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neuorobiol Dis 7:429–447
Hirsh E, Hunot S, Damier P, Faucheux B (1998) Glial cells and inflammation in Parkinson's disease: a role in neurodegeneration? . Ann Neurol 44:S115–S120
Hunter R, Dragicevic N, Seifert K, Choi D, Liu M, Kim H, Cass W, Sullivan P, Bing G (2007) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100:1375–1386
Iravani M, Kashell K, Rose S, Jenner P (2002) Involvement of inducible nitric oxide synthase in inflammation-induced dopaminergic neurodegeneration. Neuroscience 110:49–58
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–S36
Junn E, Mouradian M (2001) Apoptotic signaling in dopamine-induced cell death: the role of oxidative stress, p38 mitogen activated protein kinase, cytochrome c and caspases. J Neurochem 78:374–383
Keeny P, Xie J, Capaldi R, Bennett J Jr (2006) Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits, is functionally impaired and misassembled. J Neurosci Res 26:5256–5264
Kim R, Peters M, Jang Y, Shi W, Pintilie M, Fletcher G, DeLuca C, Liepa J, Zhou L, Snow B, Binari R, Manoukian A, Bray M, Liu F, Tsao M, Mak T (2005) DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 7:263–273
Kim S, Kong P, Kim B, Sheen D, Nam S, Chun W (2004) Inhibitory action of minocycline on lipopolysaccharide-induced release of nitric oxide and prostaglandin E2 in BV2 microglial cells. Arch Pharm Res 27:314–318
Kitamura Y, Kosaka T, Kakimura J, Matsouka Y, Nomura Y, Tan-guchi T (1998) Protective effects of the antiparkinsonian drugs talipexole and pramipexole against 1-methyl 4-phenylpyridinium-induced apoptotic death in human neuroblastoma SH-SY5Y cells. Mol Pharmacol 54:1046–1054
Kraytsberg Y, Kudryavtseva E, McKee A, Geula C, Kowall N, Khrapko K (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38:507–508
Kulich S, Chu C (2003) Role of reactive oxygen species in ERK phosphorylation and 6-hydroxydopamine cytotoxicity. J Biosci 28:83–89
Kulich S, Chu C (2001) Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson’s disease. J Neurochem 77:1058–1066
Kulisz A, Chen N, Chandel N, Shao Z, Schumacker P (2002) Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol 282:L1324–L1329
Kuroda Y, Mitsui T, Kunishige M, Matsumoto T (2006) Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet 15:883–895
Langston J, Ballard P, Tetrud J, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980
Le W, Rowe D, Xie W, Ortiz I, He Y, Appel S (2001) Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson’s disease. J Neurosci Res 21:8447–8455
Liberatore G, Jackson-Lewis V, Vukosavic S, Mandir A, Vila M, McAuliffe W, Dawson V, Dawson T, Przedborski S (1999) Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson’s. Nat Med 5:1403–1409
Lin M, Beal M (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Lindersson E, Beedholms R, Hojrup P, Moos T, Gai W, Gendil K, Jensen P (2004) Proteasomal inhibition by α-synuclein filaments and oligomers. J Biol Chem 279:12924–12934
Liu B, Gao H, Wang J, Jeohn GH, Cooper CL, Hong JS (2002) Role of nitric oxide in inflammation-mediated neurodegeneration. Ann NY Acad Sci 991:80–92
Liu B, Jiang J, Wilson B, Du L, Yang S, Wang JY, Wu G, Cao X, Hong J (2000) Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J Pharmacol Exp Ther 295:125–132
Liu Y, Qin L, Li G, Zhang W, An L, Liu B (2003) Dextromethorplan protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. J Pharmacol Exp Ther 305:212–218
Lu X, Bing G, Hagg T (2000) Naloxone prevents microglia-induced degeneration of dopaminergic substantia nigra neurons in adult rats. Neuroscience 97:285–291
Ma J, Ma J (2002) The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J Environ Carcinog Ecotoxicol Rev 20:117–147
Maguire-Zeiss K, Federoff H (2003) Convergent pathobiologic model of Parkinson’s disease. Ann NY Acad Sci 991:152–166
McGeer P, Yasojima K, McGeer E (2001) Inflammation in Parkinson’s disease. Adv Neurol 86:83–89
Menzies F, Yenisetti S, Min K-T (2005) Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr Biol 15:1578–1582
Meulener M, Whitworth AJ, Armstrong-Gold C, Rizzu P, Heutink P, Wes P, Pallanck L, Bonini N (2005) Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr Biol 15:1572–1577
Minghetti L, Levi G (1998) Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol 54:99–125
Moore D, West A, Dawson V, Dawson T (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87
Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Changes in cytokines and neurotrophins in Parkinson’s disease. J Neural Transm Suppl 60:277–290
Nemoto S, Takeda K, Yu Z, Ferrans V, Finkel T (2000) Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol 20:7311–7318
Niehaus I (2004) Lipopolysaccharides induce inflammation-mediated neurodegeneration in the substantia nigra and cerebral cortex (a case report). In: Hanin I, Cacabelos R (eds) New trends in Alzheimer and Parkinson related disorders. Monduzzi Editore, Bologna, pp 36–39
Olanow C, McNaught K (2006) Ubiquitin-proteasome system and Parkinson’s disease. Mov Disord 21:1806–1823
Onyango IG, Tuttle JB, Bennett JP Jr (2005) Brain derived growth factor and Glial cell line-derived growth factor have different survival promoting effects and use distinct intracellular signaling pathways to protect PD cybrids from H2O2 induced apoptotic death. Neurobiol Dis 20:141–154
Onyango IG, Tuttle JB, Bennett Jr JP (2005) Activation of p38 and N-acetyl cycteine sensitive c-Jun NH2-terminal kinase signaling cascades is required for induction of apoptosis in Parkinson’s disease cybrids. Mol Cell Neurosci 28(3):452–461
Park J, Lee S, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim J, Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161
Parker WD Jr, Boyson SJ, Parks JK (2007) Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol 26(6):719–723
Perier C, Bove J, Wu DC, Dehay B, Choi DK, Jackson-Lewis V, Rathke-Hartlieb S, Bouillet P, Strasser A, Schulz JB, Przedborski S, Vila M (2007) Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease. Proc Natl Acad Sci USA 104:8161–8166
Petit A, Kawarai T, Paitel E, Sanjo N, Maj M, Scheid M, Chen F, Gu Y, Hasegawa H, Salehi-Rad S, Wang L, Rogaeva E, Fraser P, Robinson B, St George-Hyslop P, Tandon A (2005) Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem 280:34025–34032
Petrovitch H, Ross GW, Abbott R, Sanderson W, Sharp D, Tanner C, Masaki K, Blanchette P, Popper J, Foley D, Launer L, White L (2002) Plantation work and risk of Parkinson disease in a population-based longitudinal study. Arch Neurol 59:1787–1792
Schapira A (2006) Etiology of Parkinson’s disease. Neurology 66:S10–S23
Schapira A (2006) Mitochondrial disease. Lancet 368:70–82
Schapira A, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54:823–827
Shen J, Cookson M (2004) Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron 43:301–304
Sherer T, Betarbet R, Greenamyre J (2002) Environment, mitochondria, and Parkinson’s disease. Neuroscientist 8:192–197
Sherer T, Betarbet R, Stout AK, Lund S, Baptista M, Panov A, Cookson M, Greenamyre J (2002) An in vitro model of Parkinson’s disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci 22:7006–7015
Sian J, Dexter D, Jenner P, Marsden C (1991) Decreased in nigral glutathione in Parkinson’s disease. Br J Pharmacol 104:281
Siciliano G, Mancuso M, Ceravolo R, Lombardi V, Iudice A, Bonuccelli U (2001) Mitochondrial DNA rearrangements in young onset parkinsonism: two case reports. J Neurol Neurosurg Psychiat 71:685–687
Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente E, Casari G (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 14:3477–3492
Singh M, Patel S, Dikshit M, Gupta Y (2006) Contribution of genomics and proteomics in understanding the role of modifying factors in Parkinson’s disease. Indian J Biochem Biophys 43:69–81
Smeyne R, Jackson-Lewis V (2005) The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res 134:57–66
Smigrodzki R, Parks J, Parker W (2004) High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol Aging 25:1273–1281
Song D, Shults C, Sisk A, Rockenstein E, Masliah E (2004) Enhanced substantia nigra mitochondrial pathology in human α-synuclein transgenic mice after treatment with MPTP. Exp Neurol 186:158–172
Sriram K, Matheson J, Benkovic S, Miller D, Luster M, O’Callaghan J (2002) Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. FASEB J 16:474–476
Strauss K, Martins L, Plun-Favreau H, Marx F, Kautzmann S, Berg D, Gasser T, Wszolek Z, Muller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz J, Kruger R (2005) Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet 14:2099–2111
Sulzer D, Zecca L (2000) Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res 1:181–195
Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R (2001) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol cell Neurosci 8:613–621
Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP Jr, Davis RE, Parker WD Jr (1996) Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol 40(4):663–671
Takai N, Nakanishi H, Tanabe K, Nishioku T, Sugiyama T, Fujiwara M, Yamamoto K (1998) Involvement of caspase-like pin apoptosis of neural PC12 cells and primary cultures microglia induced by 6-hydroxydopamine. J Neurosci Res 54:214–222
Teismann P, Tieu K, Cohen O, Choi D, Wu D, Marks D, Vila M, Jackson-Lewis V, Przedborski S (2003) Pathologic role of glial cells in Parkinson’s disease. Movement Dis 18:121–129
Teismann P, Vila M, Choi D, Tieu K, Wu D, Jackson-Lewis V, Przedborski S (2003) Cylooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc Natl Acad Sci USA 100:5473–5478
Thyagarajan D, Bressman S, Bruno C, Przedborski S, Shanske S, Lynch T, Fahn S, DiMauro S (2000) A novel mitochondrial 12S rRNA point mutation in Parkinsonism, deafness and neuropathy. Ann Neurol 48:730–736
Trifunovic A, Hansson A, Wredenberg A, Rovio A, Dufour E, Khvorostov I, Spelbrink J, Wibom R, Jacobs H, Larsson N (2005) Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA 102:17993–17998
Trimmer PA, Borland MK, Keeney PM, Bennett JP Jr, Parker WD Jr (2004) Parkinson’s disease transgenic mitochondrial cybrids generate Lewy inclusion bodies. J Neurochem 88(4):800–812
van der Walt J, Nicodemus K, Martin E, Scott W, Nance M, Watts R, Hubble J, Haines J, Koller W, Lyons K, Pahwa R, Stern M, Colcher A, Hiner B, Jankovic J, Ondo W, Allen FJ, Goetz C, Small G, Mastaglia F, Stajich J, McLaurin A, Middleton L, Scott B, Schmechel D, Pericak-Vance M, Vance J (2003) Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet 72:804–811
Vijitruth R, Liu M, Choi D, Nguyen X, Hunter R, Bing G (2006) Cyclooxygenase-2 mediates microglial activation and secondary dopaminergic cell death in the mouse MPTP model of Parkinson’s disease. J Neuroinflammation 3:1742–2094
von Bohlen und Halbach O, Schober A, Krieglstein K (2004) Genes, proteins, and neurotoxins involved in Parkinson’s disease. Prog Neurobiol 73:151–177
Waragai M, Wei J, Fujita M, Nakai M, Ho G, Masliah E, Akatsu H, Yamada T, Hashimoto M (2006) Increased level of DJ-1 in the cerebrospinal fluids of sporadic Parkinson's disease. Biochem Biophys Res Commun 345(3):967–972
Welty-Wolf K, Simonson S, Huang Y, Fracica P, Patterson J, Piantadosi C (1996) Ultrastructural changes in skeletal muscle mitochondria in Gram-negative sepsis. Shock 5:378–384
West A, Moore D, Biskup S, Bugayenko A, Smith W, Ross C, Dawson V, Dawson T (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 102:16842–16847
Wood-Kaczmar A, Gandhi S, Wood N (2006) Understanding the molecular causes of Parkinson’s disease. Trends Mol Med 12:521–528
Wu D, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci USA 100:6145–6150
Xie Z, Smith C, Van Eldik L (2004) Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia 45:170–179
Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman E, Mizuno Y (1996) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA 93:2696–2701
Zhang J, Stanton D, Nguyen X, Liu M, Zhang Z, Gash D, Bing G (2005) Intrapallidal lipopolysaccharide injection increases iron and ferritin levels in glia of the rat substantia nigra and induces locomotor deficits. Neuroscience 135:829–838
Zhu J-H, Guo F, Shelburne J, Watkins S, Chu C (2003) Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol 13:473–481
Zhu J-H, Kulich S, Oury T, Chu C (2002) Cytoplasmic aggregates of phosphorylated extracellular signal-regulated kinase in Lewy body diseases. Am J Pathol 161:2087–2098
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Onyango, I.G. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Neurochem Res 33, 589–597 (2008). https://doi.org/10.1007/s11064-007-9482-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-007-9482-y