
Abstract
Manganese (Mn) is neurotoxic: the underlying mechanisms have not been fully elucidated. l-Buthionine-(S,R)-sulfoximine (BSO) is an irreversible inhibitor of γ-glutamylcysteine synthetase, an important enzyme in glutathione (GSH) synthesis. To test the hypothesis that BSO modulates Mn toxicity, we investigated the effects of treatment of U-87 or SK-N-SH cells with MnCl2, BSO, or MnCl2 plus BSO. We monitored cell viability using MTT assay, staining with HO-33342 to assess live and/or apoptotic cells, and staining with propidium iodide (PI) to assess necrotic cells; we also measured cellular glutathione. Our results indicate decreased viability in both cell types when treated with MnCl2 or BSO: Mn was more toxic to SK-N-SH cells, whereas BSO was more toxic to U-87 cells. Because BSO treatment accentuated Mn toxicity in both cell lines, GSH may act to combat Mn toxicity. Thus, further investigation in oxidative stress mediated by glutathione depletion will unravel new Mn toxicity mechanism(s).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Manganese (Mn) is an important trace element needed for normal development and function [1]. Deficiency of Mn, although rare, may lead to improper bone and cartilage development, faulty carbohydrate metabolism, and other growth problems (see [1] and references therein). Chronic occupational exposure to Mn mostly by inhalation raises its accumulation in the central nervous system, especially in the basal ganglia [2]. Although Mn is a cofactor or required metal ion for many enzymes [e.g., superoxide dismutase (SOD), glutamine synthetase, arginase, etc.] and has important physiological roles, elevated levels of Mn in brain may induce neurotoxicity leading to manganism, a condition showing extrapyramidal signs and symptoms similar to those of Parkinson’s disease (PD) [3].
Oxidative stress is one of the putative mechanisms by which Mn can induce cell death [4]. Upon entering cells via facilitated diffusion and receptor-gated cation channels [5], Mn avidly accumulates in mitochondria [6] and may interfere with oxidative metabolism thereby leading to enhanced production of reactive oxygen species (ROS) such as superoxide radical. Mn also induced dose-dependent inhibition of respiratory complexes and ROS generation in mitochondria isolated from rat brain: this ROS generation could be decreased by treatment with antioxidants [7]. Treatment of PC12 cells with Mn also renders their mitochondrial complex I vulnerable [8]. There is evidence that Mn aids in autooxidation of dopamine with subsequent generation of ROS in the basal ganglia [9]. Chronic Mn exposure in rats can also elevate levels of iron and copper in synaptosomes and nuclei, thereby inducing neurotoxicity [10]. Moreover, allopurinol treatment protected against Mn induced oxidative stress in striatum in vivo [11]. Contrasting evidence also exists showing that Mn did not generate ROS in catecholaminergic CATH.a cells though prior administration of Mn increased H2O2-generated ROS more than H2O2 alone [12] and also Mn did not produce oxidative stress in developing brains of neonatal rats [13].
Glutathione (GSH, γ-glutamylcysteinylglycine) is one of the most important antioxidants of cells and accounts for almost 90% of nonprotein intracellular sulfur [14]. It is essential for maintaining intracellular redox homeostasis. It also plays an important role in eliminating ROS and thus protects cells from harmful consequences of oxidative stress. Normal mitochondrial electron transport chain activities generate free radicals and ultimately ROS [15]. Overload of reactive oxygen/nitrogen species in cells is implicated in various neurodegenerative diseases (e.g., PD and Alzheimer’s disease) and also in cancer, ischemia, and atherosclerosis [15]. Glutathione can be depleted from cells by treating them with l-buthionine-(S,R)-sulfoximine (BSO), an irreversible inhibitor of γ-glutamylcysteine synthetase (an important rate-limiting enzyme for glutathione synthesis) [16]. Thus, treatment with BSO can induce oxidative stress in cells and serves as a model for investigating cellular and molecular mechanisms underlying oxidative stress.
ROS such as hydroxyl and superoxide radicals generated by various biochemical reactions are harmful to cells. Superoxide radical can be converted enzymatically to H2O2 by SOD [17]. Subsequently, the enzyme glutathione peroxidase (GPx) converts H2O2 into water and concomitantly oxidizes GSH (reduced) to GSSG (oxidized), thereby preventing the formation of the highly reactive hydroxyl radical from H2O2. Hydroxyl radicals can damage DNA, lipids, and proteins by oxidizing them. Consequently, because of its functional coupling to the above antioxidant enzymes (namely, SOD and GPx, as discussed above), glutathione plays significant role in cellular defense against ROS-mediated oxidative stress [14].
Although various hypotheses were proposed, the exact molecular mechanisms of Mn neurotoxicity have not been fully elucidated [18]. Mn is implicated in oxidative stress [19], and imbalance of activities of enzymes of oxidative metabolism [20, 21] and neurotransmitter metabolism [21, 22]. Upon entering the brain, Mn selectively accumulates in astrocytes [23, 24] and is known to induce gliosis in basal ganglia subregions such as globus pallidus where astrocytes show morphological features similar to those of Alzheimer type II astrocytosis [24]. In this connection, it is noteworthy that cultured astrocytes were shown to possess greater capacity of synthesizing glutathione compared to cultured neurons [25, 26]. Thus, these observations, taken together, suggest that Mn neurotoxicity may show different patterns in neurons and astrocytes [20].
As the cellular and molecular mechanisms underlying Mn neurotoxicity have not been fully characterized, we considered it important and opportune to further investigate the oxidative stress hypothesis for Mn neurotoxicity. Thus, this study aimed to test the hypothesis that inhibition of glutathione synthesis modulates Mn neurotoxicity differentially in astrocytoma and neuroblastoma cells. We employed human U87 astrocytoma and SK-N-SH neuroblastoma cells for this study because these two cell lines have been extensively characterized by our group [20].
Experimental procedure
Materials
U-87 and SK-N-SH cells were obtained from ATCC (Manassas, VA, USA). Manganese chloride and EDTA were bought from Fisher chemicals (Fairlawn, NJ, USA). BSO, 3-(4,5-dimethylthia-zol,2-yl)-2,5-diphenyltetrazolium bromide (MTT dye), dimethyl sulfoxide, 5,5′-dithio-bis (2-nitrobenzoic acid) (DTNB), modified Eagle’s medium, fetal bovine serum, and 5-sulfosalicylic acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). The dyes, Hoechst 33342, and propidium iodide (PI) were from Molecular Probes (Eugene, OR, USA). Bicinchoninic acid protein assay kit from Pierce (Rockford, IL, USA) was used to determine protein levels in cell homogenates.
Cell culture
U-87 (Human Astrocytoma) and SK-N-SH (Human Neuroblastoma) cells were cultured in modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum and were incubated at 37°C and 5% (v/v) CO2. Cells were seeded with equal density in each well of 96-well plates. Manganese chloride and BSO, individually and in combination at different concentrations, were added the next day and cells were incubated for 48 h. Various assays were performed on the treated and untreated cells at the end of the incubation.
Cell viability assay
Cellular viability was determined using the MTT assay [27]. Cells were set up and treated in 96-well plates as described in the previous section. At the end of the incubation period, MTT dye (0.5%, w/v, in phosphate-buffered saline) was added to each well and the plates were incubated for 4 h at 37°C. Purple-colored insoluble formazan crystals in viable cells were dissolved using dimethyl sulfoxide and the subsequent absorbance of the content of each well was measured at 567 nm using a multidetection microplate reader (Bio-Tek Synergy HT, Winooski, VT, USA).
Cell survival/death assays
PI (5 μl of 1 mg/ml to each well) and Hoechst 33342 (HO) (1.5 μl of 1.5 mg/ml to each well) were added to cells in 96-well plates after initial incubation with MnCl2 with and without BSO: the plates were set up as depicted above. Blue-fluorescent HO dye stained live and apoptotic cells whereas red-fluorescent PI dye stained necrotic cells [28]. Images of fluorescent cells were captured using a fluorescence microscope (Leica DM IRB, Bannockburn, IL, USA) equipped with a digital camera (Leica DFC 300 FX, Bannockburn, IL, USA) and fluorescent cells were counted using the image pro plus (5.0) software. The color photomicrographs were digitally converted to grey-scale images for presentation herein.
GSH assay
Cellular GSH content was determined employing the method of Griffith [29]. Briefly, cells were homogenized using a solution containing sulphosalicylic acid (4.31%, w/v) and 0.25 mM EDTA. The GSH in cell homogenates was then determined chemically by reacting the GSH therein with Ellmann’s reagent (DTNB) and measuring the absorbance of the reaction product at 412 nm using a multidetection microplate reader (Bio-Tek Synergy HT, Winooski, VT, USA). GSH content was normalized to total cellular protein content determined by bicinchoninic acid protein assay kit.
Statistical analysis of data
Data are presented as mean ± standard error of the mean (SEM). Data analysis was performed by two-way ANOVA (using the SPSS software 13.0) with Dunnett’s post hoc test for multiple comparisons. Significance level was set at P < 0.05.
Results
Both manganese chloride and BSO exerted dose-dependent toxicity on U-87 and SK-N-SH cells
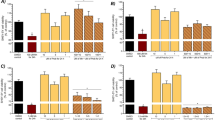
Treatment with Mn or BSO induced a decline in viability in U-87 and SK-N-SH cells in a dose-related manner (Fig. 1). Mn induced decrease in cell viability was more pronounced in SK-N-SH cells than that in U-87 cells. By itself, BSO was effective in lowering the viability of U-87 cells and also SK-N-SH cells. When used in combination, BSO enhanced the toxic effects of Mn in both cell types (Fig. 1).

Effects of Mn with and without BSO on viability of U-87 and SK-N-SH cells. The viability of U-87 (a) and SK-N-SH (b) cells was determined by MTT assay after treatment of cells with manganese chloride in the presence and absence of BSO for 48 h. All values are mean ± SEM of at least three different experiments. **P < 0.001 for manganese chloride and # P < 0.05, ## P < 0.001 for BSO by two-way ANOVA followed by Dunnett’s post hoc test
Mn- and BSO-induced cell death in U-87 and SK-N-SH cells as assessed by cell survival/death assays
Based on the results of cell viability determination by MTT assay (Fig. 1), concentrations of Mn and BSO were decreased so as to detect both apoptosis and necrosis in both types of cells. Mn treatment at 1.0 mM induced a decrease in the number of live/apoptotic U-87 and SK-N-SH cells (Fig. 2). BSO treatment at high concentrations also induced a decrease in the number of cells stained with HO (Fig. 2b). The Mn-induced effects were more pronounced in SK-N-SH cells, whereas BSO exerted more pronounced effects on U-87 cells (Fig. 3): the decrease in the number of live and apoptotic cells was accompanied by corresponding increase in the number of necrotic cells as illustrated by cells stained with PI (Fig. 2c). Mn was also tested at concentrations higher than 1 mM and the results, based on the number of cells stained with HO and PI as described above, indicated that the Mn-induced effect was dose-related and more pronounced in SK-N-SH cells (data not shown).

Brightfield and fluorescence images of U-87 and SK-N-SH cells after the following treatments—control, Mn 1.0 mM, or BSO 2.5 mM. Brightfield images (a) are followed by images of HO stained cells (b) and then images of PI stained cells (c). These are representative data derived from at least three separate experiments. The photomicrographs were taken at a magnification of 200X

Count of live/apoptotic U-87 and SK-N-SH Cells after treatment with various concentrations of Mn in the absence and presence of BSO. Determination of cell survival/death using fluorescent dye HO with fluorescence microscopy in (a) U-87 and (b) SK-N-SH cells. Cells incubated with various concentrations of manganese chloride and BSO for 48 h. All values are means ± SEM. of at least three different experiments. **P < 0.001 and # P < 0.05, ## P < 0.001 for BSO by two-way ANOVA followed by Dunnett’s post hoc test
Effects of Mn and BSO on GSH content in U-87 and SK-N-SH cells
Endogenous levels of glutathione (in the absence of any treatments) were almost two times higher in U-87 (astrocyte-like) than in SK-N-SH (neuron-like) cells. Treatment with BSO induced a lowering of GSH in both cell types and this lowering of GSH was prominent at high dose of BSO (2.5 mM) (Fig. 4). Mn treatment induced decrease in glutathione levels of astrocytoma cells, whereas Mn alone did not alter glutathione levels in neuroblastoma cells.

GSH levels in (a) U-87 cells and in (b) SK-N-SH cells. Cells were treated with various concentrations of manganese chloride and BSO for 48 h. The treated cells were then homogenized and their glutathione content measured. All values are mean ± SEM of at least three different experiments. **P < 0.001 for manganese chloride and # P < 0.05, ## P < 0.001 for BSO by two-way ANOVA followed by Dunnett’s post hoc test
Discussion
Our results show that glutathione depletion enhances Mn-induced neurotoxicity and that these toxic effects are differentially expressed in astrocytoma (U-87) and neuroblastoma (SK-N-SH) cells. Treatment with manganese chloride and BSO induced a decrease in viability in U-87 and SK-N-SH cells. Moreover, treatment of both cell types with BSO combined with Mn induced a more marked decrease in cellular viability compared to that with either of the treatments alone. The Mn treatment-induced cell death in both cell types (as determined by microscopic analysis) was predominantly necrotic whereas that induced by BSO treatment tended to be apoptotic in nature.
Constitutive glutathione levels were higher in astrocytoma cells compared to those in neuroblastoma cells and this difference in glutathione content may explain differential susceptibilities of these cells towards Mn induced neurotoxicity [20]. Furthermore, treatment with Mn and BSO depleted the levels of glutathione in both cell lines (Fig. 4). Thus, our results suggest oxidative stress induced by glutathione depletion may be an important mechanism underlying Mn induced neurotoxicity.
Mn-induced neurotoxicity
Chronic occupational (mainly welding and mining) exposure to Mn leads to a condition of Mn neurotoxicity referred to as manganism. Upon entering the body Mn binds to an iron binding protein in plasma, transferrin, and thereby can displace iron [30]. Transferrin-mediated endocytosis in brain raises level of Mn with differential distribution in various parts of brain [10] and this excess brain Mn accumulation leads to neurotoxicity.
Manganism shows signs and symptoms similar to those observed in PD but differ in the target site, with Mn toxicity affecting globus pallidus more than substantia nigra [31]. Neverthless, Mn neurotoxicity is considered as one of the contributing factors in the development of idiopathic PD and is associated with pathogenesis of chronic hepatic encephalopathy in which high Mn concentration was noted in caudate nucleus and globus pallidus [32].
Pathophysiology of manganese neurotoxicity
T-1 weighted magnetic resonance imaging in patients with chronic Mn exposure showed bilateral hyperintensities in basal ganglia structures [33]. Neuronal degeneration with diminished myelinated fibers and irregular presence of Lewy bodies are also noted in postmortem brain of patients with manganism [34]. A distinguishing feature of Mn neurotoxicity is the presence of Alzheimer type II astrocytes in the basal ganglia [35].
We used in vitro neural cell culture model systems to study Mn neurotoxcity as they offer distinct advantages and bypass complications arising from the presence of the blood–brain barrier, peripheral metabolism, and other peripheral effects known to occur in vivo [36]. Also BSO is used to deplete glutathione in animal models and thus the experimental paradigm of this study can be translated to normal cells [37]. The MTT assay used in our studies reveal the extent of mitochondrial toxicity as it measures the ability of mitochondria to metabolize the dye employed in the assay [27]. MnCl2 or BSO treatment induced dose-related decreases in viability of both U-87 and SK-N-SH cells (Figs. 2, 4) and the toxicity of Mn was also time-dependent in these cell lines [20]. A combination treatment with both Mn and BSO also induced significant decrease in viability of these cells, suggesting that BSO treatment accentuated the Mn-induced effect (Figs. 1–4).
Glutathione depletion enhances Mn neurotoxicity
Our results from the MTT assay and HO/PI staining show that Mn toxicity is enhanced by glutathione depletion in both U-87 and SK-N-SH cells (Figs. 1–4). Glutathione depletion by BSO can induce mitochondrial damage and may induce oxidative stress in cells [38, 39]. Furthermore, the effects of BSO were more pronounced in U-87 (astrocyte-like) cells than SK-N-SH (neuron-like) cells (Fig. 2) indicating their differential susceptibility towards oxidative stress. Thus, our data suggest one mechanism underlying Mn neurotoxicity may be Mn-induced oxidative stress.
In brain Mn accumulates more in astrocytes than neurons [40] but neurons are highly susceptible to Mn assault. Primary cultures of astrocytes are known to possess higher capacity to produce the antioxidant glutathione than their neuronal counterparts [25, 26] and this difference may explain the differential susceptibility of neurons towards Mn neurotoxicity. Our data, obtained with U-87 (astrocyte-like) and SK-N-SH (neuron-like) cells (Fig. 4), are consistent with earlier findings that astrocytes have higher endogenous levels of glutathione than neurons [25, 26]. Furthermore, administration of Mn induced decreased levels of glutathione in U-87 cells with maximum depletion observed at higher doses (Fig. 4). As we had anticipated, administration of BSO also depleted levels of GSH in these cells (Fig. 4). As glutathione depletion can induce oxidative stress, all the above-mentioned observations taken together suggest that Mn treatment can provoke oxidative stress and the oxidative stress augmented by glutathione depletion by BSO can and does enhance Mn-induced neurotoxicity.
Manganese and neurodegeneration
Oxidative stress is implicated as a putative mechanism for various neurodegenerative conditions like PD and Alzheimer’s disease. Moreover, Mn neurotoxicity is associated with oxidative stress and impairment of mitochondrial function. The aging brain shows accumulation of a variety of heavy metals including Mn and this chronic accumulation of heavy metals may lead to development of neurodegenerative conditions [41]. Our data support the link between Mn and oxidative stress as we have shown that glutathione depletion accentuates neurotoxic effects of Mn. Thus, taken together, the different lines of evidence discussed above suggests that studying brain Mn accumulation and Mn neurotoxicity may shed new light on pathophysiological mechanisms underlying neurodegenerative disorders.
Conclusion
Manganese chloride and BSO treatment produced a dose-related decrease in cell viability in U-87 and SK-N-SH cells. SK-N-SH cells were more susceptible to Mn toxicity whereas U-87 cells were more susceptible to BSO treatment. The results obtained with apoptosis/necrosis assays confirmed these findings and also suggested that the mechanisms of cell death induced by MnCl2 and BSO may be different although such mechanisms require further study. Constitutive levels of GSH in U-87 cells were higher than those of SK-N-SH cells and this difference in their GSH contents may account for their differential susceptibility towards oxidative stress. The results of this study thus point to the importance of cellular GSH in combating Mn-induced toxicity and suggest oxidative stress may be one of the mechanisms underlying Mn toxicity. Finally, Mn-induced oxidative stress mechanism may assume pathophysiological importance in many neurodegenerative diseases and as such merits further investigation.
References
Lai JCK, Chan AWK, Minski MJ, Lim L (1985) Roles of metal ions in brain development and aging. In: Gabay S, Harris J, Ho BT (eds) Metal ions in neurology and psychiatry. Alan Liss, New York, pp 49–67
Lai JCK, Chan AWK, Leung TKC, Minski MJ, Lim L (1992) Neurochemical changes in rats chronically treated with a high concentration of manganese chloride. Neurochem Res 17:841–847
Cotzias GC, Horiuchi K, Fuenzalida S, Mena I (1968) Chronic manganese poisoning: clearance of tissue manganese concentrations with persistance of the neurological picture. Neurology 18:376–382
Erikson KM, Dobson AW, Dorman DC, Aschner M (2004) Manganese exposure and induced oxidative stress in the rat brain. Sci Total Environ 334–335:409–416
Lai JCK, Minski MJ, Chan AWK, Lim L (2000) Interrelations between manganese and other metal ions in health and disease. In: Sigel A, Sigel H (eds) Metal ions in biological systems. Dekker, New York, pp 123–156
Lai JCK, Chan AWK, Minski MJ, Leung TKC, Lim L, Davison AN (1985) Application of instrumental neutron activation analysis to the study of trace metals in brain and metal toxicity. In: Gabay S, Harris J, Ho BT (eds) Metal ions in neurology and psychiatry, Alan Liss, New York, pp 323–343
Zhang S, Fu J, Zhou Z (2004) In vitro effect of manganese chloride exposure on reactive oxygen species generation and respiratory chain complexes activities of mitochondria isolated from rat brain. Toxicol In Vitro 18:71–77
Galvani P, Fumagalli P, Santagostino A (1995) Vulnerability of mitochondrial complex I in PC12 cells exposed to manganese. Eur J Pharmacol 293:377–383
Donaldson J, LaBella FS, Gesser D (1981) Enhanced autooxidation of dopamine as a possible basis of manganese neurotoxicity. Neurotoxicology 2:53–64
Lai JCK, Minski MJ, Chan AWK, Leung TKC, Lim L (1999) Manganese mineral interactions in brain. Neurotoxicology 20:433–444
Desole MS, Esposito G, Migheli R, Fresu L, Sircana S, Miele M, De Natale G, Miele E (1995) Allopurinol protects against manganese-induced oxidative stress in the striatum and in the brainstem of the rat. Neurosci Lett 192:73–76
Worley CG, Bombick D, Allen JW, Suber RL, Aschner M (2002) Effects of manganese on oxidative stress in CATH.a cells. Neurotoxicology 23:159–164
Weber S, Dorman DC, Lash LH, Erikson K, Vrana KE, Aschner M (2002) Effects of manganese (Mn) on the developing rat brain: oxidative-stress related endpoints. Neurotoxicology 23:169–175
Meister A (1995) Glutathione metabolism. Methods Enzymol 251:3–7
Dröge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82:47–95
Griffith OW, Meister A (1979) Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J Biol Chem 254:7558–7560
Schulz JB, Lindenau J, Seyfried J, Dichgans J (2000) Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 267:4904–4911
Verity MA (1999) Manganese neurotoxicity: a mechanistic hypothesis. Neurotoxicology 20:489–497
Stokes AH, Lewis DY, Lash LH, Jerome WG III, Grant KW, Aschner M, Vrana KE (2000) Dopamine toxicity in neuroblastoma cells: role of glutathione depletion by l-BSO and apoptosis. Brain Res 858:1–8
Malthankar GV, White BK, Bhushan A, Daniels CK, Rodnick KJ, Lai JCK (2004) Differential lowering by manganese treatment of activities of glycolytic and tricarboxylic acid (TCA) cycle enzymes investigated in neuroblastoma and astrocytoma cells is associated with manganese-induced cell death. Neurochem Res 29:709–717
Lai JCK, Leung TKC, Lim L (1984) Differences in the neurotoxic effects of manganese during development and aging: some observations on brain regional neurotransmitter and non-neurotransmitter metabolism in a developmental rat model of chronic manganese encephalopathy. Neurotoxicology 5:37–47
Miele M, Serra PA, Esposito G, Delogu MR, Migheli R, Rocchitta G, Desole MS (2000) Glutamate and catabolites of high-energy phosphates in the striatum and brainstem of young and aged rats subchronically exposed to manganese. Aging 12:393–397
Wedler FC, Ley BW (1990) Ca(II) and Zn(II) ions alter the dynamics and distribution of Mn(II) in chick cultured glial cells. Neurochem Res 15:1221–1228
Normandin L, Hazell AS (2002) Manganese Neurotoxicity: an update of pathophysiologic mechanisms. Metab Brain Dis 17:375–387
Raps SP, Lai JCK, Hertz L, Cooper AJL (1989) Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res 493:398–401
Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJL (1994) Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem 62:45–53
Mossman T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
White MG, Emery M, Nonner D, Barrett JN (2003) Caspase activation contributes to delayed death of heat-stressed striatal neurons. J Neurochem 87:958–968
Griffith OW (1980) Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem 106:207–212
Takeda A (2003) Manganese action in brain function. Brain Res Rev 41:79–87
Olanow CW, Good PF, Shinotoh H, Hewitt KA, Vingerhoets F, Snow BJ, Beal MF, Calne DB, Perl DP (1996) Manganese intoxication in the rhesus monkey: a clinical, imaging, pathologic, and biochemical study. Neurology 46:492–498
Krieger D, Krieger S, Jansen O, Gass P, Theilmann L, Lichtnecker H (1995) Manganese and chronic hepatic encephalopathy. Lancet 346:270–274
Nelson K, Golnick J, Korn T, Angle C (1993) Manganese encephalopathy: utility of early magnetic resonance imaging. Br J Ind Med 50:510–513
Yamada M, Ohno S, Okayasu I, Okeda R, Hatakeyama S, Watanabe H, Ushio K, Tsukagoshi H (1986) Chronic manganese poisoning: a neuropathological study with determination of manganese distribution in the brain. Acta Neuropathol (Berl) 70:273–278
Hazell AS, Normandin L, Norenberg MD, Kennedy G, Yi JH (2005) Alzheimer type II astrocytic changes following sub-acute exposure to manganese and its prevention by antioxidant treatment. Neurosci Lett 396:167–171
Lai JCK, Leung TKC, Lim L (1985) Effects of metal ions on neurotransmitter function and metabolism. In: Gabay S, Harris J, Ho BT (eds) Metal ions in neurology and psychiatry. Alan Liss, New York, pp 177–197
Jain A, Martensson J, Stole E, Auld PA, Meister A (1991) Glutathione deficiency leads to mitochondrial damage in brain PNAS 88:1913–1917
Meister A (1995) Mitochondrial changes associated with glutathione deficiency. Biochim Biophys Acta 1271:35–42
Desole MS, Esposito G, Migheli R, Sircana S, Delogu MR, Fresu L, Miele M, de Natale G, Miele E (1997) Glutathione deficiency potentiates manganese toxicity in rat striatum and brainstem and in PC12 cells. Pharmacol Res 36:285–292
Aschner M, Gannon M, Kimelberg HK (1992) Manganese uptake and efflux in cultured rat astrocytes. J Neurochem 58:730–735
Zatta P, Lucchini R, Van Rensburg SJ, Taylor A (2003) The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res Bull 62:15–28
Acknowledgments
This study was supported by a grant from Idaho Biomedical Research Infrastructure Network (NIH NCRR BRINIP20RR016454) and an Idaho State University FRC grant. The authors thank Isaac Alfred Orina for his helpful suggestions. V.V. Dukhande thanks the Idaho INBRE NIH program (grant # P20RR016454) for a research fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dukhande, V.V., Malthankar-Phatak, G.H., Hugus, J.J. et al. Manganese-Induced Neurotoxicity is Differentially Enhanced by Glutathione Depletion in Astrocytoma and Neuroblastoma Cells. Neurochem Res 31, 1349–1357 (2006). https://doi.org/10.1007/s11064-006-9179-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-006-9179-7