
Abstract
Chronic arsenic exposure is associated with nervous system damage, vascular disease, hepatic and renal damage as well as different types of cancer. Alterations of nitric oxide (NO) in the periphery have been detected after arsenic exposure, and we explored here NO production in the brain. Female Wistar rats were exposed to arsenite in drinking water (4–5 mg/kg/day) from gestation, lactation and until 4 months of age. NOS activity, NO metabolites content, reactive oxygen species production (ROS) and lipid peroxidation (LPx) were determined in vitro in the striatum, and NO production was estimated in vivo measuring citrulline by microdialysis. Exposed animals showed a significantly lower response to NMDA receptor stimulation, reduction of NOS activity and decreased levels of nitrites and nitrates in striatum. These markers of NO function were accompanied by significantly higher levels of LPx and ROS production. These results provide evidence of NO dysfunction in the rat brain associated with arsenic exposure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A significant association between chronic low-level arsenic exposure and cognitive dysfunction has been revealed in three epidemiological studies performed in children and adolescents from different parts of the world [1–3]. These findings raised concern because neuropathies and neural tube defects were previously reported only under conditions of high level arsenic exposure [4–6], but if arsenic neurotoxicity is present at lower exposure levels, then the central nervous system (CNS) of millions of people worldwide could be affected through contaminated drinking water.
An important progress concerning the identification of the cellular targets of arsenic has been accomplished through in vitro studies, looking at the effects of micromolar arsenic concentrations similar to those found in the blood of exposed individuals [7]. Nevertheless, one of the main limitations of these studies is that they do not allow the exploration of functions such as those of the CNS, where the impairments associated with arsenic exposure involve the interaction of many cell types located in different brain regions. Therefore, for the study of important arsenic effects such as carcinogenesis, cardiovascular dysfunction and neurotoxicity, animal models have been employed and they have provided insights into arsenic toxicity. This approach has allowed the demonstration of behavioral and neurochemical effects [8–10] as well as carcinogenesis [11, 12] in arsenic exposed rodents that do not show weight loss or intoxication signs. However, the main shortcoming of in vivo studies is that the arsenic doses employed to observe effects are much higher than those in which human exposure takes place.
In rats, arsenic exposure is associated with changes in neurotransmitter levels and release in the striatum, and the exposed animals present impairments in behavioral tasks that involve this region, such the T-maze test [8, 9]. Further studies in the striatum demonstrated the in vivo production of hydroxyl radical, which pointed to oxidative damage as the cellular changes underlying the neurochemical and behavioral alterations observed in arsenic exposed animals [13]. Nevertheless, other regions besides the striatum are also affected; an increased presence of oxidative stress markers has been reported in whole brain of rats [10, 14], and even in the blood of human population exposed to arsenic [15, 16]. Despite the diversity of arsenic targets in the brain and other organs, the striatum remains an area of interest because of its role in locomotor activity, which is also affected by arsenic, and the higher vulnerability of the basal ganglia to oxidative damage, exemplified by its involvement in disorders such as Parkinson and Huntington diseases.
Recently, the involvement of nitric oxide (NO) in arsenic toxicity has raised considerable interest, and it is the subject of numerous current studies in humans [17, 18], animal models [19, 20] and in vitro preparations [21]. Furthermore, nitrergic dysfunction can also be linked to oxidative stress, since inadequate levels of tetrahydrobiopterin, one of the cofactors for NO synthesis are thought to contribute to superoxide formation by nitric oxide synthase (NOS) [22, 23]. In the brain, NO is involved in neuronal transmission [24], long term potentiation processes related with some types of learning and memory [25, 26], and its production depends mainly on calcium entry through NMDA receptors for glutamate [27, 28]. The predominant NOS isoform in neurons is the nNOS, although endothelial and inducible isoforms are also present in nervous tissue.
In light of the considerations above, our objective was to define whether NO production in the brain is altered by chronic arsenic exposure. For this purpose, we undertook a study in rats and we quantified, in vivo and in vitro, NO production, NOS activity, and oxidative stress markers in the striatal region of the rat brain. Arsenic exposure was initiated from the fetal stage throughout lactation and development, since human exposure takes place in this way, in the places where only arsenic contaminated drinking water is available.
Materials and methods
Animal model and reagents
The experiments described here were performed according to the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research [29]. In order to guarantee a sufficient amount of female rats to obtain experimental groups of 6 animals belonging to different litters, 9 female adult Wistar rats (200–250 g) were placed in separate cages with one adult male rat each and received drinking water containing sodium arsenite (37 ppm of arsenic), which resulted in a daily intake between 4 and 5 mg/kg of arsenic, according to previous reports of biochemical and behavioral changes in absence of body weight loss observed in these conditions [10, 19]. Ten days later the males were discarded and the dams continued the exposure throughout pregnancy and lactation. After 1 month the pups were weaned, separated by sex and they continued exposure until reaching 4 months of age. In parallel, control rats from nine dams were raised without arsenic exposure. All the animals tested in a experimental group belonged to different litters and were maintained in the animal facilities under controlled conditions of temperature, humidity and light/dark cycle. Arsenic exposure neither resulted in mortality of dams or pups, nor in decreased litter size or weight loss of adult or developing animals.
Sodium arsenite was obtained from Sigma (St. Louis, MO, USA) as well as most of the reagents used in this protocol, unless otherwise stated.
In vitro studies
When the animals reached 4 months of age, 6 control and 6 exposed rats arising from different litters were sacrificed by decapitation, the brains extracted, the striata dissected in a cold plate and immediately frozen in liquid nitrogen. The left side was destined for quantification of NOS activity, while ROS production and lipid peroxidation were analyzed in the right side. Two parallel groups of 6 exposed and 6 control rats underwent blood extraction from the tail vein for nitrite and nitrate (NOx) quantification in serum. After sacrifice, the dissected striata were destined for NOx analysis, and total arsenic concentration was quantified in the rest of the brain.
In vivo study
Six exposed and seven control rats underwent stereotaxic surgery for implantation of a guide-canula in the left striatum, which allowed the sampling of extracellular fluid by means of microdialysis, in order to measure citrulline levels as an estimate of NO production.
Evaluation of oxidative stress and nitrergic function
Lipid peroxidation was determined according to Santamaría and Ríos [30] using a chloroform extraction of fluorescent products of peroxidized lipids. Striata were homogenized in buffered saline and 1 ml aliquots were added to 4 ml of a 2:1 chloroform–methanol mixture (Merck, Darmstadt, Germany) and briefly shaken. After 30 min fluorescence was measured in the chloroform phase using a Perkin Elmer spectrophotometer (Model LS50B) at 370 and 430 nm wavelengths for excitation and emission, respectively. The spectrophotometer sensitivity was calibrated with quinine (0.001 mg/ml) in order to obtain units of fluorescence intensity for expressing the results. ROS production was measured as the formation of 2′7′-dichlorofluorescein (DCF) according to Ali et al. [31], and the results were expressed as μmol/mg protein-hour. Ten milliliter homogenate aliquots were diluted in TRIS–HEPES buffer and incubated with 2′7′-dichlorofluorescein diacetate (5 mM) for 1 h at 37°C in a shaking bath. Fluorescence was recorded at 488 and 525 nm wavelengths for excitation and emission, respectively, and values were interpolated in a DCF standard curve by lineal regression. In the standardized validation procedure, we obtained a lineal regression coefficient of 0.99 and coefficients of variation ranging between 6.2 and 13% for repeatability and 6.2 and 13.5% for intra-laboratory reproducibility, which fall within the acceptable levels for these concentrations [32]. We determined protein content of homogenates according to the method of Lowry [33] for all the assays.
Nitrate and nitrite analysis was performed according to Miranda et al. and Tenorio et al. [34, 35]. For the blood analysis, serum was separated by centrifugation and deproteinized with methanol–diethylether (3:1), while striatal tissue was homogenized in 100% methanol. Vanadium chloride (500 μl, 0.8%) was added to 100 μl sample of either deproteinized serum or striatal homogenate supernatant, followed by addition of the Griess reagents (250 μl sulfanilamide, 2% and 250 μl N-(1-Naphthyl)ethylenediamine dihydrochloride, 0.1%). The reaction mixture was incubated for 45 min at 37°C and absorbance at 540 nm was measured in a Beckman spectrophotometer (model 25). The results were expressed as μM nitrites for the serum samples and μmol/mg tissue for the content in striatum.
Arsenic quantification
Arsenic determination was performed by means of atomic fluorescence in a Excalibur Millennium System (PS Analytical, Kent, England). For this purpose, we set up and standardized a technique for determination of total arsenic in brain tissue using acid digestion (6:1 mixture of nitric and perchloric acids). Calibrations curves were constructed with dilutions of an arsenic standard (High Purity, Charleston, SC, USA). After digestion, the samples were resuspended in HCl (3%) and a solution of 50% KI in 10% ascorbic acid as reductor agent. The final reaction mixture that allowed atomic arsenic fluorescence contained NaBH4 that formed gaseous AsH3. Using a standardized validation procedure, we obtained a linear quantification procedure in a range of 1–30 ppb arsenic (slope = 6.8 ± 1.9) with coefficients of variation ranging between 2.9–17.4% and 10–16.4% for repeatability and intra-laboratory reproducibility. In parallel with each assay, the recovery of a standard added to a control sample of brain was evaluated, obtaining a recovery of 106%.
NOS activity
Using [3H] l-arginine, NOS activity was measured based on its stoichiometric conversion to NO and l-citrulline as reported by Pérez-Severiano et al. [36]. Striata were dissected out on ice-cold plate and immediately frozen at −70°C until assays were performed. The tissue was homogenized on ice in 250 μl buffer (50 mM Tris–HCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% β-mercaptoethanol, pH 7.5, all reagents from MERCK, Darmstadt, Germany), and a cocktail of protease inhibitors (100 μM leupeptin, 1 mM phenyl methylsulphonyl fluoride, 2 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor) and 0.1% Nonidet P-40. Volumes of homogenates containing 500 μg of protein were incubated for 30 min at 37°C in the presence of 10 μM l-arginine–HCl, 1 mM NADPH, 100 nM calmodulin, 30 μM tetrahydrobiopterin, 2.5 mM CaCl2 and 0.2 μCi of [3H] l-arginine (Amersham). In order to measure the activity of inducible NOS (iNOS), the incubation was carried out in presence of EGTA and without CaCl2. Reactions were stopped by adding a buffer containing 2 mM EGTA, 2 mM EDTA, 20 mM Hepes, pH 5.5. The reaction mixture was loaded in a cation exchange column (Dowex-50W), previously equilibrated with stop solution and that retains arginine and allows [3H] l-citrullline to flow through the column. Labeled l-citrulline was measured in a Beckman scintillation counter (model LS6500) and expressed as ng l-citrulline/500 μg protein/min.
Estimation of in vivo NO production
In preparation for microdialysis sampling, the animals were anesthetized by an i.p. injection of sodium pentobarbital (50 mg/kg) and topic xylocaine. After that, we placed them in a stereotaxic frame. A guide-cannula was permanently fixed to the skull with dental acrylic and screws in the left striatum (coordinates: AP + 2 mm, ML 2.5, DV—4 mm measured from bregma according to the atlas of Paxinos and Watson [37]). After the surgery, paracetamol was administered orally and the animals were allowed to recover about 48 h before sampling began. During the perfusion experiments, a microdialysis probe (2 mm membrane length) was inserted through the guide-cannula and the animals were connected to a syringe infusion pump through a liquid selector and placed in a Raturn system (BAS, WestLafayette, IN, USA). A solution containing 147 mM NaCl, 4 mM KCl and 1.2 mM CaCl2, pH 7.4 was perfused through the probe at a flow rate of 2 μl/min. Sample collection started after 1 h perfusion and it was performed every 30 min during 4.5 h. We collected samples in two conditions, basal (4 samples) and after stimulation of the NMDA receptor (6 samples), by means of the administration of 1 mM NMDA in the perfusion fluid through the microdialysis probe during 30 min. In a series of preliminary experiments in three additional control animals, NMDA, glutamate, glutamine, GABA and l-NAME were perfused through the probe in different experiments in order to analyze the response in terms of increase or decrease of extracellular citrulline.
For HPLC quantification of citrulline, we modified a previously reported method for aminoacid quantification in brain samples [38]. The samples were prederivatized with O-pthaladehyde and injected in a C18 column (Alltech Associates, Deerfield, IL, USA) for separation and posterior compound identification in a fluorescence detector (Perkin Elmer, LC240). The elution was accomplished with a sodium acetate buffer (50 mM, pH = 5.9) containing 1.5% tetrahydrofuran and using a methanol gradient. Citrulline concentration was calculated using external standard curves. Using a standardized validation procedure, we obtained a linear quantification procedure with coefficients of variation ranging between 6 and 15% for repeatability and 8.3 and 14.6% for intra-laboratory reproducibility.
Statistical analysis
Most data were expressed as means ± standard deviation, except for brain arsenic concentrations, which was expressed as median and their respective range. Variance homogeneity and normality of data were tested by means of Levene and Shapiro-Wilk test, respectively, and those data that did not show normality were log transformed. Comparisons between the control and the exposed groups (LP, ROS, NOx and NOS activity) were performed by means of Student t-test for independent samples. A general lineal model (GLM) for repeated measures were used to compare citrulline levels along the microdialysis experiment. The between-subjects factor was the treatment (exposed and control groups) and the within subjects factor was the sample (nine samples). This analysis was preceded by the Mauschly sphericity test for the raw data. In order to adjust the degrees of freedom in the within subjects variations for the fact that measurements were not independent, but they arise from the same subjects in this repeated measures design, we considered the Greenhouse and Geisser correction [39]. The Wilcoxon test was used to compare brain arsenic concentrations. A probability level of P < 0.05 was considered to indicate significant differences.
Results
Oxidative stress and nitrergic function
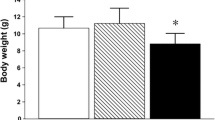
Chronic exposure to arsenite did not result in weight loss of the animals (Fig. 1A) Brain arsenic levels were very variable among exposed animals (median: 707 ng/g, range 421–839 ng/g, n = 6), but more than 50 times higher than in control animals (median: 12 ng/g, range 1–18, n = 6, Fig. 1B). Arsenite exposure resulted in a significant increase of LP and ROS production, as shown in Fig. 1C, D. Control rats had a basal LP level of 0.49 ± 0.17 fluorescence units/mg protein while in exposed rats this level increased to 1.28 ± 0.48 fluorescence units/mg protein (P = 0.001, n = 6). Arsenite exposure enhanced the production of ROS from 0.86 ± 0.25 μmol/mg protein-hour to 1.47 ± 0.25 μmol/mg protein-hour (P < 0.01, n = 6).

Characteristics of the animal model, oxidative stress and nitrergic function markers. (A) Body weight at 4 months of age (mean ± SD, n = 18), (B) brain arsenic content (median ± range). Arsenic exposure increases lipid peroxidation (C) and ROS production (D) in striatum homogenates, while it decreases NOx in serum (E) and in striatum homogenates (F). Bars from (B) through (F) represent means ± SD obtained from six animals per group, * P < 0.05
The results of nitrites and nitrates quantification in serum and striatal tissue are displayed in Fig. 1E, F. Arsenite exposure resulted in a significantly decreased concentrations of these markers from 265.9 ± 19.6 to 186.2 ± 25.5 μM in serum and from 22 ± 3 to 13.4 ± 3.9 μmol/mg protein in striata (P < 0.001, n = 6 in both cases).
NOS activity
Enzymatic activity obtained through this assay in presence of calcium includes the total activity of the present isoforms, namely, constitutive isoforms (nNOS and eNOS) and inducible isoform (iNOS), while the assay in absence of calcium reveals the activity of iNOS. We found a significant decrease of Ca-dependent NOS activity in the exposed group (11.0 ± 0.6 vs. 7.8 ± 1.0 ng l-citrulline/500 μg/min in control and exposed animals, respectively, P < 0.001), while the assay in presence of EGTA resulted in similar values of NOS activity for both groups. Therefore, these experiments showed a significant decrease of constitutive NOS activity in the exposed animals (7.3 ± 1.3 ng l-citrulline/500 μg/min) compared to controls (10.8 ± 0.8 ng l-citrulline/500 μg/min; P < 0.001, n = 6, Fig. 2).

Significant decrease of Ca-dependent NOS activity and no change of inducible NOS activity in arsenic exposed rats. Bars represent the mean ± SD (n = 6), * P < 0.001
In vivo estimation of NO production
In order to find out whether the stimulation of NMDA receptors is associated with an increase of extracellular citrulline as a result of NO production, in a series of experiments we perfused GABA and glutamine through the microdialysis probe, which did not result in a significant change of extracellular citrulline, while the addition of glutamate to the perfusion fluid resulted in a 62 ± 16% augmentation of this variable (Fig. 3A). The stimulation with 10, 100 and 500 μM NMDA did not change citrulline levels in dialysates; however, 1 mM increased citrulline outflow more than 250%. The perfusion with the NOS inhibitor N-nitro-l-arginine–methyl ester (l-NAME) resulted in citrulline levels that were 75.9 ± 8.6% of basal levels. The temporal course of the experiments is shown in Fig. 3B, where 1 mM was used to stimulate NO production. Four basal samples were destined to calculate the mean basal release, designed as 100%, followed by a 30 min perfusion with 1 mM NMDA and four further 30 min samples were collected with the initial perfusion solution. The comparison of extracellular citrulline between control and exposed animals reveals that basal values do not show significant differences and both groups respond to NMDA stimulation with significant increases of extracellular citrulline (F 8,88 = 6.6, P = 0.001); however, the response of the arsenite group is significantly lower than that of control animals (F 1,11 = 6, P = 0.032). When the highest point of the response to NMDA is compared (Fig. 3C), we found that citrulline levels increased in the exposed groups only 170 ± 24%, while control rats reached a 278 ± 27% (P < 0.001) increase.

(A) Effect of 30 min perfusion with different aminoacids on citrulline extracellular levels. The highest increase is observed with NMDA and l-NAME decreases citrulline presence to 75.9 ± 8.6%. Bars represent means ± SD of three experiments. (B) Comparison between control and arsenic exposed groups. Although basal citrulline values are similar, NMDA stimulation produces a significantly lower response in exposed animals. Values are mean ± SD, (n = 6–7) See text for description of statistical analysis. (C) Peak citrulline concentrations in microdialysates reached after 1 mM NMDA stimulation. The exposed group shows lower values in peak citrulline than the control group. Values are mean ± SD, (n = 6–7), * P = 0.01
Discussion
The main contribution of this work is the in vivo and in vitro demonstration of a decreased NO production in the rat brain associated with chronic arsenite exposure, particularly as a response to one of the main factors that regulate its production in neurons, namely NMDA receptor stimulation. The activity of NOS displayed a significant decrease in this animal model, which also presented the characteristic effects of lipid peroxidation and increase of ROS production associated with arsenic exposure.
The analysis of NOS activity was performed by means of two complementary test, in vitro activity measurements and in vivo assessment of basal and stimulated response, obtaining by both approaches a decrease of the co-product of NO synthesis, l-citrulline. We consider these results strong evidence, because under the conditions of the in vivo experiment, the system responded adequately to physiological stimuli such as glutamate and NMDA, but not to other compounds perfused through the probe. It can be argued that extracellular citrulline reflects other cellular processes in addition to NOS activity, since perfusion with l-NAME did not make citrulline disappear from perfusates, but only a decrease to 75 ± 8.6%. This response is in agreement with that reported by Hagioka et al. [40] and we attribute the presence of citrulline in the perfusate even under NOS inhibition to its relatively slow recapture for recycling to arginine since the presence of citrulline in the brain is mostly due to NOS activity [41].
These in vivo results are supported by the in vitro NOS assay, which reveals that the constitutive nNOS activity decreases approximately 30% while the inducible isoform does not seem to be affected. According to Tsou et al. [21], eNOS from porcine aortic endothelial cells presents a dose- and time-dependent decrease of activity and expression in presence of arsenite in the micromolar range. They proposed that arsenite decreases NO production by increasing eNOS degradation through the ubitiquin-proteasome pathway and also through direct inhibition of eNOS activity, and the recent report by Kamada and collaborators [42] contributes to our understanding of NOS depletion associated with arsenic exposure. Using two experimental approaches, they demonstrate that inadequate tetrahydrobiopterin levels enhance the ubiquitylation of nNOS and consequent degradation of the protein. Decreased tetrahydrobiopterin levels have been found in arsenic exposed rabbits [19] and this event can also be associated with decreased monoamine levels in arsenic exposed animals [43, 44] since this cofactor is necessary for tyrosine hydroxylase activity.
The arsenic quantification procedure revealed that our chronic exposure protocol resulted in micromolar concentrations of arsenic in the brain, therefore we found an event of NOS inhibition through oral arsenic exposure at similar concentrations that those reported in vitro. In this same direction, the diminution of NO metabolites in striatal tissue showed in Fig. 1D points to NOS dysfunction.
Concerning the markers of oxidative stress, although arsenite exposed animals presented a modest increase of ROS formation, this amount of oxygen species was enough to trigger a significant increase of lipid peroxidation. Previous experiments where arsenite was infused directly in the brain through a microdialysis probe showed that an inhibitor of the respiratory chain, 3-nitropropionic acid, triggers a higher production of hydroxyl radical than arsenite [13]. Together with those previous results, we consider that ROS formation associated with arsenite is due to dysfunction of antioxidant systems and depletion of antioxidant defenses rather than to direct interference of mitochondrial activity in this concentration range. The dysfunction of antioxidant defenses associated with arsenic exposure has been widely reported, and it arises from different factors. Arsenic decreases the available pool of reduced glutathione through direct union to its sulfhydryl groups, but more importantly through its metabolism, since glutathione is required for several steps of arsenic methylation and excretion [7]. In addition, dimethylated metabolites can either react with molecular oxygen or release iron from ferritin, and arsenic induces heme oxygenase activity [45–47]. All these reactions can induce free radicals generation. Therefore, the activity reduction of antioxidant enzymes such as superoxide dismutases, catalase and glutathione peroxidase associated with arsenic exposure that have been reported in the rat brain [10, 14, 48], can be attributed to ROS-induced changes in protein structure, and these reduced activities are in turn a further source of free radicals, even in absence of higher NO, and hence, peroxynitrite production.
In conclusion, we demonstrated alterations of nitrergic function in the brain of rats chronically exposed to arsenic in drinking water. The employed dose is similar to those associated with behavioral deficits in other studies with rodents and does not produce signs of toxicity or weight loss [8, 19] and the resulting arsenic concentration is in the range of doses that reduce NOS activity in in vitro assays. These results support the participation of NO in arsenic neurotoxicity.
References
Calderón J, Navarro ME, Jiménez-Capdeville ME et al (2001) Exposure to arsenic and lead and neuropsychological development in Mexican children. Environ Res Sect A 85:69–76
Tsai SY, Chou HY, The HW et al (2003) The effects of chronic arsenic exposure from drinking water on the neurobehavioral development in adolescence. Neurotoxicology 24:747–753
Wasserman GA, Liu X, Parvez F et al (2004) Water arsenic exposure and children’s intellectual function in Araihazar, Bangladesh. Environ Health Perspect 112:1329–1333
Rodriguez VM, Jiménez-Capdeville ME, Giordano M (2003) The effects of arsenic exposure on the nervous system. Toxicol Lett 145:1–18
Chhuttani PN, Chopra JS (1979) Arsenic poisoning. In: Vinken PJ, Bruyn GW (eds) Handbook of Clinical Neurology, Intoxications of the Nervous system, Part I, chapter 7, Vol. 36. Elsevier/North Holland Biomedical Press, Amsterdam, pp 199–216
Goebel HH, Schmidt PF, Bohl J et al (1990) Polyneuropathy due to acute arsenic intoxication: biopsy studies. J Neuropathol Exp Neurol 49:137–149
Styblo M, Drobná Z, Jaspers I et al (2002) The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ Health Perspect 110S:767–771
Rodríguez VM, Carrizales L, Jiménez-Capdeville ME et al (2001) The effects of sodium arsenite exposure on behavioral parameters in the rat. Brain Res Bull 55:301–308
Rodríguez VM, Dufour L, Carrizales L et al (1998) Effects of oral exposure to mining waste on in vivo dopamine release from rat striatum. Environ Health Perspect 106:487–491
Flora SJS (1999) Arsenic-induced oxidative stress and its reversibility following combined administration of N-acetylcysteine and meso 2,3-dimercaptosuccinic acid in rats. Clin Exp Pharmacol Physiol 26:865–869
Waalkes MP, Ward JM, Liu J et al (2003) Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol Appl Phamacol 186:116–126
Shen J, Wanibuchi H, Salim EI et al (2003) Liver tumorigenicity of trimethylarsine oxide in male Fischer 344 rats-association with oxidative DNA damage and enhanced cell proliferation. Carcinogen 24:1827–1835
García-Chavez E, Santamaría A, Díaz-Barriga F et al (2003) Arsenite-induced formation of hydroxyl radical in the striatum of awake rats. Brain Res 976:82–89
Chaudhuri AN, Basu S, Chattopadhyay S et al (1999) Effect of high arsenic content in drinking water on rat brain. Indian J Biochem Biophys 36:51–54
Pi J, Yamauchi H, Kumagai Y et al (2002) Evidence for induction of oxidative stress caused by chronic exposure of chinese residents to arsenic contained in drinking water. Environ Health Perspect 110:331–336
Kumagai Y, Pi J (2004) Molecular basis for arsenic-induced alteration in nitric oxide production and oxidative stress: implication of endothelial dysfunction. Toxicol Appl Pharmacol 198:450–457
Pi J, Kumagai Y, Sun G et al (2000) Decreased serum concentrations of nitric oxide metabolites among Chinese in an endemic area of chronic arsenic poisoning in inner Mongolia. Free Rad Biol Med 28:1137–1142
Pi J, Yamauchi H, Sun G et al (2005) Vascular dysfunction in patients with chronic arsenosis can be reversed by reduction of arsenic exposure. Environ Health Perspect 113:339–341
Pi J, Horiguchi S, Sun Y et al (2003) A potential mechanism for the impairment of nitric oxide formation caused by prolonged oral exposure to arsenate in rabbits. Free Rad Biol Med 35:102–113
Lee MY, Jung BI, Chung SM et al (2003) Arsenic-induced dysfunction in relaxation of blood vessels. Environ Health Perspect 111:513–517
Tsou TC, Tsai FY, Hsieh YW et al (2005) Arsenite induces endotelial cytotoxicity by down-regulation of vascular endothelial nitric oxide synthase. Toxicol Appl Pharmacol 208:277–284
Rosen GM, Tsai P, Weaver J et al (2002) The role of tetrahydrobiopterin in the regulation of neuronal nitric-oxide synthase-generated superoxide. J Biol Chem 277:40275–40280
Xia Y, Tsai AL, Berka V et al (1998) Superoxide generation from endothelial nitric-oxide synthase. J Biol Chem 274:25804–25808
Gally JA, Montague PR, Reeke GN Jr et al (1990) The NO hypothesis: possible effects of a short-lived, rapidly diffusible signal in the development and function of the nervous system. Proc Natl Acad Sci USA 87:3547–3551
Edelman GM, Gally JA (1992) Nitric oxide: linking space and time in the brain. Proc Natl Acad Sci USA 89:11651–11652
Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39
Bredt DS, Snyder SH (1989) Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci USA 86:9030–9033
Bredt DS, Snyder SH (1990) Isolation of nitric oxide synthase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA 87:682–685
Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research (2003) National research council of the national academies. The National Academies Press, Washington DC
Santamaria A, Rios C (1993) MK-801, an N-methyl-d-aspartate receptor antagonist, blocks quinolinic acid-induced lipid peroxidation in rat corpus striatum. Neurosci Lett 159:51–54
Ali SF, LeBel CP, Bondy SC (1992) Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxicology 13:637–648
Horwitz W (1982) Evaluation of analytical methods used for regulation of foods and drugs. Anal Chem 54:67–76
Lowry OH, Rosenbrough NJ, Farr L et al (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Miranda KM, Espey MG, Wink DA (2001) A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide: Biol Chem 5:62–71
Tenorio FA, del Valle L, Pastelín G (2005) Validación de un método analítico espectrofotométrico para la cuantificación de metabolitos estables de óxido nítrico en fluidos biológicos. Revista Mexicana de Ciencias Farmacéuticas 36:31–41
Pérez-Severiano F, Escalante B, Vergara P et al (2002) Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington’s disease mutation. Brain Res 951:36–42
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Academic Press Inc., San Diego
Castillo CG, Montante M, Dufour L et al (2002) Behavioral effects of exposure to endosulfan and methyl parathion in adult rats. Neurotoxicol Teratol 24:797–804
Armitage P, Berry G (1997) Estadística para la investigación biomedical, 3rd edn. Harcourt Brace, Madrid, pp 359–363
Hagioka S, Takeda Y, Zhang S et al (2005) Effects of 7-nitroindazole and N-nitro-l-arginine methyl ester on changes in cerebral blood flow and nitric oxide production preceding development of hyperbaric oxygen-induced seizures in rats. Neuroscience Lett 382:206–210
Morris SM Jr (2000) Regulation of arginine availability and its impact on NO synthesis. In: Ignarro LJ (ed) Nitric oxide, biology and pathobiology. Academic Press, San Diego, pp 187–197
Kamada Y, Jenkins GJ, Lau M et al (2005) Tetrahydrobiopterin depletion and ubiquitylation of neuronal nitric oxide synthase. Brain Res Mol Brain Res 142:19–27
Mejía JJ, Díaz-Barriga F, Calderón J et al (1997) Effects of lead–arsenic combined exposure on central monoaminergic systems. Neurotoxicol Teratol 19:489–497
Rodriguez VM, Carrizales L, Mendoza MS et al (2002) Effects of sodium arsenite exposure on development and behavior in the rat. Neurotoxicol Teratol 24:743–750
Ahmad S, Kitchin KT, Cullen WR (2000) Arsenic species that cause release of iron from ferritin and generation of activated oxygen. Arch Biochem Biophys 382:195–202
Yamanka K, Hoshino M, Okamoto M et al (1990) Induction of DNA damage by dimethylarsine, a metabolite of inorganic arsenic, is for the major part likely due to its peroxyl radical. Biochem Biophys Res Commun 168:58–64
Bernstam L, Nriagu J (2000) Molecular aspects of arsenic stress. J Toxicol Environ Health 3:293–322
Shila S, Kokilavani V, Subathra M et al (2005) Brain regional responses in antioxidant system to α-lipoic acid in arsenic intoxicated rat. Toxicology 210:25–36
Acknowledgments
This work was supported by the grants 40627-M from Consejo Nacional de Ciencia y Tecnología (CONACYT), Programa Integral de Fortalecimiento a la Investigación (PIFI 3.1), and a CONACYT fellowship (186322) to S.Z. The authors thank Paula Vergara and Melissa Bocanegra for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zarazúa, S., Pérez-Severiano, F., Delgado, J.M. et al. Decreased Nitric Oxide Production in the Rat Brain after Chronic Arsenic Exposure. Neurochem Res 31, 1069–1077 (2006). https://doi.org/10.1007/s11064-006-9118-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-006-9118-7