
Abstract
Post-transplant lymphoproliferative disorder (PTLD) is a serious complication after allogeneic hematopoietic stem cell transplantation (HSCT). Extra nodal involvement is common in PTLD, but isolated involvement of the central nervous system (CNS) is extremely rare. Given the rarity of primary CNS-PTLD there is no consensus on optimal treatment. We report a patient who developed Epstein-Barr virus related primary CNS-PTLD following allogeneic HSCT who was treated with the monoclonal anti-CD20 antibody rituximab and reduction of immunosuppression. In addition, we review the literature and discuss treatment options for patients with primary CNS-PTLD following allogeneic HSCT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Post-transplant lymphoproliferative disorders (PTLD) are a heterogeneous group of lymphoid or plasmacytic proliferations occurring in recipients of solid organ transplants and hematopoietic stem cell transplantation (HSCT) [1]. The clinical presentation of PTLD is highly variable, and ranges from an indolent self-limited form of lymphoproliferation to fulminant disease and from localized to widely disseminated disease. PTLD based on the revised classification by the World Health Organization are grouped into four categories; early lesions, polymorphic PTLD, monomorphic PTLD, and classic Hodgkin lymphoma-type PTLD [2]. The pathogenesis of PTLD in most patients is related to B-cell proliferation induced by infection with Epstein-Barr virus (EBV) in the setting of chronic immunosuppression [3, 4].
Extra nodal involvement is common in PTLD, but isolated involvement of the central nervous system (CNS) is extremely rare. In solid organ transplant recipients the median time from transplantation to primary CNS-PTLD is 4.4 years [5]. The cerebral hemispheres are the most common site of CNS-PTLD, with the the subcortical white matter and basal ganglia, being most commonly affected. CNS-PTLD after solid organ transplants is usually monomorphic EBV positive disease of B-cell origin and median survival is 47 months from diagnosis.
Primary CNS-PTLD after HSCT is also rare and has not been well described. Here we report an adult allogeneic HSCT recipient who developed primary CNS-PTLD, review the literature and discuss treatment options.
Case report
A 27-year old female underwent human leukocyte antigen-matched unrelated donor HSCT for acute leukemia of ambiguous lineage. The conditioning regimen consisted of cyclophosphamide and total body irradiation. Cyclosporine and methotrexate were given for prophylaxis of graft-versus-host disease (GVHD). On day 25 post-transplant the patient developed acute GVHD of the gut and skin that responded to high-dose steroids. On day 34 post-transplant the cyclosporine was changed to mycophenolate mofetil due to thrombotic microangiopathy. Post-transplant evaluation of the bone marrow did not demonstrate evidence of leukemia. By day 152 post-transplant the patient’s steroids were discontinued and the dose of mycophenolate mofetil was tapered to 500 mg per day orally.
On day 173 post-transplant the patient developed bilateral aching headache pain, worse when recumbent and exacerbated by eye movements. The headaches were present constantly, and were not associated with visual, sensory, or motor symptoms, but were associated with photophobia and phonophobia. The patient reported intermittent blurred vision but not diplopia. Cognitive testing using the MMSE was normal. The cranial nerve exam was unremarkable and fundoscopy normal. Muscle tone was normal, reflexes were depressed throughout. Motor strength was full in all muscle groups tested. Cerebellar testing and sensory examination were normal.
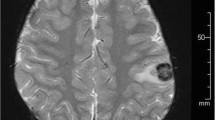
MRI of the brain with and without gadolinium contrast demonstrated multiple intraparenchymal contrast enhancing lesions with surrounding T2Flair hyperintensities. Several of the lesions were adjacent to the leptomeningeal surface but there was no leptomeningeal enhancement at other sites (Fig. 1). MRI of the total spine demonstrated no evidence of leptomeningeal, intradural, or intraparenchymal spinal cord abnormalities.

a T1 weighted post-gadolinium MRI at diagnosis demonstrating enhancing lesions in the right frontal and left occipital lobes. b T1 weighted post-gadolilnium MRI of comparable axial level after 4 weekly doses of rituximab demonstrating partial response with subgyral enhancement remaining in the right frontal lobe and left occipital lobes. c T1 weighted post-gadolilnium MRI obtained 3 months after completion of treatment demonstrating only small residual focus of enhancement in the right frontal lobe. d T1 weighted post-gadolinium MRI obtained 6 months after completion of treatment demonstrating complete resolution of all lesions
A diagnostic lumbar puncture demonstrated a CSF protein of 90 μg/dl, glucose 47 mg/dl, 7 WBC/μl, 75% lymphocytes, 25% mononuclear cells. Cerebrospinal fluid (CSF) cytology demonstrated no malignant cells. CSF analysis was also negative for bacterial, fungal, viral, or parasitic organisms. Real time PCR for EBV DNA on whole blood and CSF were negative. Brain biopsy was performed, which demonstrated a perivascular and interstitial infiltrate of abnormal large, mononuclear cells, some with irregular nuclear contours, dense chromatin and/or prominent nucleoli. Numerous mitotic figures were seen. Immunohistochemical stains confirmed a B-cell origin (PAX-5 positive) with positive staining for CD20 and CD79a. In situ hybridization demonstrated positive staining for latent EBV-associated early RNA (EBER) (Fig. 2). A diagnosis of post-transplant lymphoproliferative disorder, monomorphic-type, with diffuse large B-cell morphology was made. CT/PET scan of the chest, abdomen, and pelvis showed no evidence of nodal or organ involvement. Bone marrow biopsy showed no evidence of leukemia or PTLD.

a Brain biopsy demonstrated an abnormal, pleomorphic infiltrate of large centroblastic cells (×1000, H & E stain), which showed strong nuclear staining for the B-cell transcription factor PAX-5 (b, ×500, immunohistochemical stain), nuclear staining for EBER (c, ×1000, in situ hybridization), and a high proliferative index on Ki67/Mib-1 staining (d, ×500, immunohistochemical stain)
The patient was treated with rituximab administered intravenously weekly for 4 weeks. The dose of rituximab was escalated after the first dose from 375 to 500 mg/m2. In addition, the mycophenolate mofetil was rapidly tapered and discontinued within 4 weeks with no exacerbation of GVHD. The patient’s symptoms improved after the first dose of rituximab and resolved by the completion of therapy. Decrease in size of all of the lesions was seen on MRI imaging at the completion of therapy. Follow up brain MRI 3 months after treatment began demonstrated near complete resolution of all of the previously seen lesions and complete resolution by 6 months (Fig. 1).
Discussion
Although PTLD represent a relatively uncommon complication in HSCT patients, this complication is a significant cause of morbidity and mortality. Incidence rates for PTLD peak at 2–3 months after allo-HSCT, and then decline sharply with increasing time since transplantation. The risk of development of PTLD following allo-HSCT is associated with T cell depletion of the donor marrow, antithymocyte globulin use, unrelated or HLA-mismatched grafts, acute and chronic GVHD and advanced age at transplantation [6].
Treatment options for systemic PTLD include reduction of immunosuppression, EBV-specific cytotoxic T lymphocytes (CTL), monoclonal antibodies, chemotherapy, and radiation therapy [7–9]. Primary CNS-PTLD following allo-HSCT presents a particular challenge as there is limited clinical experience, few reported cases and lack of prospective studies. Only a small number of cases isolated CNS-PTLD following allo-HSCT have been reported [10–18] (Table 1). The latency between transplantation and development of CNS-PTLD ranged from 2 to 23 months. CNS-PTLD presented as multiple bilateral lesions in the majority of cases.
Given the rarity of primary CNS-PTLD following allo-HSCT there is no consensus on optimal treatment. Reduction of immunosuppression to restore immune function should be considered if the patient’s symptoms are not life threatening and the risk of developing or exacerbating GVHD is low. However, reduction of the immunosuppression alone may not be sufficient to restore immune responses to EBV in the early post-HSCT period as patients may have not fully reconstituted their immune cells and their ability to mount immune responses [19]. If a significant response is not achieved with reduction of immunosuppression or there is a need for immediate therapy based on the patient’s symptomatology, choice of additional therapy depends on the patient’s performance status and organ function. High-dose methotrexate may be of particular value of PTLD involving the CNS. Multiple clinical trials have demonstrated the efficacy of high-dose methotrexate in treatment of primary CNS lymphoma [20]. In addition, chemotherapy with high-dose methotrexate has also induced responses in patients with CNS-PTLD after liver and renal transplantation [21, 22]. For our patient, concerns about hepatotoxicity and renal toxicity for the high-dose methotrexate, as well as concern about potential bone marrow suppression in a allo-HSCT recipient who was already immunocompromised were reasons not to use high-dose methotrexate as first-line treatment. The use of EBV-specific CTL in allo-HSCT recipients has been proven to be safe with no appreciable alloreactivity and without the development of acute GVHD [23]. However, the availability and time needed for generation of EBV-specific CTL was a limiting factor in treating our patient at presentation of her symptoms.
As most cases of PTLD after HSCT are derived from donor B-cells, monoclonal anti-B-cell antibodies have been used to treat PTLD. The most widely antibody used for the treatment of PTLD is rituximab, a chimeric murine/human monoclonal anti-CD20 antibody [24–28].
In a prospective multicenter phase II trial of 60 patients with PTLD after solid organ transplants treated with rituximab after not responding to reduction of immunosuppression the 1 year progression free survival was 42% [25]. The median time to progression was 6.0 months with 38% of patients being primarily refractory and 12% experiencing progression after a partial response, stable disease, or complete response. In patients that received solid organ transplants and developed primary CNS-PTLD rituximab either at standard doses or higher doses has been used with some success [5, 29]. In addition, rituximab as a single agent as well as in combination with cytotoxic chemotherapy, has been employed in the treatment of primary CNS lymphoma [20, 30–34] and intra-ventricular rituximab has been used in patients with lymphomatous meningitis [35–37].
In our patient, with a negative CSF cytology and no MRI evidence of leptomeningeal lymphoma, we decided to use first systemic rituximab with concomitant reduction of the immunosuppression that resulted in complete resolution of the patient’s symptoms and radiographic findings. Whether maintenance rituximab would be effective after the contrast enhancing lesions have resolved, and the blood brain barrier is presumably reconstitituted, is unclear.
Post-transplant lymphoproliferative disorder isolated to the CNS following allo-HSCT is a rare presentation of an uncommon disease. The optimal therapy for treatment of CNS-PTLD after allo-HSCT remains to be determined. Prospective trials that incorporate stepwise treatment approaches i.e., first reduction of immunosuppression followed with rituximab and then intensive chemotherapy are needed to further define effective therapies for primary CNS-PTLD.
References
Kaushansky K, Williams WJ (2010) Williams hematology. McGraw-Hill Medical, New York
Swerdlow S, Campo E, Lee Harris N, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, World Health Orgnization, International Agency for Research on Cancer (2008) WHO classification of tumours of haematopoietic and lymphoid tissues. World Health Organization International Agency for Research on Cancer, Lyon
Gottschalk S, Rooney CM, Heslop HE (2005) Post-transplant lymphoproliferative disorders. Annu Rev Med 56:29–44
Tsao L, Hsi ED (2007) The clinicopathologic spectrum of posttransplantation lymphoproliferative disorders. Arch Pathol Lab Med 131:1209–1218
Cavaliere R, Petroni G, Lopes MB, Schiff D (2010) Primary central nervous system post-transplantation lymphoproliferative disorder: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Cancer 116:863–870
Landgren O, Gilbert ES, Rizzo JD, Socie G, Banks PM, Sobocinski KA, Horowitz MM, Jaffe ES, Kingma DW, Travis LB, Flowers ME, Martin PJ, Deeg HJ, Curtis RE (2009) Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 113:4992–5001
DiNardo CD, Tsai DE (2010) Treatment advances in posttransplant lymphoproliferative disease. Curr Opin Hematol 17:368–374
Parker A, Bowles K, Bradley JA, Emery V, Featherstone C, Gupte G, Marcus R, Parameshwar J, Ramsay A, Newstead C (2010) Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients - BCSH and BTS Guidelines. British journal of haematology 149:693–705
Evens AM, Roy R, Sterrenberg D, Moll MZ, Chadburn A, Gordon LI (2010) Post-transplantation lymphoproliferative disorders: diagnosis, prognosis, and current approaches to therapy. Current oncology reports 12:383–394
Aisa Y, Mori T, Nakazato T, Suzuki S, Suzuki N, Ikeda Y, Okamoto S (2009) Primary central nervous system post-transplant lymphoproliferative disorder presenting as cerebral hemorrhage after unrelated bone marrow transplantation. Transpl Infect Dis 11:438–441
Castellano-Sanchez AA, Li S, Qian J, Lagoo A, Weir E, Brat DJ (2004) Primary central nervous system posttransplant lymphoproliferative disorders. Am J Clin Pathol 121:246–253
Hamadani M, Martin LK, Benson DM, Copelan EA, Devine SM, Hofmeister CC (2007) Central nervous system post-transplant lymphoproliferative disorder despite negative serum and spinal fluid Epstein-Barr virus DNA PCR. Bone Marrow Transplant 39:249–251
Kordelas L, Trenschel R, Koldehoff M, Elmaagacli A, Beelen DW (2008) Successful treatment of EBV PTLD with CNS lymphomas with the monoclonal anti-CD20 antibody rituximab. Onkologie 31:691–693
Nagafuji K, Eto T, Hayashi S, Oshima K, Maeda Y, Gondo H, Inamura T, Niho Y (1998) Donor lymphocyte transfusion for the treatment of Epstein-Barr virus-associated lymphoproliferative disorder of the brain. Bone Marrow Transplant 21:1155–1158
Nozzoli C, Bartolozzi B, Guidi S, Orsi A, Vannucchi AM, Leoni F, Bosi A (2006) Epstein-Barr virus-associated post-transplant lymphoproliferative disease with central nervous system involvement after unrelated allogeneic hematopoietic stem cell transplantation. Leuk Lymphoma 47:167–169
Pakakasama S, Eames GM, Morriss MC, Huls MH, Rooney CM, Heslop HE, Krance RA (2004) Treatment of Epstein-Barr virus lymphoproliferative disease after hematopoietic stem-cell transplantation with hydroxyurea and cytotoxic T-cell lymphocytes. Transplantation 78:755–757
Terasawa T, Ohashi H, Tsushita K, Utsumi M, Mukai E, Nakamura S, Shimoyama M (2002) Failure to detect Epstein-Barr virus (EBV) DNA in plasma by real-time PCR in a case of EBV-associated posttransplantation lymphoproliferative disorder confined to the central nervous system. Int J Hematol 75:416–420
Verschuur A, Brousse N, Raynal B, Brison O, Rohrlich P, Rahimy C, Vilmer E (1994) Donor B cell lymphoma of the brain after allogeneic bone marrow transplantation for acute myeloid leukemia. Bone Marrow Transplant 14:467–470
Heslop HE (2009) How I treat EBV lymphoproliferation. Blood 114:4002–4008
Gerstner ER, Batchelor TT (2010) Primary central nervous system lymphoma. Arch Neurol 67:291–297
Taj MM, Messahel B, Mycroft J, Pritchard-Jones K, Baker A, Height S, Hadzic N, Pinkerton CR (2008) Efficacy and tolerability of high-dose methotrexate in central nervous system positive or relapsed lymphoproliferative disease following liver transplant in children. British journal of haematology 140:191–196
Nabors LB, Palmer CA, Julian BA, Przekwas AM, Kew CE (2009) Isolated central nervous system posttransplant lymphoproliferative disorder treated with high-dose intravenous methotrexate. Am J Transplant 9:1243–1248
Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, Bollard CM, Liu H, Wu MF, Rochester RJ, Amrolia PJ, Hurwitz JL, Brenner MK, Rooney CM (2010) Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 115:925–935
Choquet S, Leblond V, Herbrecht R, Socie G, Stoppa AM, Vandenberghe P, Fischer A, Morschhauser F, Salles G, Feremans W, Vilmer E, Peraldi MN, Lang P, Lebranchu Y, Oksenhendler E, Garnier JL, Lamy T, Jaccard A, Ferrant A, Offner F, Hermine O, Moreau A, Fafi-Kremer S, Morand P, Chatenoud L, Berriot-Varoqueaux N, Bergougnoux L, Milpied N (2006) Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood 107:3053–3057
Choquet S, Oertel S, LeBlond V, Riess H, Varoqueaux N, Dorken B, Trappe R (2007) Rituximab in the management of post-transplantation lymphoproliferative disorder after solid organ transplantation: proceed with caution. Ann Hematol 86:599–607
Kuehnle I, Huls MH, Liu Z, Semmelmann M, Krance RA, Brenner MK, Rooney CM, Heslop HE (2000) CD20 monoclonal antibody (rituximab) for therapy of Epstein-Barr virus lymphoma after hemopoietic stem-cell transplantation. Blood 95:1502–1505
Milpied N, Vasseur B, Parquet N, Garnier JL, Antoine C, Quartier P, Carret AS, Bouscary D, Faye A, Bourbigot B, Reguerre Y, Stoppa AM, Bourquard P, de Hurault Ligny B, Dubief F, Mathieu-Boue A, Leblond V (2000) Humanized anti-CD20 monoclonal antibody (Rituximab) in post transplant B-lymphoproliferative disorder: a retrospective analysis on 32 patients. Ann Oncol 11(1):113–116
Oertel SH, Verschuuren E, Reinke P, Zeidler K, Papp-Vary M, Babel N, Trappe RU, Jonas S, Hummel M, Anagnostopoulos I, Dorken B, Riess HB (2005) Effect of anti-CD 20 antibody rituximab in patients with post-transplant lymphoproliferative disorder (PTLD). Am J Transplant 5:2901–2906
Patrick A, Wee A, Hedderman A, Wilson D, Weiss J, Govani M (2011) High-dose intravenous rituximab for multifocal, monomorphic primary central nervous system posttransplant lymphoproliferative disorder. J Neurooncol 103(3):739–743
Doolittle ND, Jahnke K, Belanger R, Ryan DA, Nance RW Jr, Lacy CA, Tyson RM, Haluska M, Hedrick NA, Varallyay C, Neuwelt EA (2007) Potential of chemo-immunotherapy and radioimmunotherapy in relapsed primary central nervous system (CNS) lymphoma. Leuk Lymphoma 48:1712–1720
Enting RH, Demopoulos A, DeAngelis LM, Abrey LE (2004) Salvage therapy for primary CNS lymphoma with a combination of rituximab and temozolomide. Neurology 63:901–903
Santisteban M, Nieto Y, De la Cruz S, Aristu J, Zubieta JL, Fernandez Hidalgo O (2007) Primary central nervous system lymphoma treated with rituximab plus temozolomide in a second line schedule. Clin Transl Oncol 9:465–467
Wong ET (2005) Monoclonal antibody therapy for central nervous system lymphomas: an emerging treatment paradigm. Expert opinion on pharmacotherapy 6:1107–1114
Wong ET, Tishler R, Barron L, Wu JK (2004) Immunochemotherapy with rituximab and temozolomide for central nervous system lymphomas. Cancer 101:139–145
Akyuz C, Aydin GB, Cila A, Akalan N, Soylemezoglu F, Cengiz M, Yazici N, Buyukpamukcu M (2007) Successful use of intraventricular and intravenous rituximab therapy for refractory primary CNS lymphoma in a child. Leuk Lymphoma 48:1253–1255
Canova F, Marino D, Trentin C, Solda C, Ghiotto C, Aversa SM (2011) Intrathecal chemotherapy in lymphomatous meningitis. Crit Rev Oncol Hematol 79:127–134
Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, Ignoffo R, Aldape K, Shen A, Lee D, Grillo-Lopez A, Shuman MA (2003) Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood 101:466–468
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lieberman, F., Yazbeck, V., Raptis, A. et al. Primary central nervous system post-transplant lymphoproliferative disorders following allogeneic hematopoietic stem cell transplantation. J Neurooncol 107, 225–232 (2012). https://doi.org/10.1007/s11060-011-0739-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-011-0739-6