
Abstract
Approximately one-third of patients with advanced, HER2-positive breast cancer develop brain metastases. A significant proportion of women experience central nervous system (CNS) progression after standard radiation therapy. The optimal treatment in the refractory setting is undefined. This study evaluated the toxicity and efficacy of lapatinib in combination with chemotherapy among patients with HER2-positive, progressive brain metastases. Patients with HER2-positive breast cancer with progressive brain metastases after trastuzumab and cranial radiotherapy were included. The primary endpoint was CNS objective response, defined as a ≥50% volumetric reduction of CNS lesion(s) in the absence of new or progressive CNS or non-CNS lesions, or increasing steroid requirements. The study was closed early after 22 of a planned 110 patients were enrolled due to excess toxicity and lack of efficacy in the lapatinib plus topotecan arm. The objective response rate (ORR) in the lapatinib plus capecitabine arm was 38% (exact 95% confidence interval [CI] 13.9–68.4). No responses were observed in the lapatinib plus topotecan arm. Although the study was stopped prior to full enrollment, some promising indications of CNS activity were noted for lapatinib plus capecitabine. The combination of lapatinib plus topotecan was not active and was associated with excess toxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among patients treated with trastuzumab for HER2-positive, advanced breast cancer, approximately one-third will develop brain metastases [1–5]. In contrast to the historical experience, overall survival after central nervous system (CNS) diagnosis in patients with HER2-positive disease now approaches 2 years, largely due to improvements in extracranial control [4, 6, 7]. Initial treatment with whole brain radiotherapy (WBRT) and/or stereotactic radiosurgery (SRS) is often effective. However, as patients live longer, subsequent CNS progression events are becoming an increasing problem. Unfortunately, the optimal treatment for patients with recurrent brain metastases remains undefined.
Lapatinib is an orally bioavailable, 4-aniloquinazoline tyrosine kinase inhibitor of the epidermal growth factor and HER2. In animal models, lapatinib inhibits CNS outgrowth of HER2-positive cell lines [8]. CNS therapeutic levels have been demonstrated in tumor resection specimens in patients with glioblastoma multiforme [9]. Two phase 2 trials have evaluated lapatinib monotherapy in patients with progressive HER2-positive, brain metastases [10, 11]. In the larger study (EGF105084), the CNS objective response (CNS ORR) was 6% (95% confidence interval [CI] 3.6–10.2) [11]. In the extension phase of the trial, patients who progressed either intra- or extracranially on lapatinib monotherapy were given the option of receiving lapatinib plus capecitabine. Among 50 evaluable patients, the CNS ORR was 20%. An important limitation of the extension study was that an effect of capecitabine alone could not be excluded. On the other hand, support for the potential CNS activity of the combination comes from EGF100151, a randomized trial of capecitabine with or without lapatinib [12]. In an exploratory analysis of this trial, fewer CNS progression events were observed among patients assigned combination therapy [13].
Topotecan is a topoisomerase I inhibitor approved for the treatment of patients with small cell lung cancer and ovarian cancer. Therapeutic CNS levels can be achieved with intravenous administration, and CNS objective responses have been reported in breast cancer [14–16]. One postulated mechanism of resistance to topotecan is drug efflux via the breast cancer resistance protein (BCRP). Of note, 4-aniloquinazoline tyrosine kinase inhibitors may potentially enhance topotecan cytotoxicity via inhibition of BCRP-mediated drug efflux [17]. The combination of lapatinib and topotecan was evaluated in a phase I study in solid tumor patients [18, 19]. The most commonly observed toxicities were nausea, diarrhea, fatigue, alopecia, and neutropenia. Dose limiting toxicity consisted of nausea, vomiting, and diarrhea and a recommended phase II dose has been established. These data provided the rationale for evaluating the combination of lapatinib and topotecan in brain metastasis patients.
The current study, EGF107671, was conducted to evaluate the toxicity and efficacy profile of two different lapatinib-based chemotherapy regimens (lapatinib plus capecitabine, lapatinib plus topotecan) in patients with progressive brain metastases from HER2-positive breast cancer despite standard therapy with WBRT and/or SRS.
Patients and methods
Patients
Patients were enrolled from 14 centers in the United States, Canada, the European Union, and Israel between May 2007 and December 2008. Eligible patients had HER2-positive breast cancer (defined as 3+ immunohistochemistry or evidence of gene amplification by fluorescence in situ hybridization) and unequivocal radiographic evidence of new and/or progressive metastases in the brain despite prior standard treatment with WBRT and/or SRS. Prior trastuzumab exposure was also required. Patients had at least one measurable brain lesion (≥10 mm on T1-weighted, gadolinium-enhanced magnetic resonance imaging [MRI]). Other eligibility criteria included age ≥18 years, Eastern Cooperative Oncology Group [ECOG] performance status 0 or 1, life expectancy >12 weeks, cardiac ejection fraction within institutional normal range, ability to swallow and retain oral medications, and adequate organ function. All radiotherapy, chemotherapy, hormonal therapy, and/or trastuzumab had to be discontinued at least 2 weeks before initiation of protocol treatment. Concurrent administration of other antineoplastic agents, radiotherapy, or inducers or inhibitors of CYP3A4 were not permitted, with the exception of corticosteroids as clinically indicated. Concurrent use of enzyme inducing anti-epileptic agents was also not allowed. Patients with prior exposure to lapatinib, capecitabine, or topotecan were excluded, as were patients with evidence of leptomeningeal carcinomatosis at screening.
The study was conducted in compliance with the International Conference on Harmonization of Good Clinical Practice guidelines, in accordance with the Declaration of Helsinki, and with GlaxoSmithKline Standard Operating Procedures for all processes involved, including the archiving of essential documents. The institutional review board for each participating institution approved the study protocol and informed consent documents. All patients gave written informed consent prior to the performance of any study-specific procedures. The trial was registered at the ClinicalTrials.gov web site (no. NCT00437073).
Treatment
Patients on the lapatinib plus capecitabine arm received lapatinib 1,250 mg orally once daily and capecitabine 2,000 mg/m2 orally divided twice daily on days 1–14 of a 21-day cycle. Patients on the lapatinib plus topotecan arm received lapatinib 1,250 mg orally once daily and topotecan 3.2 mg/m2 intravenously on days 1, 8, and 15 of a 28-day cycle. Lapatinib could be delayed for up to 2 weeks to allow for resolution of toxicity. In the event of toxicity, one dose reduction to 1,000 mg once daily was allowed. Lapatinib was discontinued permanently in patients with grade 3 or 4 left ventricular ejection fraction (LVEF) decline, grade 3 or 4 interstitial pneumonitis, or who met the stopping criteria for a liver chemistry adverse event. Capecitabine dosing was managed according to a pre-defined algorithm. Up to two dose reductions were allowed (to 75 and 50% of the initial prescribed dose, respectively). Topotecan dose delays and reductions were also managed according to a pre-defined algorithm. In patients who required a delay of 8–14 days for adequate count recovery, the topotecan dose was subsequently reduced to 2.7 mg/m2. A second delay required the use of hematopoietic growth factors in all subsequent cycles. No further dose reductions were allowed. Dose delays were also required in patients with grade ≥2 or higher peripheral neuropathy, grade ≥2 renal toxicity, grade ≥3 liver function tests, fatigue, or diarrhea. Dose reductions of lapatinib, topotecan, and capecitabine could occur independently. Patients requiring delays of more than 2 weeks were taken off study. Patients remained on protocol therapy until disease progression, unacceptable toxicity, or withdrawal of consent. At the time of radiographically documented progression, crossover was allowed to the other arm.
Efficacy assessments
Brain MRIs were obtained every 6 weeks on the lapatinib plus capecitabine arm, and every 8 weeks on the lapatinib plus topotecan arm, and included T1-weighted, contrast-enhanced images at 3.0 mm slice thickness without gaps in the axial dimension. All brain MRIs were evaluated by central, independent review according to previously described methods [10]. A CNS objective response was defined as either a complete response (CR) or partial response (PR, ≥50% reduction in the volumetric sum of CNS target lesions), provided there was no progression of extracranial disease, or increasing steroid requirement. Progressive disease was defined as any of the following: >40% increase in volumetric sum of all evaluable brain lesions compared to the nadir, progression of non-measurable brain lesions, new brain lesions ≥6 mm in longest diameter, increasing steroid requirement, progression of extracranial disease, or progressive tumor-related symptoms resulting in study withdrawal. Neurological signs and symptoms (NSS) were evaluated at baseline and Day 1 of each cycle using a physician reported worksheet, as described previously [11].
Response in extracranial sites was evaluated by the investigator with MRI or computed tomography scans of the chest, abdomen, and pelvis, according to Response Evaluation Criteria in Solid Tumors, version 1.0.
Safety assessments
Adverse events, complete blood count, chemistry profile, and liver function tests were assessed on Day 1 of each cycle and graded according to NCI CTCAE v3.0. For patients receiving lapatinib plus topotecan, complete blood counts were also obtained on Days 8 and 15 and reviewed prior to dosing. LVEF was evaluated with echocardiogram or multigated acquisition scan every two cycles.
Study design and statistical analysis
This was an open-label, randomized phase II study. The primary endpoint was objective response in the CNS. Secondary endpoints included safety and toxicity profile, percentage of patients with disease stabilization ≥6 months, clinical benefit rate (CR + PR + SD ≥6 months), duration of CNS objective response, percentage of patients with improvements in NSS, ORR in non-CNS sites, initial site of disease progression, time to progression, and overall survival.
The study was designed to distinguish between a CNS ORR ≤10% versus ≥25% in each arm separately. The study was not designed to compare the two arms. Within each arm, if 10 or more of 55 patients responded, the treatment would be deemed worthy of further study (α level 0.04; power 91%).
On June 13, 2008, the study was amended to halt accrual to the lapatinib plus topotecan arm and to disallow crossover to lapatinib plus topotecan in patients who progressed on lapatinib plus capecitabine. Due to slow accrual, the study was permanently closed on August 11, 2008.
Role of the sponsor
The study was designed by both the academic investigators and employees of the sponsor (GlaxoSmithKline). Data collection and analysis were supervised by an employee of the sponsor and reviewed along with the raw data by both academic investigators and employees of the sponsor. Interpretation of the data was performed by the academic investigators in conjunction with employees of the sponsor. The authors had full access to all data. The initial draft of the manuscript was written by the first author. All authors participated in the conception and design or analysis and interpretation of the data, drafting and critical revisions of the manuscript, and approval of the final submitted version. All authors reviewed and amended the manuscript and vouch for the completeness and integrity of the reported data.
Results
Patient characteristics
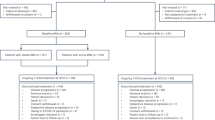
A total of 22 patients were enrolled in the intent-to-treat population (13 enrolled on the lapatinib plus capecitabine arm, and 9 on the lapatinib plus topotecan arm). All patients had previously received WBRT and prior systemic chemotherapy for metastatic disease (Table 1). Ten patients (45%) had measurable extracranial disease at study entry. Fewer patients in the lapatinib plus topotecan had a baseline performance status of zero. Mean sum volume of target lesions was similar between arms.
Treatment
As of the data cut-off date (December 26, 2008), all patients had discontinued therapy. The most common reason for discontinuation in the lapatinib plus capecitabine arm was disease progression. Reasons for treatment discontinuation in the lapatinib plus topotecan arm were more variable (Table 2).
Dose reductions of capecitabine for diarrhea or palmar-plantar erythrodysesthesia (PPE) were required in 4 (31%) patients assigned lapatinib plus capecitabine (Table 3). No dose reductions of lapatinib were required in this arm. Dose reductions of topotecan, primarily for hematologic toxicity, were required in 3 (33%) of patients assigned lapatinib plus topotecan. In 3 (33%) patients assigned to this arm, lapatinib dose reduction was also required, primary for diarrhea. In addition, topotecan dose delays were common, and reported in 8/9 (89%) patients randomized to this treatment arm.
Hematologic and non-hematologic toxicities
Table 4 summarizes the toxicity data for both arms. Consistent with the literature, the most commonly reported AEs on the lapatinib plus capecitabine arm were diarrhea, PPE, and fatigue. Grade 3 or 4 diarrhea was observed in 4 (31%) patients; grade 3 PPE was reported in 2 patients. One patient was withdrawn from the study for grade 3 hyperbilirubinemia. Other grade 3/4 toxicities were rare. One fatal SAE of multi-organ failure was reported. This patient presented with grade 3 hypokalemia 55 days after start of protocol therapy. She was hospitalized for IV hydration and electrolyte repletion. Five days following hospital admission, she expired. In the investigator’s opinion, there was a reasonable possibility that the multiorgan failure was due to study medication. No further details were available and an autopsy was not performed.
Among patients treated with lapatinib plus topotecan, the most commonly reported AEs were diarrhea, nausea, fatigue, and thrombocytopenia. One grade 5 SAE of sepsis was reported. The patient developed neutropenic sepsis 17 days after the initiation of protocol therapy. In retrospect, symptoms were likely masked by concurrent corticosteroid use. Despite broad-spectrum antibiotics, the patient expired 3 days following her presentation of staphylococcal and E. coli bacteremia. This event triggered a thorough investigation by the study investigators and lapatinib global safety team and, in concert with lack of demonstrated efficacy as described below, led to closure of the lapatinib plus topotecan arm of the study.
Efficacy
Table 5 summarizes the efficacy data. Among the 13 patients randomized to lapatinib plus capecitabine, 5 CNS PRs were observed (ORR 38%, exact 95% CI 13.9–68.4). Of patients with PR, time on study was 58, 86, 164, 166, and 252 days, respectively. No objective responses were observed among the 9 patients treated with lapatinib plus topotecan.
Three patients initially assigned to lapatinib plus topotecan were crossed over to lapatinib plus capecitabine at the time of progression and one CNS PR was observed. No responses were observed in the two patients who crossed over to lapatinib plus topotecan.
Planned secondary endpoints included ORR in non-CNS sites, initial site of disease progression, time to progression, and overall survival. 10 patients had measurable non-CNS disease at study baseline. Only one non-CNS objective response was observed (in a single patient who had best response of SD in the brain). CNS was the most common site of initial disease progression. Because of the small number of patients enrolled to each arm at the time of study closure, time-to-event endpoints were not formally evaluated.
Discussion
We evaluated the toxicity and efficacy profile of two different lapatinib-based chemotherapy regimens for the treatment of progressive brain metastases from HER2-positive breast cancer. The study was closed early to accrual due to tolerability issues in the lapatinib plus topotecan arm, in combination with lack of early efficacy. Despite an attempt to keep the lapatinib plus capecitabine arm open, study accrual slowed considerably after closure of the topotecan arm, and a decision was made to close the study in its entirety.
Among patients randomized to lapatinib plus capecitabine, the CNS ORR was 38% (exact 95% CI 13.9–68.4), with time on study ranging from 58 to 252 days. While the number of patients treated on this study was small, the results are consistent with data from the extension phase of the EGF105084 trial (n = 50), in which the CNS ORR was 20%. Of note, despite the wide CI, the lower bound of the 95% CI we observed in the current trial exceeds the upper bound of the 95% CI for CNS response to monotherapy lapatinib in the EGF105084 study (6%, 95% CI 3.6–10.2), supporting the hypothesis that the combination of lapatinib plus capecitabine is associated with a higher level of CNS activity in comparison to lapatinib alone. Within the lapatinib expanded access program (LEAP) and French Authorisation Temporaire d’Utilisation (ATU), 138 patients with progressive brain metastases on study entry were identified. Complete or partial CNS responses were reported in 25 (18%) of patients, including patients who had received prior capecitabine [20]. The experience within the expanded access program in the United Kingdom was similar, with a response rate of 21% (7/34 patients) [21]. In a recently published single-institution series, lapatinib plus capecitabine was associated with a CNS response rate of 31.8% (7/22 patients) [22]. Taken together, the data indicate that lapatinib plus capecitabine is a viable option for patients with progressive brain metastases after standard radiotherapy approaches and support investigation of the combination in patients with previously untreated CNS disease. Indeed, an ongoing French study (NCT00967031) is evaluating the combination of lapatinib plus capecitabine in patients with newly diagnosed brain metastases with objectives including response rate and time to need for radiotherapy. From a prevention standpoint, an ongoing study in the metastatic setting is evaluating the incidence of CNS as site of first relapse among patients treated with lapatinib plus capecitabine versus trastuzumab plus capecitabine. In the adjuvant setting, the ALLTO trial includes site of first recurrence as a secondary endpoint, with a specific focus upon CNS events.
This study had several limitations. First, because of the small size of the study, the CI around the lapatinib/capecitabine response rate is wide. Next, the study was not designed to ask whether lapatinib definitively adds to the reported efficacy of capecitabine. In a retrospective experience of seven patients treated with capecitabine for breast cancer brain metastases, three patients achieved a CR [23]. In a phase I study of capecitabine and temozolomide, four objective responses were observed among 24 patients with brain metastases from breast cancer [24]. However, in the non-CNS metastatic setting, the combination of lapatinib and capecitabine was found to be superior to capecitabine alone with respect to time to progression (HR 0.49, 95% CI 0.34–0.71; P < 0.001) and with a trend towards improvement in response rate (22% vs. 14%, P = 0.09). Furthermore, there were fewer CNS progression events among patients treated on the combination arm (4 vs. 13 events, P = 0.045) [13]. In concert with the modest single agent activity of lapatinib in the CNS, these data support, though do not prove, that the combination of lapatinib and capecitabine is likely to be more active than either agent alone with respect to treatment of brain metastases [10, 11].
The high frequency of CNS involvement is thought to reflect an intrinsic propensity of HER2-positive breast cancer for CNS dissemination, in concert with improved systemic control with trastuzumab, an agent that does not efficiently cross the blood–brain barrier. A number of novel agents are now in clinical development. Elucidation of the potential CNS efficacy of these agents will be of interest, either singly, or in combination with chemotherapy or other targeted agents.
In summary, the present study provides a promising indication of CNS activity with the combination of lapatinib and capecitabine in patients with refractory brain metastases from HER2-positive breast cancer. Definitive conclusions cannot be made as the study was stopped prior to full accrual. The combination of lapatinib and topotecan is not recommended given the excess toxicity observed. Ongoing and planned studies are evaluating the activity of lapatinib and other HER2-targeted agents in patients with previously untreated brain metastases as an alternative to radiotherapy, or for the prevention of brain metastases in patients at high risk.
References
Bendell JC, Domchek SM, Burstein HJ, Harris L, Younger J, Kuter I, Bunnell C, Rue M, Gelman R, Winer E (2003) Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 97:2972–2977
Stemmler HJ, Kahlert S, Siekiera W, Untch M, Heinrich B, Heinemann V (2006) Characteristics of patients with brain metastases receiving trastuzumab for HER2 overexpressing metastatic breast cancer. Breast (Edinb, Scotl) 15:219–225
Lin NU, Winer EP (2007) Brain metastases: the HER2 paradigm. Clin Cancer Res 13:1648–1655
Gori S, Rimondini S, De Angelis V, Colozza M, Bisagni G, Moretti G, Sidoni A, Basurto C, Aristei C, Anastasi P, Crino L (2007) Central nervous system metastases in HER-2 positive metastatic breast cancer patients treated with trastuzumab: incidence, survival, and risk factors. Oncologist 12:766–773
Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, Nielsen TO, Gelmon K (2010) Metastatic behavior of breast cancer subtypes. J Clin Oncol 28:3271–3277
Melisko ME, Moore DH, Sneed PK, De Franco J, Rugo HS (2008) Brain metastases in breast cancer: clinical and pathologic characteristics associated with improvements in survival. J Neurooncol 88:359–365
Eichler AF, Kuter I, Ryan P, Schapira L, Younger J, Henson JW (2008) Survival in patients with brain metastases from breast cancer: the importance of HER-2 status. Cancer 112:2359–2367
Gril B, Palmieri D, Bronder JL, Herring JM, Vega-Valle E, Feigenbaum L, Liewehr DJ, Steinberg SM, Merino MJ, Rubin SD, Steeg PS (2008) Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst 100:1092–1103
Kuhn J, Robins HI, Mehta M, Fine H, Cloughesy T, Wen P, Chang SM, DeAngelis L, Lieberman F, Reardon D, Abrey LE, Lassman AB, Adalpe K, Yung WKA, Grabinski J, Lamborn K, Prados M (2008) Tumor sequestration of lapatinib (NABTC 04-01). Neurooncology 10:abstr ET-05
Lin NU, Carey LA, Liu MC, Younger J, Come SE, Ewend M, Harris GJ, Bullitt E, Van den Abbeele AD, Henson JW, Li X, Gelman R, Burstein HJ, Kasparian E, Kirsch DG, Crawford A, Hochberg F, Winer EP (2008) Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol 26:1993–1999
Lin NU, Dieras V, Paul D, Lossignol D, Christodoulou C, Stemmler HJ, Roche H, Liu MC, Greil R, Ciruelos E, Loibl S, Gori S, Wardley A, Yardley D, Brufsky A, Blum JL, Rubin SD, Dharan B, Steplewski K, Zembryki D, Oliva C, Roychowdhury D, Paoletti P, Winer EP (2009) Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res 15:1452–1459
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355:2733–2743
Geyer CE, Martin A, Newstat B, Casey MA, Berger MS, Oliva CR, Rubin SD, Stein S, Cameron D (2007) Lapatinib (L) plus capecitabine (C) in HER2+ advanced breast cancer (ABC): updated efficacy and biomarker analysis. J Clin Oncol 25:40s (abstr 1035)
Sung C, Blaney SM, Cole DE, Balis FM, Dedrick RL (1994) A pharmacokinetic model of topotecan clearance from plasma and cerebrospinal fluid. Cancer Res 54:5118–5122
Baker SD, Heideman RL, Crom WR, Kuttesch JF, Gajjar A, Stewart CF (1996) Cerebrospinal fluid pharmacokinetics and penetration of continuous infusion topotecan in children with central nervous system tumors. Cancer Chemother Pharmacol 37:195–202
Oberhoff C, Kieback DG, Wurstlein R, Deertz H, Sehouli J, van Soest C, Hilfrich J, Mesrogli M, von Minckwitz G, Staab HJ, Schindler AE (2001) Topotecan chemotherapy in patients with breast cancer and brain metastases: results of a pilot study. Onkologie 24:256–260
Erlichman C, Boerner SA, Hallgren CG, Spieker R, Wang XY, James CD, Scheffer GL, Maliepaard M, Ross DD, Bible KC, Kaufmann SH (2001) The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7-ethyl-10-hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein-mediated drug efflux. Cancer Res 61:739–748
Erlichman C, Adjei AA, Joy ME, Rubin SD, Friedman RA, Ames MM, Reid JM, Mandrekar S, Felten S, Molina JR (2005) A phase I study of lapatinib (GW572016) and topotecan in patients with solid tumors. Clin Cancer Res 11:8994s (abstr A8122)
Molina JR, Kaufmann SH, Reid JM, Rubin SD, Galvez-Peralta M, Friedman R, Flatten KS, Koch KM, Gilmer TM, Mullin RJ, Jewell RC, Felten SJ, Mandrekar S, Adjei AA, Erlichman C (2008) Evaluation of lapatinib and topotecan combination therapy: tissue culture, murine xenograft, and phase I clinical trial data. Clin Cancer Res 14:7900–7908
Boccardo F, Kaufman B, Baselga J, Dieras V, Link J, Casey MA, Fittipaldo A, Oliva C, Zembryki D, Rubin SD (2008) Evaluation of lapatinib (Lap) plus capecitabine (Cap) in patients with brain metastses (BM) from HER2+ breast cancer (BC) enrolled in the Lapatinib Expanded Access Program (LEAP) and French Authorisation Temporaire d’Utilisation (ATU). J Clin Oncol 26:abstract 1094
Sutherland S, Ashley S, Miles D, Chan S, Wardley A, Davidson N, Bhatti R, Shehata M, Nouras H, Camburn T, Johnston SR (2010) Treatment of HER2-positive metastatic breast cancer with lapatinib and capecitabine in the lapatinib expanded access programme, including efficacy in brain metastases—the UK experience. Br J Cancer 102:995–1002
Metro G, Foglietta J, Russillo M, Stocchi L, Vidiri A, Giannarelli D, Crino L, Papaldo P, Mottolese M, Cognetti F, Fabi A, Gori S (2010) Clinical outcome of patients with brain metastases from HER2-positive breast cancer treated with lapatinib and capecitabine. Ann Oncol 22:625–630
Ekenel M, Hormigo AM, Peak S, Deangelis LM, Abrey LE (2007) Capecitabine therapy of central nervous system metastases from breast cancer. J Neurooncol 85:223–227
Rivera E, Meyers C, Groves M, Valero V, Francis D, Arun B, Broglio K, Yin G, Hortobagyi GN, Buchholz T (2006) Phase I study of capecitabine in combination with temozolomide in the treatment of patients with brain metastases from breast carcinoma. Cancer 107:1348–1354
Acknowledgments
The authors would like to acknowledge the work of Trinity Urban and Gordon J. Harris, M.D. (Tumor Imaging Metrics Core, Dana-Farber/Harvard Cancer Center) for independent radiology review including volumetric analyses of brain MRIs. The authors would also like to acknowledge the following investigators and study sites for their contributions to the study: Steve Chui, Oregon Health and Science University, Portland, OR; Catherine Doyle, Hopital du Saint Sacrement du CHAUQ-Quebec-Canada; Gabrielle Doering, Praxis fuer Haematologie und Onkologie, Bremen, Germany; Ella Evron, Assaf Harofeh MC, Zrifin, Israel; Karen Gelmon, BC Cancer Agency, Vancouver Centre, Vancouver, Canada; Henrik Lindman, Akademiska Sjukhuset, Uppsala, Sweden; Minetta Liu, Georgetown University Hospital, Washington, D.C.; Rafael Lopez, Clinico de Santiago—Santiago de Compostela-Spain; Franco Nole, Instituto Europeo di Oncologia (IRCCS) di Milano, Milan, Italy; Anna Maria Storniolo, Indiana University, Indianapolis, IN. The study was supported by the Breast Cancer Research Foundation (to E.P.W. and N.U.L.), American Society of Clinical Oncology Cancer Foundation (Career Development Award to N.U.L.), and GlaxoSmithKline.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lin, N.U., Eierman, W., Greil, R. et al. Randomized phase II study of lapatinib plus capecitabine or lapatinib plus topotecan for patients with HER2-positive breast cancer brain metastases. J Neurooncol 105, 613–620 (2011). https://doi.org/10.1007/s11060-011-0629-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-011-0629-y