
Abstract
Purpose Anaplastic gangliogliomas (AGG) are gangliogliomas with areas of pronounced hypercellularity, vascular proliferation, necrosis, and many mitotic figures. As very few pediatric patients have been studied, we analyzed the cases registered in the HIT-GBM database. Patients and Methods Patient data were obtained from the German HIT-GBM database. Inclusion criteria were diagnosis of AGG proven by a central neuropathological review and patient age 0 to 17 years. Eight patients (five male and three female) were identified. Results Patients’ median age was 10 years. The median history of disease was 9 months (range, 1.0–43.0 months). Initial symptoms included signs of raised intracranial pressure, seizures, and, in the case of spinal cord tumor, bladder dysfunction. In five cases, AGGs were localized supratentorially with three patients having multiple lobes involved. The tumors affected the frontal (n = 3 cases), parietal (n = 2), temporal (n = 2), and occipital lobes (n = 1), as well as the brainstem (n = 1) and the spinal cord (n = 2). Gross total tumor resection was achieved in six patients. The estimated 5-year overall survival rate ± standard error was 88 ± 12%, and the event-free survival rate was 63 ± 17%. While gender and tumor location did not affect survival rates, gross total tumor resection provided a better overall survival than non-total resection. Conclusion The prognosis of pediatric patients with AGG is good, especially for those who undergo gross total tumor resection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gangliogliomas (GGs) occur mainly in children and young adults [1–3] and account for 1–4% of all central nervous system (CNS) neoplasms in pediatric patients [4]. GGs are characterized by a biphasic morphologic pattern composed of neuronal and glial cells, and range from a predominantly neuronal phenotype to variants with a prominent glial population [5]. Both the glial and neuronal cells are believed to evolve from a common precursor cell in a hamartomatous glioneuronal lesion that subsequently differentiates to form neoplastic glial and neuronal elements [6, 7]. The most recent World Health Organization (WHO) classification system for brain tumors categorizes most GGs as low-grade tumors, but anaplastic GGs (AGGs; ICD-O code 9505/3), which are an aggressive subtype of GGs, are categorized as grade III tumors [8]. AGGs feature areas of pronounced hypercellularity, vascular proliferation, necrosis, and many mitotic figures mainly found in the glial component (Fig. 1) [9]. Typically, GGs have a good prognosis [4], but the prognosis for AGGs is less clear because these tumors are rare [5, 10] and their clinical course has not been well described, particularly for children [10–12]. As differences in molecular pathogenesis and clinical behavior of several histological defined brain tumors have recently been identified in this age group, this study retrospectively analyzed data on pediatric patients diagnosed with AGG who were enrolled in the HIT-GBM® trials (HIT-GBM is a registered brand name for the studies originated from an acronym of “Hirntumor Studien––Glioblastoma multiforme”) in Germany, Austria, and Switzerland from 1994 to 2007 [14–17] to determine what factors, if any, affect prognosis.

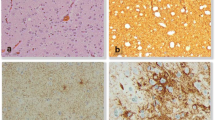
Histology of case no. 2. a, conventional HE staining shows frequent glomeruloid microvascular proliferations (arrows); b, the glial tumor cells strongly express glial fibrillary acidic protein (GFAP); c, the neuronal nuclear marker NeuN illustrates dysmorphic ganglionic cells with some binucleated cells (arrow); d, the proliferation marker Ki-67 (Mib-1) labels a high fraction of tumor cells, aberrant mitotic figures can be seen (arrow)
Patients and methods
Patient characteristics and inclusion criteria:
Patient data were obtained from the HIT-GBM database, which is maintained by the Brain Tumor Study Group of the Society of Pediatric Oncology and Hematology in Germany, Austria, and Switzerland (Gesellschaft fuer Paediatrische Haematologie und Onkologie). At the time of their enrollment in the various HIT-GBM trials, all patients and their parents gave written informed consent for their information to be included in future statistical analyses, in accordance with the Declaration of Helsinki and confirmed by the local institutional review boards.
From this database, which contained data from 1,009 patients, we identified all patients who were less than 18 years old at the time of their histopathological diagnosis with AGG, which had been confirmed by central neuropathological review (conducted by the German Brain Tumor Reference Center, Department of Neuropathology, University of Bonn, Bonn, Germany). Tissues had been characterized by immunohistochemistry with antibodies against GFAP, neurofilaments, synaptophysin, Ki-67 (MIB-1), and CD34. By applying these criteria, we identified eight patients (Fig. 1). The clinical characteristics of these patients are listed in Tables 1 and 2.
HIT-GBM treatment protocols
Eligible pediatric patients with high-grade gliomas and/or diffuse intrinsic pontine gliomas had been enrolled in the various HIT-GBM trials since 1994. For patients in these trials who were 3 years or older but younger than 18 years of age, the best feasible tumor resection was recommended before chemo- and/or radiotherapy was started [13]. Standard fractionated radiotherapy (54.0–59.4 Gy total dose; doses of 1.8 Gy/day, 5 days/week over 6–7 weeks) was common for all HIT-GBM trials [13]. The chemotherapy regimens for this group of patients were HIT 91 [14], HIT-GBM-A [15], HIT-GBM-B [16, 17], HIT-GBM-C [18], and HIT-GBM-D [19]. Children younger than 3 years of age were primarily treated with surgery and chemotherapy alone, following the HIT-SKK protocol [20] for infant patients with brain tumors.
Neuropathology and neuroradiology reports were reviewed centrally and recorded in the database. The extent of tumor resection was determined on the basis of early post-surgical imaging and/or the neurosurgical reports. Gross total tumor resection was defined as 100% macroscopic removal of the tumor mass (Fig. 2). Non-gross total tumor resection included subtotal (<100% but ≥90%) and partial (<90%) resection as well as open or stereotactic biopsy without tumor debulking.

Typical MRI (T1 with contrast) image of a pediatric patient with anaplastic ganglioglioma before (a) and after (b) tumor resection
Statistical analysis
In the current review of patients with AGG, statistical analysis was performed using SPSS 12.0 (Statistical Package for Social Studies, SPSS Inc., San Francisco, CA, USA). The Kaplan–Meier method was used to estimate event-free survival (EFS) and overall survival (OS). Possible events were tumor relapse or progression, occurrence of a secondary malignancy, or death from any cause. EFS was defined as time from initial diagnosis until occurrence of an event, and OS was defined as time from initial diagnosis until death (Fig. 3).

Event-free survival and overall survival in anaplastic ganglioglioma. Events were only reported in the first year of follow-up. Of the patients studied, 63% remained event-free and as many as 88% were alive at the end of follow-up
Results
Five boys and three girls, with a median age of 10 years (range, 2–16), were identified in the database. The median history of disease was 9 months (range, 1.0 to 43.0 months). Clinical evidence of disease for some time before diagnosis was reported in seven patients. The most common signs were seizures (in three patients) and signs of raised intracranial pressure, including vomiting and/or headache (in three patients). Other presenting signs are listed in Table 2.
In five patients, AGGs were localized supratentorially, and three patients had multiple lobes involved and gross total tumor resection was achieved in six of eight cases (Fig. 2).
The median follow-up time was 28.0 months (range, 6.0–61.0 months). The mean 5-year OS estimation and standard error was 88 ± 12%, and the mean 5-year EFS estimation and standard error was 63 ± 17% (Fig. 3). It is notable, that all events occurred within 1 year of therapy.
A detailed summary of epidemiological data and survival is given in Tables 1 and 2.
OS appeared to be superior in patients who underwent gross total tumor resection versus non-gross total resection. Of the six patients who underwent gross total resection (Fig. 2), none died during a median follow-up period of 36.5 months (range, 7.0–61.0 months). Two patients underwent non-total resection. The patient who underwent a partial resection died after 6 months; the patient who underwent a subtotal resection showed stable disease, but the follow-up period was only 8 months (Table 2). The extent of resection did not have a significant affect on EFS, and neither gender nor tumor location had prognostic relevance to OS or EFS.
Discussion
This study represents data for the largest cohort of pediatric patients with AGG to date. Our most important observation is the relatively good survival rate of these patients (Fig. 3), although no specific factors were found to be statistically significantly prognostic in these patients.
In our cohort, AGG was found at all ages within the pediatric population. The tumor showed a male predominance (5:3), which was also true in a series of pediatric GG [12] and has been found for most pediatric high-grade gliomas.
In contrast to low-grade GG [4, 12, 21], AGG did not show a strong predilection for the temporal lobe, which was only affected in two of our patients. We thus conclude that pediatric AGG can be found in all parts of the CNS without a distinct predilection.
The median clinical history was 9 months (1.0–43 months). Two patients had a surprisingly long history of disease (33 and 43 months). Both patients reported focal seizures, and AGG affected the frontal and temporal lobes, respectively. We suspect that the AGG in these patients might have developed from a low-grade GG with typical location and typical symptoms (focal seizures). Previous reports on malignant transformation in the glial component of a low-grade GG or gray matter nodules into an AGG support this hypothesis [9, 22–24]. Chromosomal aberrations and candidate genes involved in this process have been described and linked to tumor growth and seizures [6, 25, 26].
Probably because of the different locations at which AGG tumors arise, initial disease symptoms vary widely. Unlike in low-grade GG, focal seizures do not predominate in AGG [4, 12, 21, 27]. In AGG, signs of increased intracranial pressure (such as vomiting or headache) were as frequent as seizures. Less frequent symptoms included psychomotor slowing, tremor, and diplopia. In the case in which the tumor was located in the spinal cord, the symptoms of back pain and bladder dysfunction led to the diagnosis.
One of the central goals of our study was to elucidate the clinical outcome of AGG in children. Despite the malignancy of the tumor (WHO grade III), we found distinctly good survival rates reported in previous case reports [10, 28, 29] as well as in a small series [12]. Only one of the patients in our study died during follow-up and as many as five of the patients had survived for more than 2 years at this writing (Fig. 3). In fact, our AGG patients had—with intensive treatment—survival rates comparable to those of children with GG (WHO grades I and II). However, clinical outcome does not always correlate with the degree of histological malignancy, and cytogenetics, proliferation markers, and growth indices may allow better prediction of outcomes in the future [12, 30]. One patient survived after total resection without further treatment. Notably, he was only 2 years old, which parallels the apparently better prognosis in very young children with diffuse high-grade glioma.
Probably because of the location and relatively non-infiltrative nature of AGG (Fig. 2), gross total tumor resection was feasible in six of eight of patients. None of these patients died during follow-up (7–61 months), while one of the two patients with non-total resection died 6 months after diagnosis from tumor progression. The other patient had stable disease, but the follow-up period of 8 months was short. We conclude that in pediatric AGG, gross total tumor resection is a powerful positive prognostic indicator. Other clinical variables such as gender and tumor location, did not have an impact on survival. All these findings are consistent with those from several previous studies in which gross total tumor resection represented a predictor of favorable outcome in children with both high-grade gliomas [13, 31–36] and low-grade GG [4, 12, 21]. Thus, we strongly recommend performing a gross total tumor resection in pediatric patients with AGG whenever possible.
In conclusion, our study encompassed the largest cohort of pediatric patients with AGG to date. We found that AGG develops in all parts of the CNS without a specific predilection. Its prognosis is good and OS is comparable to that of low-grade GG. Patients who underwent gross total resection along with radiochemotherapy had excellent survival rates, so whenever possible, efforts should be made to completely resect these tumors.
References
Selch MT, Goy BW, Lee SP, El-Sadin S, Kincaid P, Park SH, Withers HR (1998) Gangliogliomas: experience with 34 patients and review of the literature. Am J Clin Oncol 21:557–564. doi:10.1097/00000421-199812000-00006
Nair V, Suri VS, Tatke M, Saran RK, Malhotra V, Singh D (2004) Gangliogliomas: a report of five cases. Indian J Cancer 41:41–46
Liauw SL, Byer JE, Yachnis AT, Amdur RJ, Mendenhall WM (2007) Radiotherapy after subtotally resected or recurrent ganglioglioma. Int J Radiat Oncol Biol Phys 67:244–247. doi:10.1016/j.ijrobp.2006.08.029
Johnson MD, Jennings MT, Toms ST (2001) Oligodendroglial ganglioglioma with anaplastic features arising from the thalamus. Pediatr Neurosurg 34:301–305. doi:10.1159/000056042
Luyken C, Blumcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J (2004) Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer 101:146–155. doi:10.1002/cncr.20332
Becker AJ, Lobach M, Klein H, Normann S, Nothen MM, von Deimling A, Mizuguchi M, Elger CE, Schramm J, Wiestler OD, Blumcke I (2001) Mutational analysis of TSC1 and TSC2 genes in gangliogliomas. Neuropathol Appl Neurobiol 27:105–114. doi:10.1046/j.0305-1846.2001.00302.x
Zhu JJ, Leon SP, Folkerth RD, Guo SZ, Wu JK, Black PM (1997) Evidence for clonal origin of neoplastic neuronal and glial cells in gangliogliomas. Am J Pathol 151:565–571
Becker AJ, Wiestler O, Figarella-Branger D, Blümcke I (2007) Ganglioglioma and gangliocytoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) WHO classification of tumors of the central nervous system. IARC Press, Lyon, pp 103–105
Mekni A, Chelly I, Haouet S, Zitouna M, Kchir N (2006) Malignant cerebellar ganglioglioma. A case report and review of the literature. Neurochirurgie 52:119–122. doi:10.1016/S0028-3770(06)71206-1
Karabekir HS, Balci C, Tokyol C (2006) Primary spinal anaplastic ganglioglioma. Pediatr Neurosurg 42:374–378. doi:10.1159/000095568
Nakajima M, Kidooka M, Nakasu S (1998) Anaplastic ganglioglioma with dissemination to the spinal cord: a case report. Surg Neurol 49:445–448. doi:10.1016/S0090-3019(97)00293-0
Chintagumpala MM, Armstrong D, Miki S, Nelson T, Cheek W, Laurent J, Woo SY, Mahoney DH Jr (1996) Mixed neuronal-glial tumors (gangliogliomas) in children. Pediatr Neurosurg 24:306–313. doi:10.1159/000121060
Wolff JE, Classen CF, Wagner S, Kortmann RD, Palla SL, Pietsch T, Kuhl J, Gnekow A, Kramm CM (2008) Subpopulations of malignant gliomas in pediatric patients: analysis of the HIT-GBM database. J Neurooncol 87:155–164. doi:10.1007/s11060-007-9495-z
Wolff JE, Gnekow AK, Kortmann RD, Pietsch T, Urban C, Graf N, Kuhl J (2002) Preradiation chemotherapy for pediatric patients with high-grade glioma. Cancer 94:264–271. doi:10.1002/cncr.10114
Wolff JE, Molenkamp G, Westphal S, Pietsch T, Gnekow A, Kortmann RD, Kuehl J (2000) Oral trofosfamide and etoposide in pediatric patients with glioblastoma multiforme. Cancer 89:2131–2137. doi:10.1002/1097-0142(20001115)89:10<2131::AID-CNCR14>3.0.CO;2-J
Wolff JE, Wagner S, Reinert C, Gnekow A, Kortmann RD, Kuhl J, Van Gool SW (2006) Maintenance treatment with interferon-gamma and low-dose cyclophosphamide for pediatric high-grade glioma. J Neurooncol 79:315–321. doi:10.1007/s11060-006-9147-8
Wolff JE, Wagner S, Sindichakis M, Pietsch T, Gnekow A, Kortmann RD, Strater R, Kuehl J (2002) Simultaneous radiochemotherapy in pediatric patients with high-grade glioma: a phase I study. Anticancer Res 22:3569–3572
Wagner S, Leuthold U, Schmid H-J, Wolff JE (2003) Pilotstudie mit hochdosiertem Methotrexat und nachfolgender simultaner Radiochemotherapie bei neun Kindern mit hochgradig-malignen Gliomen. Monatsschr Kinderheilkd 151:467–476. doi:10.1007/s00112-003-0693-2 abstract
Wagner S, Reinert C, Schmid HJ, Liebeskind AK, Jorch N, Langler A, Graf N, Warmuth-Metz M, Pietsch T, Peters O, Wolff JE (2005) High-dose methotrexate prior to simultaneous radiochemotherapy in children with malignant high-grade gliomas. Anticancer Res 25:2583–2587
Rutkowski S, Bode U, Deinlein F, Ottensmeier H, Warmuth-Metz M, Soerensen N, Graf N, Emser A, Pietsch T, Wolff JE, Kortmann RD, Kuehl J (2005) Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352:978–986. doi:10.1056/NEJMoa042176
Haddad SF, Moore SA, Menezes AH, VanGilder JC (1992) Ganglioglioma: 13 years of experience. Neurosurgery 31:171–178. doi:10.1097/00006123-199208000-00001
Jay V, Squire J, Becker LE, Humphreys R (1994) Malignant transformation in a ganglioglioma with anaplastic neuronal and astrocytic components. Report of a case with flow cytometric and cytogenetic analysis. Cancer 73:2862–2868. doi:10.1002/1097-0142(19940601)73:11<2862::AID-CNCR2820731133>3.0.CO;2-5
Demaerel P, Droessaert M, Lammens M, Menten J, Nuttin B, Verbeeck R, Wilms G, Baert AL (1996) Anaplastic (malignant) ganglioglioma arising from heterotopic grey matter nodules. J Neurooncol 30:237–242. doi:10.1007/BF00177274
Kurian NI, Nair S, Radhakrishnan VV (1998) Anaplastic ganglioglioma: case report and review of the literature. Br J Neurosurg 12:277–280. doi:10.1080/02688699845159
Samadani U, Judkins AR, Akpalu A, Aronica E, Crino PB (2007) Differential cellular gene expression in ganglioglioma. Epilepsia 48:646–653. doi:10.1111/j.1528-1167.2007.00925.x
Pandita A, Balasubramaniam A, Perrin R, Shannon P, Guha A (2007) Malignant and benign ganglioglioma: a pathological and molecular study. Neurooncology 9:124–134. doi:10.1215/15228517-2006-029
Tarnaris A, O’Brien C, Redfern RM (2006) Ganglioglioma with anaplastic recurrence of the neuronal element following radiotherapy. Clin Neurol Neurosurg 108:761–767. doi:10.1016/j.clineuro.2005.09.005
Hall WA, Yunis EJ, Albright AL (1986) Anaplastic ganglioglioma in an infant: case report and review of the literature. Neurosurgery 19:1016–1020. doi:10.1097/00006123-198612000-00019
Tamiya T, Takao S, Ichikawa T, Chayama K, Date I (2006) Successful chemotherapy for congenital malignant gliomas: a report of two cases. Pediatr Neurosurg 42:240–244. doi:10.1159/000092362
Lang FF, Epstein FJ, Ransohoff J, Allen JC, Wisoff J, Abbott IR, Miller DC (1993) Central nervous system gangliogliomas. Part 2: clinical outcome. J Neurosurg 79:867–873
Bucci MK, Maity A, Janss AJ, Belasco JB, Fisher MJ, Tochner ZA, Rorke L, Sutton LN, Phillips PC, Shu HK (2004) Near complete surgical resection predicts a favorable outcome in pediatric patients with nonbrainstem, malignant gliomas: results from a single center in the magnetic resonance imaging era. Cancer 101:817–824. doi:10.1002/cncr.20422
Finlay JL, Wisoff JH (1999) The impact of extent of resection in the management of malignant gliomas of childhood. Childs Nerv Syst 15:786–788. doi:10.1007/s003810050471
Kramm CM, Wagner S, Van Gool S, Schmid H, Strater R, Gnekow A, Rutkowski S, Wolff JE (2006) Improved survival after gross total resection of malignant gliomas in pediatric patients from the HIT-GBM studies. Anticancer Res 26:3773–3779
Heideman RL, Kuttesch J Jr, Gajjar AJ, Walter AW, Jenkins JJ, Li Y, Sanford RA, Kun LE (1997) Supratentorial malignant gliomas in childhood: a single institution perspective. Cancer 80:497–504. doi:10.1002/(SICI)1097-0142(19970801)80:3<497::AID-CNCR18>3.0.CO;2-S
Campbell JW, Pollack IF, Martinez AJ, Shultz B (1996) High-grade astrocytomas in children: radiologically complete resection is associated with an excellent long-term prognosis. Neurosurgery 38:258–264. doi:10.1097/00006123-199602000-00006
Artico M, Cervoni L, Celli P, Salvati M, Palma L (1993) Supratentorial glioblastoma in children: a series of 27 surgically treated cases. Childs Nerv Syst 9:7–9. doi:10.1007/BF00301926
Acknowledgments
The ongoing support of the Deutsche Kinderkrebsstiftung, Bonn, Germany, is greatly appreciated. We also thank our colleagues who contributed patient data to the HIT-GBM studies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Christof M. Kramm and Johannes E. A. Wolff are contributed equally to this work.
Rights and permissions
About this article
Cite this article
Karremann, M., Pietsch, T., Janssen, G. et al. Anaplastic ganglioglioma in children. J Neurooncol 92, 157–163 (2009). https://doi.org/10.1007/s11060-008-9747-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-008-9747-6