
Abstract
Gangliogliomas are rare tumors of the central nervous system that are thought to arise from a glioneuronal precursor and consist of both neuronal and glial elements. Grade III, or anaplastic ganglioglioma (AGG), most commonly affects children and young adults, generally arises in a supratentorial location, is highly epileptogenic, and often results in diffuse local and distant failure within the craniospinal axis. Pathologically, these tumors are graded by the degree of malignancy in their glial portion and radiologic diagnosis is difficult due to the wide variation in its degree of solid and cystic components, contrast uptake, and calcification patterns. This report presents three cases of AGG, with initial treatment including subtotal resection followed by conformal radiotherapy. In the case where the AGG developed in the setting of an existent low-grade astrocytoma, the patient received no chemotherapy. Both of the other de novo cases were managed with adjuvant chemoradiotherapy with temozolomide. Recurrence occurred at 6, 16, and 20 months following therapy. Two of the three patients experienced symptomatic decline at recurrence, but experienced Karnofsky performance status (KPS) improvement after salvage therapy, including the reduction of cranial neuropathy and balance. All patients had a significant reduction in presenting symptoms following salvage therapy. Patients died at 23, 20, and 22 months following initial surgical management, respectively. A review of anaplastic and malignant gangliogliomas is presented in the context of these three cases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gangliogliomas (GGs) are rare, highly epileptogenic [1, 2] malignancies that make up approximately1.3 % of all primary brain tumors [3, 4]. The World Health Organization (WHO) Grade III variant [5] or anaplastic ganglioglioma (AGG), represents 1-5 % of all GGs [3, 6, 7] and is associated with significantly worse local control rates and shorter overall survival [8]. Patients are generally children or young adults at presentation [9–13], Caucasian [13], and more likely to be male, as there is slight gender predominance [13]. AGGs are most commonly unifocal, supratentorial in location, and located in the temporal lobes [13], but cases have also been reported within the spinal cord [14], as well as in intraventricular locations [15]. The present case series is a retrospective review of three unique cases of AGG treated at the Comprehensive Cancer Center of Wake Forest University (CCCWFU) between January 2011 and May 2014 (Supplementary Fig. 4).
Case reports
Case 1: juvenile anaplastic ganglioglioma with leptomeningeal spread
History
This 12-year-old Caucasian male presented to his primary care physician after a 1-week course of unexplained progressive headaches and nausea. MRI of the brain revealed a 2.0 × 3.0 × 3.0 cm left temporoparietal mass (Supplementary Fig. 1) approximating the angular gyrus with edema and localized mass effect. He was prescribed steroids that mildly alleviated his headaches. It was recommended that he undergo resection and he thus underwent a left temporoparietal craniotomy and was found to have residual tumor along the anterior/superior surgical margin measuring approximately 0.7 × 2.2 × 0.7 cm on his postoperative MRI (Fig. 1, Supplementary Fig. 1).

Leptomeningeal failure. In each case, enhancement on T1 gadolinium enhanced scans leptomeningeal disease was documented. In case 1 this was most apparent at multiple levels within the spinal canal. Case 2 displayed leptomeningeal enhancement along the anteromedial aspect of the prior resection bed as well as then tentorium cerebelli. In case 3, the anterior horn of the temporal lobe showed enhancement across the entire anterior surface of the temporal lobe as well as the undersurface of the right frontal lobe. Arrows indicate sites of leptomeningeal enhancement
Pathological findings
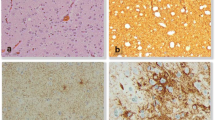
Histopathologic examination of the surgical specimen revealed highly pleomorphic cells with gangliocytoid features (Fig. 2). Focally, the tumor cells had a more typical glial appearance (Fig. 2b), including oligodendroglial-like areas with vascular proliferation (Fig. 2a). MIB-1 index was greater than 40–50 % and areas of necrosis were present (Fig. 2c). There were inflammatory foci within the tumor, but no well-formed Rosenthal fibers or eosinophilic granular bodies. The gangliocytoid areas were immunoreactive for neurofilament protein and GFAP stain highlighted scattered tumor cells (Fig. 2d and e). The final pathologic diagnosis was an anaplastic ganglioglioma with features of an anaplastic pleomorphic xanthoastrocytoma without an associated WHO grade.

Anaplastic ganglioglioma Histopathology. a Case 1 the tumor is composed of clusters of neoplastic ganglion cells with large nuclei, prominent nucleoli, and abundant vacuolated cytoplasm (arrow) (H&E, X600). b Some areas of the tumor display oligodendroglial features (i.e., cells with perinuclear halos) and admixed ganglion cells (arrow) (H&E, X600). c The tumor has regions of necrosis (arrow), consistent with a high-grade classification (H&E, X400). d The neoplastic ganglion cells are immunoreactive for neurofilament protein (X600). e The neoplastic glial cells are immunoreactive for glial fibrillary acidic protein (X600)
Postoperative course
His headaches and nausea initially resolved but 3 weeks post-resection, he developed impaired speech. An MRI revealed rapid progression of the residual tumor, now measuring 2.7 × 3.6 × 3.1 cm. He received concurrent Temozolomide and 59.4 Gy of conformal radiation in 33 fractions followed by adjuvant Temozolomide, Irinotecan, and Bevacizumab. During his second cycle of chemotherapy, he was hospitalized for nausea, vomiting, and diarrhea, and MRI showed evidence of increased perfusion and nodular enhancement along the resection bed suggestive of primary tumor failure approximately 4 months after completion of radiotherapy.
The decision was made to proceed with repeat resection. Review of the pathology once again demonstrated findings consistent with anaplastic ganglioglioma including a mixed population of atypical gangliocytoid and glial cells with multifocal necrosis. Postoperative MRI revealed residual tumor along the medial wall of the resection cavity and consequently he resumed his regimen of Temozolomide, Irinotecan, and Bevacizumab. During the last two cycles, Bevacizumab was discontinued due to scalp wound healing failure requiring excision of the chronic wound, removal of skull hardware, and creation of a flap for scalp closure.
Five months after his second resection he developed right-sided numbness, headache, and aphasia. MRI of the brain revealed foci of linear and nodular enhancement in the parasagittal left parieto-occipital region, as well as subtle nodular areas of enhancement along the surface of the right aspect of the cervicomedullary junction. Follow-up MRI revealed increasing enhancement along the left parietal resection cavity with no changes of the size of the primary residual tumor. Soon after, he developed low back pain and MRI of the spine revealed diffuse plaque-like enhancement throughout the spinal cord, confirming leptomeningeal failure.
The previously re-resected primary site tissue was assayed and found to be positive for the BRAF V600 mutation by PCR. He underwent craniospinal irradiation to a dose of 36 Gy of 24 fractions with concurrent and maintenance Vemurafenib at a dose of 720 mg PO BID. Initially his leptomeningeal disease burden decreased, but 4 months after the completion of craniospinal irradiation, he developed radiographic evidence of tumor progression and was escalated to an adult Vemurafenib dose of 960 mg PO BID. He had no evidence of progression for an additional 4 months. His leptomeningeal disease subsequently worsened and he progressed at that time; he died 23 months from the date of his first resection.
Case 2—malignant transformation of a juvenile low-grade astrocytoma
History
This Caucasian male began suffering from seizure episodes at age 11, described as staring, grasping for objects in the air, and speaking gibberish. Stereotactic biopsy revealed a fibrillary and gemistocytic astrocytoma involving the left medial temporal lobe and hypothalamus. He underwent 54 Gy in 30 fractions to a localized field, and while he had a favorable treatment response, he continued to experience intermittent complex partial seizures until he developed radiographic progression 2 years later. He underwent a left temporal craniotomy and resection of the tumor. Unfortunately, this resulted in a left CN III palsy with dilated pupil and absent left medial rectus function. He continued to take antiepileptic medications until the age of 20 and did not experience any seizures for the next 14 years after that.
At 34 years of age he again began experiencing seizures and progressive dysarthria. CT scan revealed a partially calcified mass at the margin of his previous resection cavity. Three months after radiographic evidence of recurrence, he suffered a devastating left temporal hemorrhage that left him with right-sided hemiplegia, impaired verbal communication, and impaired cognition, as well as bowel and bladder incontinence. CT scan at this time revealed a hemorrhagic lesion approximately 3.7 × 3.0 cm in the greatest transaxial dimensions, and MRI demonstrated an enhancing mass at the margin of the resection cavity extending into the cerebral peduncle and thalamus (Supplementary Fig. 1).
Operation
Two days post-hemorrhage, he underwent a repeat left temporal craniotomy under general anesthesia. The tumor was very vascular and engulfed the posterior cerebral artery. Branches supplying the tumor were coagulated and divided. Microsurgical technique with suction catheters and bipolar cautery was employed to debulk the grossly abnormal tissue. Because of the eloquent location, no additional margin around the tumor was obtained. Postoperative MRI confirmed residual tumor (Supplementary Fig. 1).
Pathological findings
Microscopic examination revealed a hypercellular glioneuronal neoplasm composed of glial cells with pleomorphic, hyperchromatic nuclei intermingled with gangliocytoid cells (highlighted by Neu-N), some of which were binucleated (Supplementary Fig. 2). Mitotic figures were frequent, with a MIB-1 index of 5–7 %. Endothelial proliferation and areas of necrosis were identified. Focal areas of calcification were noted, and PAS stain highlighted occasional eosinophilic granular bodies, consistent with a longstanding lesion, suggesting this may have arisen from a low-grade ganglioglioma (WHO Grade I). Final conclusions were that this was a malignant transformation of his prior low-grade astrocytoma to an anaplastic ganglioglioma, WHO Grade III.
Postoperative course
The patient did not improve significantly after surgery and was readmitted to the hospital 1 month after resection due to sepsis secondary to aspiration pneumonia. Due to his poor performance status, he was not a candidate for chemotherapy, and palliative-intent re-irradiation was offered. He received 45 Gy in 25 fractions of conformal radiation and he experienced only a modest improvement in his motor and cognitive function.
Two months after the completion of radiotherapy, he began experiencing worsening gait instability, significant urinary incontinence, and global memory deficits. MRI revealed communicating hydrocephalus, and 2 days later, he received a ventriculoperitoneal (VP) shunt. He was maintained on valproic acid and levetiracetam but continued to experience breakthrough seizures. In the next few months he was hospitalized several times for confusion, lethargy, dehydration and seizures, though serial MRI imaging did not reveal any appreciable changes to the tumor. Sixteen months after the completion of his radiotherapy, he was seen during an outpatient visit for cognitive decline. His follow-up MRI imaging revealed a new sub-centimeter focus of enhancement in the right aspect of the pons, and interval increase in the size of the mass in the medial left temporal lobe highly concerning for local and leptomeningeal spread along the posterior ventricle. He died 2 weeks later.
Case 3—adult anaplastic ganglioglioma
History
This 34-year-old Nigerian man was in his usual state of health until he began experiencing a 1-month history of intermittent headaches, a single episode of nausea, followed by a prolonged, grand-mal seizure while driving. MRI revealed a poorly defined, diffuse signal abnormality in the medial right temporal lobe/peri-Sylvian region with associated mass effect and a small focus of intraparenchymal hemorrhage (Supplementary Fig. 1). Presumed diagnosis at that point balanced between neoplasm and herpes encephalitis as there was minimal mass effect but prominent edema and sub-acute hemorrhage. Neurosurgical intervention was delayed due to renal failure secondary to rhabdomyolysis.
Operation
Two weeks after his seizure, he underwent a right-sided fronto-temporo-parietal craniotomy under general anesthesia. Microsurgical technique was employed to resect most of the grossly abnormal tissue via separate frontal and temporal corticotomies. Antero-mesial temporal lobectomy was performed using suction catheters and bipolar cautery. Postoperative MRI revealed a poorly defined, right frontotemporal residual T2 hyperintense mass (Supplementary Fig. 1).
Pathological findings
Most of the tissue consisted of an infiltrating glioma, though a significant proportion was made up of neoplastic gemistocytic astrocytes (Supplementary Fig. 2). A smaller subset of tumor cells had smaller, rounder nuclei. Numerous mitotic figures were identified, but no microvascular proliferation or necrosis was present. Many foci of abnormal ganglion cell clusters, which were high grade, were noted (Supplementary Fig. 2B). Final conclusions were that this was an anaplastic gangliogioma, WHO Grade III.
Postoperative course
Postoperatively, the patient was conversant without evidence of intraoperative complication. He received concurrent Temozolomide and 59.4 Gy in 33 fractions with a conformal irradiation technique. After two cycles of adjuvant Temozolomide, follow-up MRI revealed pseudoprogression with local mass effect and enhancement. Follow-up MRI 1 month later showed an interval decrease in the size of the tumor. He completed six cycles of adjuvant Temozolomide.
Two months after the completion of his sixth cycle of Temozolomide, he developed sudden headaches and right-sided neck pain. MRI revealed increased size and mass effect of the dominant right frontal lobe tumor with central necrosis consistent with progression 12.1 months following initial resection. The lesion extended posteriorly along the arcuate fasciculus to the anterior right temporal lobe causing right to left midline shift with subfalcine herniation. He declined repeat resection.
He suffered another grand-mal seizure and in the next month, the patient began carboplatin (AUC = 4), with monthly CPT-11 (125 mg/m2), and q2 weekly Bevacizumab (10 mg/kg). After 4 months, the tumor remained radiographically stable and his carboplatin was reduced to AUC = 2 and Bevacizumab was decreased to 5 mg/kg due to a slight increase in serum creatinine to 1.4 mg/dL. Over the next month the patient began experiencing progressive left hemiparesis and falls, and MRI revealed further progression at the primary site as well as leptomeningeal spread in the anterior cranial fossa. His Carboplatin and CPT-11 were discontinued and he began CCNU. He continued to worsen clinically and radiographically over the next month and VP-16 was substituted for the CCNU. He died 1 month later, 22 months after his first resection.
Discussion
AGGs, whether de novo or arising from a low-grade lesion, consist of neoplastic ganglionic and glial cells [16], which are most often astrocytes [17]. In AGGs, the anaplastic transformation most often occurs in the glial component [17], but transformation has also been reported in the neural component as well [18]. While the etiology and pathogenesis remains unclear, the cell of origin is noted to be a glioneuronal precursor [19, 20]. While no specific mutation has been identified as a solely causative genetic aberration, a mutation in BRAF V600E has been identified in a number of cases [21–23]. The incidence of this mutation appears to be inversely related to the age at presentation [21]. In fact, recent literature indicates that BRAF V600E is present in 13 % of pediatric low grade gliomas, with as much as 15 % of these cases demonstrating ganglioglioma histology [24]. In the present case series, only the pediatric tumor in case 1 underwent PCR testing for the BRAF V600E mutation. This patient did experience a treatment response to Vemurafenib, first at a lower dose (720 mg by mouth twice daily) and then at standard adult dosing (960 mg by mouth twice daily) at first sign of possible progression. Whereas Bautista et al. [25] initiated doses as low as 480 mg PO twice daily and observed systemic concentrations approximating 80 % of the adult dose, we initiated dosing at 720 mg twice daily due to concerns with sub therapeutic levels within the cerebrospinal fluid. Escalation to adult doses of 960 mg twice daily more closely resembled doses which approximated the adult literature [26]. Significant variation in clearance in Vemurafenib has been documented and may explain limited therapeutic response in cases where clearance is high [25]. Providers may consider this option for brain tumors that have mutations in BRAF V600E as it is currently under study in the recurrent setting (NCT01748149).
Imaging characteristics of gangliogliomas are varied and non-specific [19, 27, 28]. As a testament to how varied imaging can be in AGG, the diffuse, poorly delineated mass in our case 3 was first thought to be herpes encephalitis on the initial radiology report. Zhu et al. noted that larger solid masses with remarkable heterogeneous enhancement and ipsilateral cerebellar cortical atrophy in the infratentorial region were suggestive of GG [19]. In AGGs, tumors are typically isointense or hypointense on T1 imaging, hyperintense on T2 imaging, and contrast enhancement is generally irregular [9, 29, 30]. Some studies have noted that MR spectroscopy of gangliogliomas may reveal distinct but non-specific choline peaks which may differentiate these from benign conditions, but not necessarily from other primary brain tumors [18].
Both the treatment and prognosis of AGG is varied. In the largest evaluation of AGG outcomes using the SEER database, 5-year survival was 63 % (95 % CI 46–76) [13]. The primary prognostic factor was resectability at presentation, with a greater than 25 % difference in overall survival at 2 years. In a separate cohort of 40 malignant glioneuronal tumors, median survival was 44 months in those who underwent gross total resection, but only 15 months in those who underwent subtotal resection [31]. While maximal safe resection is regarded as the primary treatment for anaplastic gangliogliomas, the role of radiation and chemotherapy has not been established in prospective randomized trials and is poorly reported in existing case series.
In Rades et al. retrospective analysis of 60 patients with high-grade gangliogliomas [8], radiotherapy conferred a statistically significant benefit in local control following subtotal resection but no survival benefit. In addition, the authors did not find that dose escalation beyond 54 Gy improved local control, regardless of the pathologic grade. While our case series did not include a control group, and thus was not able to definitively demonstrate benefit, we observed a progression free interval in all 3 cases.
Repeat radiation was utilized in two cases when resection was not possible. The role of repeat radiotherapy is the subject of intensive investigation and is being explored prospectively in RTOG 1205. Repeat radiotherapy has shown some success in early reports from Fogh et al. with a median survival of 11 months using a hypofractionated approach independent of re-resection of use of chemotherapy [40]. Dosing and schedule range widely with the current RTOG study employing a dose of 3.5 Gy per fraction for a total dose of 10 fractions. Others have advocated the use of conventional fractionation at the time of repeat radiation in settings where the prognosis is favorable (second brain tumors) or when large volumes are required (repeat craniospinal radiotherapy) [41, 42]. Select populations appear to benefit from repeat radiation with durable local control but depending on tumor type, may be at risk for distant CNS failure [42].
While GGs almost always progress locally, [3, 10] AGGs often exhibit leptomeningeal failure, which tends to be a rare phenomenon amongst the more common high-grade gliomas, and more frequent in all pediatric gliomas [32, 33]. In Scoccianti et al. review of 19 pediatric AGGs, 10 (53 %) patients experienced tumor progression, and of these, five progressed distantly [34, 35]. In our series, all patients experienced out-of-field, leptomeningeal failures. Similar natural histories have been described in at least six different reports of patients with malignant GGs [27, 36–39]. These findings suggest that chemotherapy may be warranted as an upfront treatment modality to minimize the risk of neuraxis failure. In spite of these distant failures, most authors continue to recommend conformal, partial brain irradiation to minimize the morbidity of extended fields [30, 34].
In summary, AGGs are very rare tumors with a strong potential for distant relapse, suggesting the need for more efficacious systemic treatment regimens and potential integration of newer biologic agents. Careful review of imaging, pathology, and molecular features has the potential to improve diagnosis, management and treatment of this rare tumor.
References
Giulioni M, Galassi E, Zucchelli M, Volpi L (2005) Seizure outcome of lesionectomy in glioneuronal tumors associated with epilepsy in children. J Neurosurg 102:288–293
Giulioni M, Gardella E, Rubboli G, Roncaroli F, Zucchelli M, Bernardi B, Tassinari CA, Calbucci F (2006) Lesionectomy in epileptogenic gangliogliomas: seizure outcome and surgical results. J Clin Neurosci 13:529–535
Luyken C, Blumcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J (2004) Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer 101:146–155
Kalyan-Raman UP, Olivero WC (1987) Ganglioglioma: a correlative clinicopathological and radiological study of ten surgically treated cases with follow-up. Neurosurgery 20:428–433
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO classification of tumours of the central nervous system, 4th edn. International Agency for Research on Cancer Lyon, Lyon
Miller DC, Lang FF, Epstein FJ (1993) Central nervous system gangliogliomas. Part 1: pathology. J Neurosurg 79:859–866
Wolf HK, Muller MB, Spanle M, Zentner J, Schramm J, Wiestler OD (1994) Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol 88:166–173
Rades D, Zwick L, Leppert J, Bonsanto MM, Tronnier V, Dunst J, Schild SE (2010) The role of postoperative radiotherapy for the treatment of gangliogliomas. Cancer 116:432–442
Karremann M, Pietsch T, Janssen G, Kramm CM, Wolff JE (2009) Anaplastic ganglioglioma in children. J Neurooncol 92:157–163
Selch MT, Goy BW, Lee SP, El-Sadin S, Kincaid P, Park SH, Withers HR (1998) Gangliogliomas: experience with 34 patients and review of the literature. Am J Clin Oncol 21:557–564
Nair V, Suri VS, Tatke M, Saran RK, Malhotra V, Singh D (2004) Gangliogliomas: a report of five cases. Indian J Cancer 41:41–46
Liauw SL, Byer JE, Yachnis AT, Amdur RJ, Mendenhall WM (2007) Radiotherapy after subtotally resected or recurrent ganglioglioma. Int J Radiat Oncol Biol Phys 67:244–247
Selvanathan SK, Hammouche S, Salminen HJ, Jenkinson MD (2011) Outcome and prognostic features in anaplastic ganglioglioma: analysis of cases from the SEER database. J Neurooncol 105:539–545
Schneider C, Vosbeck J, Grotzer MA, Boltshauser E, Kothbauer KF (2012) Anaplastic ganglioglioma: a very rare intramedullary spinal cord tumor. Pediatr Neurosurg 48:42–47
Deling L, Nan J, Yongji T, Shuqing Y, Zhixian G, Jisheng W, Liwei Z (2013) Intraventricular ganglioglioma prognosis and hydrocephalus: the largest case series and systematic literature review. Acta Neurochir 155:1253–1260
Krouwer HG, Davis RL, McDermott MW, Hoshino T, Prados MD (1993) Gangliogliomas: a clinicopathological study of 25 cases and review of the literature. J Neurooncol 17:139–154
Norenberg MD (1998) Gangliogliomas: issues of prognosis and treatment. AJNR Am J Neuroradiol 19:810
Kawataki T, Sato E, Sato T, Kinouchi H (2010) Anaplastic ganglioglioma with malignant features in both neuronal and glial components–case report. Neurol Med Chir 50:228–231
Zhu JJ, Leon SP, Folkerth RD, Guo SZ, Wu JK, Black PM (1997) Evidence for clonal origin of neoplastic neuronal and glial cells in gangliogliomas. Am J Pathol 151:565–571
Becker AJ, Lobach M, Klein H, Normann S, Nothen MM, von Deimling A, Mizuguchi M, Elger CE, Schramm J, Wiestler OD, Blumcke I (2001) Mutational analysis of TSC1 and TSC2 genes in gangliogliomas. Neuropathol Appl Neurobiol 27:105–114
Koelsche C, Wohrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M, Lasitschka F, von Deimling A, Capper D (2013) Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 125:891–900
Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR (2013) BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol 125:901–910
Rush S, Foreman N, Liu A (2013) Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol 31:e159–e160
Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ (2014) Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin Cancer Res 20:5630–5640
Bautista F, Paci A, Minard-Colin V, Dufour C, Grill J, Lacroix L, Varlet P, Valteau-Couanet D, Geoerger B (2014) Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr Blood Cancer 61:1101–1103
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516
Zentner J, Wolf HK, Ostertun B, Hufnagel A, Campos MG, Solymosi L, Schramm J (1994) Gangliogliomas: clinical, radiological, and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry 57:1497–1502
Lou X, Gui QP, Sun L, Wu NZ, Lyu JH, Ma L (2014) Comparisons of MR findings between supratentorial and infratentorial gangliogliomas. Clinical Neuroradiol. doi:10.1007/s00062-014-0333-3
DeMarchi R, Abu-Abed S, Munoz D (2011) Loch Macdonald R: malignant ganglioglioma: case report and review of literature. J Neurooncol 101:311–318
Zorlu F, Selek U, Onal C, Soylemezoglu F, Gurkaynak M (2006) Postoperative radiotherapy in cranial ganglioglioma. J Neurooncol 77:321–324
Varlet P, Soni D, Miquel C, Roux FX, Meder JF, Chneiweiss H, Daumas-Duport C (2004) New variants of malignant glioneuronal tumors: a clinicopathological study of 40 cases. Neurosurgery 55: 1377–1391, discussion 1391–1372
Paulsson AK, McMullen KP, Peiffer AM, Hinson WH, Kearns WT, Johnson AJ, Lesser GJ, Ellis TL, Tatter SB, Debinski W, Shaw EG, Chan MD (2014) Limited margins using modern radiotherapy techniques does not increase marginal failure rate of glioblastoma. Am J Clin Oncol 37:177–181
Gururangan S, McLaughlin CA, Brashears J, Watral MA, Provenzale J, Coleman RE, Halperin EC, Quinn J, Reardon D, Vredenburgh J, Friedman A, Friedman HS (2006) Incidence and patterns of neuraxis metastases in children with diffuse pontine glioma. J Neurooncol 77:207–212
Scoccianti S, Giordano F, Agresti B, Detti B, Cipressi S, Franceschini D, Greto D, Mussa F, Sardi I, Buccoliero A, Arico M, Genitori L, Biti G (2012) Pediatric primary anaplastic ganglioglioma: a case report and review of the literature. Pediatr Neurosurg 48:35–41
Broniscer A, Chintagumpala M, Fouladi M, Krasin MJ, Kocak M, Bowers DC, Iacono LC, Merchant TE, Stewart CF, Houghton PJ, Kun LE, Ledet D, Gajjar A (2006) Temozolomide after radiotherapy for newly diagnosed high-grade glioma and unfavorable low-grade glioma in children. J Neurooncol 76:313–319
Nakajima M, Kidooka M, Nakasu S (1998) Anaplastic ganglioglioma with dissemination to the spinal cord: a case report. Surg Neurol 49:445–448
Kitano M, Takayama S, Nagao T, Yoshimura O (1987) Malignant ganglioglioma of the spinal cord. Acta pathologica japonica 37:1009–1018
Sutton LN, Packer RJ, Rorke LB, Bruce DA, Schut L (1983) Cerebral gangliogliomas during childhood. Neurosurgery 13:124–128
Wacker MR, Cogen PH, Etzell JE, Daneshvar L, Davis RL, Prados MD (1992) Diffuse leptomeningeal involvement by a ganglioglioma in a child. Case report. J Neurosurg 77:302–306
Fogh SE (2010) Hypofractionated stereotactic radiation therapy: an effective therapy for recurrent high-grade gliomas. J Clin Oncol 28:3048–3053
You SH, Lyu CJ, Kim DS (2013) Second primary brain tumors following cranial irradiation for pediatric solid brain tumors. Childs Nerv Syst 29(10):1865–1870
Merchant TE, Boop FA, Kun LE (2008) A retrospective study of surgery and reirradiation for recurrent ependymoma. Int J Radiat Oncol Biol Phys 71(1):87–97
Acknowledgments
The authors wish to acknowledge the support of the “Biostatistics Shared Resource”, and NCI Cancer Center Support Grant P30 CA012197, Comprehensive Cancer Center of Wake Forest University, for study design and monitoring, analysis and interpretation of data, preparation of the manuscript and presentation. We would also like to acknowledge Bonny Morris for her careful and insightful comments and review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lucas, J.T., Huang, A.J., Mott, R.T. et al. Anaplastic ganglioglioma: a report of three cases and review of the literature. J Neurooncol 123, 171–177 (2015). https://doi.org/10.1007/s11060-015-1781-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1781-6