
Abstract
B7-H4, a newly discovered member of B7 family that negatively regulates T cell-mediated immunity, may facilitate tumor progression by undermining host immunity. Recent studies show that brain tumor stem-like cells (TSCs) contribute to tumorigenesis. However, the relationship between B7-H4 and the clinical behavior of brain TSCs remains unclear. In this study, we found that B7-H4 was expressed in cultured tumor cells from human gliomas (n = 5) and medulloblastomas (n = 3). Double immunostaining indicated that B7-H4 was primarily restricted to non-dividing (Ki67−) cultured tumor cells. Tumor cells cultured under medium conditions favoring the growth of neural stem cells were able to form primary and secondary spheres, along with expression of neural stem/progenitor cell markers. These cells differentiated into different neural lineages when cultured in differentiation medium, indicating that these cells have TSCs characteristics. Double immunostaining showed that TSCs consisted of proliferative (Ki67+) and quiescent (Ki67−) cells. We also found that B7-H4 was expressed in a small population of CD133+ cells sorted by flow cytometry. Interestingly, both CD133+ and CD133− cells were tumorigenic in SCID mice in vivo. However, CD133+ cells-initiated glioblastomas showed a higher proliferation index, compared to CD133− cells-induced glioblastomas in vivo. Secondary glioma cells derived from CD133+ or CD133− cell xenografts expressed B7-H4 as well. Our data suggest B7-H4 is preferentially expressed in non-dividing brain tumor cells and in a subpopulation of brain TSCs, and CD133− tumor cells also have the capacity to initiate brain formation in vivo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although the host immune system is able to recognize tumor cells as harassment and eliminate these cells, brain tumor cells can acquire various characteristics, which allow them to evade this immunological surveillance [1]. For immunotherapy to be successful, it is essential to elucidate tumor cell immune escape mechanisms.
B7-H4, a member of the B7 family, is a type I cell surface transmembranous protein that is primarily restricted to activated T cells, B cells and antigen-presenting cells (APCs). In vitro and in vivo studies indicate that B7-H4 may deliver an inhibitory signal to T cells, abrogating the proliferation of CD4+ and CD8+ T cells, the generation of CD8+ T cell, cell cycle progression, and IL-2, IL-4, IL-10 and IFN-γ production. Therefore, B7-H4 may participate in the negative regulation of T cell-mediated immunity [2–4]. Recent studies also show that B7-H4 protein is aberrantly overexpressed in human cancers of the lung, breast, and ovary [5–7]. Our previous study found that B7-H4 was abnormally expressed on human astrocytomas and its expression level was correlated with malignancy grade of astrocytomas [8]. In addition, we also found that B7-H4 had a negative correlation with glioma infiltrating CD8+ T cells [8]. B7-H4 in malignant tumors has thus been suggested to modulate tumor apoptosis and ultimately shield tumors from cell-mediated immune surveillance [9]. Moreover, Salceda et al. [7] found that overexpression of B7-H4 in a human ovarian cancer cell line with little endogenous B7-H4 expression significantly increased tumor formation in Severe Combined Immune Deficiency (SCID) mice, suggesting that B7-H4 might have a new tumor-specific function other than its ability to modulate immune function. However, the relationship between B7-H4 and the proliferation behavior of brain tumors remains uncertain. It is important to unravel this question, because the proliferation index is strongly associated with the outcome in malignant brain tumors [10–12], and the tumor cells in the quiescent phase (G0) of cell cycle are less vulnerable to injury by radiation and/or chemotherapy [13–15].
In addition, recent studies reveal that CD133+ cells, a minority fraction of the entire brain tumor population, are tumor stem-like cells (TSCs) and contribute to tumorigensis, based on the observations that CD133+ cells exhibit increased self-renewal capacity and display the potential to differentiate into neuronal, astroglial, and oligodendroglial cells, and that CD133+ tumor cells can recapitulate the original tumor in vivo even after serial transplantation [16–20]. Our previous study showed the number of dividing TSCs was directly correlated with malignancy grade of human astrocytomas [21]. Therefore, brain TSCs play a role in tumorigensis. However, whether tumorigensis is due to immune escape of brain TSCs remains uncertain.
In the present study, we found that B7-H4 was expressed on all examined brain tumor cells from human primary gliomas and medulloblastomas, but preferentially in the non-dividing tumor cells. In addition, we also found that B7-H4 was expressed in a small population of CD133+ cells sorted by flow cytometry. Interestingly; we found that both CD133+ and CD133− cells could initiate tumor formation after implantation into SCID mice. These secondary glioma cells expressed B7-H4 as well. Our findings may have implications not only for understanding the clinical behavior of brain TSCs, but also for the potential implications for brain tumor immunology and immunotherapy.
Materials and methods
Brain tumor tissue specimens
Human astrocytic tumor specimens (n = 5) and medulloblastoma specimens (n = 3) were obtained from the patients (mean age 37 ± 28) undergoing surgical resection at the Department of Neurosurgery, Huashan Hospital, Fudan University (Shanghai, China). Histopathological diagnosis was performed by the attending neuropathologists at the Department of Neuropathology, Huashan Hospital, Fudan University, according to World Health Organization (WHO) guidelines [22]. The patients’ information is given in Table 1. The protocol for this study was approved by the Institutional Review Board.
Culture of brain tumor spheres
Brain tumor tissues were washed with cold PBS, cut into very small pieces, and incubated in Accutase solution (Chemicon, San Diego, CA, USA) at 37°C with continuous stirring for 30 min. The supernatant containing liberated cells was collected, washed with PBS, and cells at a density of 1 × 106 cells/ml were plated in 75 cm2 culture flasks containing DMEM/F12 medium (Gibco, Grand Island, NY, USA), supplemented with penicillin/streptomycin sulfate, B-27(Gibco), recombinant epidermal growth factor (EGF; 20 ng/ml; Chemicon, San Diego, CA, USA) and recombinant human fibroblast growth factor-2 (FGF-2; 20 ng/ml; Chemicon). The cultures were incubated at 37°C in a humidified 5% CO2 incubator and fresh FGF-2 and EGF were added twice every week. Tumor spheres were passaged by trituration through a fire-polished pipette and reseeded into fresh proliferative medium. Brain TSCs were also cultured in the DMEM/F12 medium supplemented with 10% serum but without FGF-2 and EGF (differentiation medium) for 7–14 days.
Immunocytochemistry
Brain tumor cells were plated onto coverslips precoated with poly-l-lysine (Sigma) and cultured in neurobasal medium for 12 h. After fixation with 4% paraformaldehyde, cells were incubated with blocking buffer (2% horse serum, 0.2% Triton X-100, 0.1% BSA in PBS) for 1 h at room temperature and with primary anti-B7-H4 antibody (H74, Ebioscience; 1:200) at 4°C for overnight. After wash, FITC-conjugated goat anti-mouse antibody (KPL; 1:50) or rhodamine-conjugated goat anti-mouse antibody (KPL; 1:150) was added and incubated for 60 min at room temperature.
Brain sections were processed for immunocytochemistry as described previously [16–19]. Primary antibodies used were (1) mouse monoclonal anti-human CD133-Biotin (Miltenyi Biotec; 1:50), (2) rabbit polyclonal anti-CD133 (Abcam; 1:250), (3) mouse monoclonal anti-human specific nestin (Chemicon; 1:200), (4) rabbit polyclonal anti-TUC-4 (Chemicon; 1:1,000), (5) rabbit polyclonal anti-Sox2 (Abcam; 1:100), (6) affinity-purified goat polyclonal anti-doublecortin (DCX) (Santa Cruz Biotechnology; 1:200), (7) rabbit polyclonal anti-GFAP (Zymed Laboratories; 1:200), (8) mouse monoclonal anti-β III-tubulin (Chemicon; 1:400) and (9) rabbit polyclonal anti-human Ki67 antigen (clone MIB-1; Zymed Laboratories; 1:50). The secondary antibodies used were FITC-conjugated goat anti-mouse, FITC-conjugated goat anti-rabbit, FITC-conjugated rabbit anti-goat, FITC-conjugated anti-biotin, rhodamine-conjugated goat anti-mouse, rhodamine-conjugated goat anti-rabbit and rhodamine-conjugated rabbit anti-mouse. 4′-6-Diamidino-2-phenylindole (DAPI; Chemicon) was used to counterstain nuclei. Fluorescence signals were detected with a Nikon fluorescent microscope at excitation/emission wavelengths of 535/565 nm (rhodamine, red) and 470/505 (FITC, green). Results were recorded with a digital camera. Controls included omitting or preabsorbing primary antibody or omitting secondary antibody.
Double-label immunohistochemistry
Double-label immunohistochemistry was performed on cultured cells and brain sections to detect expression of B7-H4 and neural stem/progenitor specific-cell protein markers, as described in our previous publication [21].
Flow cytometry
Brain tumor cells were dissociated and flow cytometry (FCM) analysis was performed as described previously [8]. Briefly, tumor spheres were dissociated with trituration and Accutase. Cells were then washed three times by PBE buffer. 1 × 106 cells were placed in FCM tubes and treated with Fix and Perm® cell permeabilization reagents (Caltag Laboratories) according to the instruction of manufacturer. Tumor cells were incubated on ice for 30 min with PE-conjugated anti-B7-H4 (Ebioscience; 1:5) or with isotype control antibodies (PE-mouse IgG1; Ebioscience; 1:5). Cells were washed twice with FCM buffer (PBS containing 0.1% NaN3 and 5% fetal bovine serum) and then resuspended in 0.5 ml 1% formalin/PBS. Analysis was performed on a FCM calibur system (Becton Dickinson Immunocytometry Systems). 1 × 104–3 × 104 cells were collected and analyzed with WinMDI software. Forward and side scatter plots were used to exclude debris/dead cells from further analysis.
CD133+ cells were sorted with FCM (Beckon Epics-ALTRA) using EXPO32-MultiCOMP software after labeled with CD133/1-PE (Miltenyi Biotech). The positive cell populations were >98% pure as confirmed by flow cytometry. FCM analysis was also performed to explore profile of B7-H4 expression in CD133+, Sox2+ and Ki67+ tumor cells. All the FCM evaluations were performed in triplicate.
Transplantation of CD133+ or CD133− cells into SCID mice
To evaluate the tumorigenicity of brain TSCs, CD133+ or CD133− cells were subcutaneously injected into the right armpit (1 × 107) or into the right striatum (1 × 105) of SCID mice (n = 3). The tumor sizes in the right armpit were monitored by palpation and caliper measurement, and tumor volumes were calculated using the formula: (length × width2)/2. Hematoxylin and eosin (H&E) staining and immunohistochemistry using antibodies against nestin, GFAP, DCX and B7-H4 were performed on the primary tumor sections and secondary xenografts 18 days after subcutaneously transplantation or 30 days after orthotopic injection. Proliferation index was defined as the number of Ki67+ cells divided by the total number of cells in the evaluated area of primary and secondary gliomas and was expressed as a percentage. Secondary tumor cells from CD133+ or CD133− cell xenografts were also cultured for B7-H4 analysis.
Results
B7-H4 was predominantly expressed in non-dividing tumor cells
To determine whether B7-H4 protein was expressed in tumor cells from human astrocytoma and medulloblastoma, we first performed immunocytochemistry using anti-B7-H4 antibody. As shown in Fig. 1a, we found that B7-H4 was expressed in cultured tumor cells derived from eight brain tumors and B7-H4 protein was predominantly located in the cytoplasm. Flow cytometry analysis showed that 8.12%–80.59% of B7-H4-positive cells were found in astrocytomas and 20.12%–45.07% in medulloblastomas (Fig. 1b; Table 1). We noted that tumor cells isolated from recurrent anaplastic astrocytoma (BT4) expressed lower levels of B7-H4 protein, compared to tumor cells from other primary astrocytoma.

B7-H4 expression in glioblastoma and medulloblastoma cells in vitro. (a) Tumor cells from glioblastoma (left) and medulloblastoma (right) were stained with anti-B7-H4 antibody (green in left panel and red in right panel). Nuclei were counterstained with DAPI (blue). Inset in right panel: high magnification of B7-H4-positive cells. (b) B7-H4 expression in glioblastoma (left) and medulloblastoma (right) were analyzed by flow cytometry. Filled red histogram: B7-H4 expression; open histogram: isotype control
We then asked whether B7-H4-immunoreactive tumor cells exhibited features associated with tumor cell proliferation. Double-immunolabeling was conducted using anti-B7-H4 and anti-Ki67, a proliferation protein marker expressed in all phases of the active cell cycle (G1, S, G2 and M phase) [23]. Interestingly, we found that B7-H4 was primarily restricted to Ki67− tumor cells (Fig. 2a). Consistent with this, FCM analysis also showed that about 93% of B7-H4+ cells were quiescent (Fig. 2b).

B7-H4 expression predominantly in non-dividing tumor cells. (a) Double immunostaining was performed on brain sections from glioblastoma (left) and medulloblastoma (right) using anti-B7-H4 and anti-Ki67 antibodies. Nuclei were counterstained with DAPI. (b) Expression profiles of B7-H4 and Ki67 in glioblastoma was analyzed by flow cytometry. Left panel: isotype control; right panel: B7-H4 and Ki67 expression in glioblastoma
Tumor stem-like cells expressed B7-H4
In order to explore the linkage between B7-H4 and brain TSCs, we cultured tumor cells isolated from brain tumors tissue under medium conditions favoring the growth of stem cells. A number of small spheres (about 5–20 cells per sphere) were observed in all tested tumor samples in 5–10 days and the cell number in spheres increased with increased incubation. The tumor spheres displayed nonadherent and adherent forms when cultured in the proliferation medium. However, tumor spheres became adherent, along with appearance of differentiated morphology, when cultured in the differentiation medium (Fig. 3a).

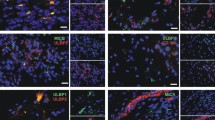
Culture and isolation of TSCs. (a) Nonadherent and adherent tumor spheres were observed when tumor cells derived from the primary brain tumors were cultured in the medium conditions favoring the growth of NSCs (left panel). Tumor spheres became adherent and began to differentiate when cultured in the differentiated medium (right panel). (b) Cultured tumor cells expressed neural stem/progenitor cell markers including nestin, TUC4 and βIII-tubulin. Some of them express proliferation marker Ki67 (right panel). (c) These tumor cells including CD133+ cells could differentiate into astrocytes (GFAP+), neuronal cells (βIII-tubulin+) and oligodentrocytes (CNPase+), when cultured in differentiation medium. (d) Tumor cells sorted using anti-CD133 antibody were immunoreactive for CD133 (left panel). Some sorted CD133+ cells expressed Ki67 (right top panel) and nestin/Ki67 (right bottom panel). (e) CD133− cells expressed nestin (left panel, red), TUC-4 (right top panel, green) and DCX (right bottom panel, green)
By immunocytochemistry analysis, we found the tumor spheres contained nestin+, TUC-4+ and nestin+/TUC-4+ cells (Fig. 3b). These TSCs could differentiate into neural lineages when cultured under differentiating conditions (Fig. 3c). FCM analysis using anti-CD133 antibody showed that 0.3%–9.85% of tumor cells were CD133+ cells (Table 1). Sorted CD133+ cells were confirmed by immunocytochemistry (Fig. 3d). CD133 protein was predominantly located in membrane of cells. Some CD133+ cells co-expressed GFAP and β III-tubulin under differentiating conditions. Double immunostaining indicated that CD133+ cells consisted of Ki67+ and Ki67− cells (Fig. 3d). In addition, sorted CD133+ cells also contained nestin+/Ki67+ cells and nestin+/Ki67− cells, suggesting that some TSCs were proliferative and others were quiescent (Fig. 3d). CD133− cells also expressed nestin, TUC-4 and DCX (Fig. 3e), suggesting that CD133− cells may contain some tumor progenitor cells.
We then examined whether brain TSCs could express B7-H4. We observed B7-H4+ cells could express nestin, CD133, TUC-4 and DCX (Fig. 4a). FCM analysis showed nearly 25% of CD133+ cells and 42% of Sox2+ cells expressed B7-H4 (Fig. 4b). Taken together, these data indicate that brain TSCs derived from gliomas and medulloblastomas are either dividing or non-dividing, and negative costimulatory molecule B7-H4 is expressed in a subpopulation of brain TSCs.

Expression of B7-H4 in brain TSCs. (a) Double-immunolabeling was performed using antibodies anti-B7-H4 and against neural stem/progenitor cell markers nestin (left panel, green), CD133 (right top panel, red), TUC-4 (right middle panel, red), and DCX (right bottom panel, red). Nuclei were counterstained with DAPI (blue). (b) Expression profile of B7-H4 and CD133 (left panel; BT3) and of B7-H4 and SOX2 in tumor cells (right panel; BT8) were analyzed by flow cytometry
Expression of B7-H4 in secondary glioma from CD133+ and CD133− xenografts
To evaluate the tumorigenicity of CD133+ and CD133− cells and to determine whether secondary tumor cells could also express B7-H4, cells isolated from the patient with left temporal glioblastoma (BT1) (Fig. 5a) were implanted into the right armpit or into the right striatum of SCID mice. A large glioblastoma-like subcutaneous mass was detected in the SCID mice implanted with either CD133+ or CD133− cells in 18 days (Fig. 5b, c), suggesting that both CD133+ and CD133− cells were tumorigenic. However, CD133+ cells-initiated glioblastoma was larger than that of CD133− cells. The average volumes of tumor initiated by CD133+ and CD133− cells were 2.386 cm3 and 1.044 cm3 in 18 days, respectively. CD133+ or CD133− cells could also initiate gliomas in 30 days when implanted into the right striatum. However, the former had enhanced tumor growth and vascularity (Fig. 5b, c). CD133+ cell-induced glioblastoma had a higher proliferation index (30%), compared with CD133− xenograft (19%). H&E staining indicated that CD133+ and CD133− xenografts resembled the primary tumor. As shown in Fig. 5d, B7-H4 protein was also expressed in the cultured tumor cells derived from CD133+ (14.83%) and CD133− (15.21%) xenografts, whereas, primary glioma cells expressed higher level of B7-H4 (44.35%).

Tumor formation after implantation of CD133+ or CD133− cells into SCID mice and B7-H4 expression in secondary glioblastoma. (a) Head axial contrast enhanced MRI image revealed the patient with left temporal space-occupying tumor with heterogeneous enhancement. H&E staining showed histological features of glioblastoma. GFAP, nestin and DCX were expressed on the primary tumor. (b) Tumor was formed in SCID mice after subcutaneously transplantation of 1 × 107 CD133+ cells in 18 days (left panel). H&E staining showed histological features of CD133+ cells-initiated tumors in the right armpit in 18 days (1 × 107 cells; middle panel) and in the right striatum in 30 days (1 × 105 cells; right panel). Secondary glioma formed in the brain displayed clear border and enhanced tumor growth and vascularity. (c) Tumor formed in SCID mice after subcutaneously transplantation of 1 × 107 CD133− cells in 18 days (left panel). H&E staining showed histological features of CD133− cells-initiated tumors in the right armpit in 18 days (1 × 107; middle panel) and in the right striatum in 30 days (1 × 105 cells; right panel). (d) B7-H4 was expressed in both primary and secondary glioma cells. Left panel: immunostaining with anti-B7-H4 in primary tumors; middle panel: flow cytometry analysis using anti-B7-H4 in primary glioma cells (44.35%); right panel: flow cytometry analysis of secondary tumor cells. Filled red histogram: CD133+ xenograft cells (14.83%); open green histogram: CD133− xenograft cells (15.21%); open black histogram: isotype control
Discussion
In this study, we found that B7-H4 was expressed in human glioma and medulloblastoma cells. Double immunostaining indicated that B7-H4 was primarily restricted to non-dividing tumor cells. In addition, B7-H4 was also expressed in a small subpopulation of CD133+ TSCs. Interestingly, both CD133+ and CD133− cells sorted by flow cytometry were tumorigenic in SCID mice in vivo. CD133+ cells-generated tumor, however, showed a higher proliferation index compared with CD133− cells-induced tumor. Secondary glioma cells derived from CD133+ or CD133− cells xenografts expressed B7-H4 as well. Our findings suggest that negative costimulatory molecules may play an important role in tumor immune surveillance mechanisms.
Although B7-H4 is the youngest member of the B7 family, growing evidence indicate that B7-H4 plays an important role in tumor immunity. Kryczek and his colleagues found that 0.5% of primary ovarian tumor cells express surface B7-H4 and 91% express intracellular B7-H4 [24], consistent with our finding. Our previous study shows that B7-H4 expression profile may be correlated with malignancy grade of human astrocytomas [8]. Other studies suggest that B7-H4 may modulate tumor apoptosis and ultimately shield tumors from cell-mediated immune surveillance [9]. Therefore, the expression of B7-H4 predominantly in the quiescent tumor cells indicates that B7-H4+ tumor cells may gain chances to deliver an inhibitory signal to T cells and facilitate themselves survival after treatments for brain tumors. It is surprising that B7-H4 is expressed relative low level in the patient with recurrent glioma (BT4), which argues the notion that recurrent tumors are often endowed with more capacities to escape from immune surveillance. Further investigation of B7-H1, another negative co-stimulatory molecule, showed that more than 60% of the tumor cells expressed B7-H1 and some tumor cells co-expressed both B7-H1 and B7-H4 (data not shown). This differential expression profiles of B7-H1 and B7-H4 in gliomas imply that costimulatory molecules play roles in tumor immune function. In consistent with this, B7-H1 protein is highly expressed in melanoma, but B7-H4 is barely detectable [25].
Our data indicate that tumor stem-like cells may be present in brain tumors based on the observations that cultured tumor cells can form tumor spheres, along with expression of neural stem/progenitor markers such as CD133, nestin, TUC-4 and DCX, and that they can differentiate into neural lineages when cultured in the differentiated medium. Although these brain TSCs share many characteristics of NSCs, the differences between brain TSCs and NSCs as well as between CD133+ cells and CD133− cells are obvious. For example, unlike normal NSCs, brain TSCs undergo aberrant proliferation (active type) and differentiation [17]. We observed that CD133+ cells showed a higher proliferation index than that of CD133− cells in vitro and in vivo [26]. In addition, we note that a subpopulation of brain TSCs is not proliferative (quiescent type) and some of them express B7-H4. It is thus possible that these B7-H4+ tumor cells may be of unresponsiveness to chemotherapy and immunotherapy.
Our findings that CD133− tumor cells are also tumorigenic in SCID mice in vivo and that secondary tumors phenotypically resemble the primary tumor suggest that CD133− tumor cells have stem or progenitor cells-like properties. Recently, Beier et al. [26] found that 0.5%–2% of CD133− cells were able to proliferate and form new adherent spheres in vitro, that these cells had the capacity of multilineage differentiation, and that large glioblastoma-like lesions were formed within 50 days when 1 × 105 or 1 × 106 CD133− cells were implanted into the SCID mouse brain. In addition, our findings showed that CD133− tumor cells express neural stem/progenitor markers. Take together, CD133− tumor cells may contain a small subpopulation of brain tumor progenitor cells, which retain the capacity of proliferation, and therefore, could initiate tumor formation in mice. We found that CD133− and CD133+-induced secondary glioblastomas have a higher proliferation index (19% and 30%), compared with primary tumors (15%). One of explanations is that SCID mice are absent T cells and lack B cell function, tumor can thereby growth rapidly without any homogeneous anti-tumor responses mediated by T cells and other cytokines. In agreement with the previous finding [26], CD133+-induced gliomas displays a higher proliferation index, compared with CD133−-induced gliomas, which may explain why CD133+ cells can produce larger malignant lesions than that of CD133− cells. Tumor cells derived from CD133+ and CD133− xenograft also express B7-H4 with similar proportion. Of note, the level of B7-H4 in secondary glioma cells declined after 18 days, compared with primary tumor. One possibility is that secondary cells growth in non-naïve matched tumor microenvironment. Another possibility is that the tumors growth microenvironment in SCID mice may induce more quiescent cells (G0-phase cells) into proliferative status, which subsequently down-regulate B7-H4 expression.
Our findings raise the possibility that brain TSCs may be resistant to adaptive and innate immune surveillance through B7-H4 pathway. Unfortunately, it remains unclear how these B7-H4+ brain TSCs really act in vivo, since microenvironment in the mouse mode for tumorigenicity evaluation is not reliable and felicitous due to lacking of all the requisite human support cells, such as APC, T cells. Nonetheless, the demonstration that B7-H4 are expressed in brain TSCs, and that B7-H4 is preferentially expressed in non-dividing tumor cells, together with recent finding that the expression of seven MHC class II proteins is upregulated in CD133− cells [26], suggest that brain TSCs can lead to immune paralysis during tumorigenesis. However, the present study has two limitations. First, clonal cultures were not performed so that a tumor sphere came from a single cell, but not from a single tumor stem cell. Second, the further functional profiles of B7-H4 protein in brain tumors were not analyzed in this study although our previous data showed that B7-H4 has negative correlation with glioma infiltrating CD8+ T cells [8].
References
Walker PR, Calzascia T, de Tribolet N, Dietrich PY (2003) T cell immune responses in the brain and their relevance for cerebral malignancies. Brain Res Rev 42:97–122
Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, Chapoval AI, Flies DB, Bajorath J, Chen L (2003) B7-H4, a molecule of the B7 family negatively regulates T cell immunity. Immunity 18:849–861
Prasad DV, Richards S, Mai XM, Dong C (2003) B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity 18:863–873
Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP (2003) B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci U S A 100:10388–10392
Krambeck AE, Thompson RH, Dong H, Christine ML, Eugene SP, Susan MK, Bradley CL, Michael LB, John CC, Eugene DK (2006) B7-H4 expression in renal cell carcinoma and tumor vasculature: associations with cancer progression and survival. Proc Natl Acad Sci U S A 103:10391–10396
Sun Y, Wang Y, Zhao J, Gua M, Giscombea R, Lefverta AK, Wanga X (2006) B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer 53:143–151
Salceda S, Tang T, Kmet M, Munteanu A, Ghosh M, Macina R, Liu W, Pilkington G, Papkoff J (2005) The immunomodulatory protein B7-H4 is overexpressed in breast and ovarian cancers and promotes epithelial cell transformation. Exp Cell Res 306:128–141
Yao Y, Zhou LF, Mao Y, Wang XM, Xiong SD, Tao R, Zhu JH, Wang Y. B7-H4 expression in human astrocytomas and CD133 positive tumor cells. Front Biosci (in press)
Choi IH, Zhu G, Sica GL, Strome SE, Cheville JC, Lau JS, Zhu Y, Flies DB, Tamada K, Chen L (2003) Genomic organization and expression analysis of B7-H4, an immune inhibitory molecule of the B7 family. J Immunol 171:4650–4654
Torp SH (2002) Diagnostic and prognostic role of Ki67 immunostaining in human astrocytomas using four different antibodies. Clin Neuropathol 21:252–257
Pollack IF, Hamilton RL, Burnham J, Holmes EJ, Finkelstein SD, Sposto R, Yates AJ, Boyett JM, Finlay JL (2002) Impact of proliferation index on outcome in childhood malignant gliomas: results in a multi-institutional cohort. Neurosurgery 50:1238–1244
Wakimoto H, Aoyagi M, Nakayama T, Nagashima G, Yamamoto S, Tamaki M, Hirakawa K (1996) Prognostic significance of Ki-67 labeling indices obtained using MIB-1 monoclonal antibody in patients with supratentorial astrocytomas. Cancer 77:373–380
Hambek M, Werner C, Baghi M, Gstottner W, Knecht R (2007) Enhancement of docetaxel efficacy in head and neck cancer treatment by G0 cell stimulation. Eur J Cancer 43:1502–1507
Banes AK, Shaw SM, Tawfik A, Patel BP, Ogbi S, Fulton D, Marrero MB (2005) Activation of the JAK/STAT pathway in vascular smooth muscle by serotonin. Am J Physiol Cell Physiol 288:C805–C812
Jedema I, Barge RM, Nijmeijer BA, Willemze R, Falkenburg JH (2003) Recruitment of leukemic cells from G0 phase of the cell cycle by interferons results in conversion of resistance to daunorubicin. Leukemia 17:2049–2951
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828
Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A 100:15178–15183
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414:105–111
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760
Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 25:67–78
Mao Y, Zhou LF, Zhu W, Wang XM, Yang GY, Xie L, Mao XQ, Jin KL (2007) Proliferative status of tumor stem cells may be correlated with malignancy grade of human astrocytomas. Front Biosci 12:2252–2259
Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK (2002) The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 61:215–225
Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H (1984) Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133:1710–1715
Kryczek I, Zou L, Rodriguez P, Zhu GF, Wei S, Mottram P, Brumlik M, Cheng P, Curiel T, Myers L, Lackner A, Alvarez X, Ochoa A, Chen L, Zou W (2006) B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med 203:871–881
Flies DB, Chen L (2007) The new B7s: playing a pivotal role in tumor immunity. J Immunother 30:251–260
Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP (2007) CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67:4010–4015
Acknowledgments
This work was supported by “National Natural Science Foundation of China” Grant 30772225 (to L.Z.), “Program for Shanghai Outstanding Medical Academic Leader” Grant 06027 (to Y.M.), “Program for the leaders in their subjects from Science and Technology Commission of Shanghai Municipality grant” 07XD14005 (to Y.M.) and “The Chinese National Postdoctoral grant” Grant 20060400145 (to Y.Y.).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yu Yao and Xiaomei Wang contributed equally to this work.
Liangfu Zhou and Ying Mao are co-corresponding authors.
Rights and permissions
About this article
Cite this article
Yao, Y., Wang, X., Jin, K. et al. B7-H4 is preferentially expressed in non-dividing brain tumor cells and in a subset of brain tumor stem-like cells. J Neurooncol 89, 121–129 (2008). https://doi.org/10.1007/s11060-008-9601-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-008-9601-x