
Abstract
Glioblastoma (GBM) remains the most common and lethal intracranial tumor. In a comparison of gene expression by A2B5-defined tumor-initiating progenitor cells (TPCs) to glial progenitor cells derived from normal adult human brain, we found that the F2R gene encoding PAR1 was differentially overexpressed by A2B5-sorted TPCs isolated from gliomas at all stages of malignant development. In this study, we asked if PAR1 is causally associated with glioma progression. Lentiviral knockdown of PAR1 inhibited the expansion and self-renewal of human GBM-derived A2B5+ TPCs in vitro, while pharmacological inhibition of PAR 1 similarly slowed both the growth and migration of A2B5+ TPCs in culture. In addition, PAR1 silencing potently suppressed tumor expansion in vivo, and significantly prolonged the survival of mice following intracranial transplantation of human TPCs. These data strongly suggest the importance of PAR1 to the self-renewal and tumorigenicity of A2B5-defined glioma TPCs; as such, the abrogation of PAR1-dependent signaling pathways may prove a promising strategy for gliomas.
Similar content being viewed by others

Introduction
Gliomas are the most common primary intracranial neoplasms in humans, accounting for 80% of all malignant brain tumors. Malignant high-grade (HG) gliomas include anaplastic astrocytoma and oligodendroglioma (WHO grade III) and glioblastoma multiforme (GBM; WHO grade IV); the latter comprises the most frequent and lethal glial neoplasm. Less malignant, low-grade (LG) forms (WHO grade II) are slow growing and infiltrating tumors that inexorably degrade into more aggressive tumors, typically and pre-terminally evolving into GBM. Current treatment strategies, including surgical resection, radiation and chemotherapy, only modestly improve patient survival, and survivors frequently suffer permanent deficits from treatment-related toxicity.1, 2
Recent studies have suggested that a discrete fraction of tumor-initiating cells may be responsible for the initiation, recurrence and treatment-resistance of gliomas.3, 4, 5, 6 These tumor-initiating cells share phenotypic and molecular traits with neural stem cells and glial progenitor cells of the normal adult central nervous system. As a result, their pharmacological targeting requires the identification of differentially expressed molecular targets by which they may be distinguished from normally resident neural and glial precursors, so as to limit treatment-associated toxicity. We have previously isolated and characterized a population of tumor precursor cells (TPCs) derived from primary human gliomas using the monoclonal antibody A2B5,7 which identifies normal glial progenitor cells (GPCs) in the adult human brain.8 We showed that GBM-derived A2B5+ cells were clonogenic, self-renewing and multipotential in vitro, induced tumor formation after transplantation into mice brain, and propagated new tumors at lower cell doses than did their negative A2B5− homologs,7 all as would be expected of tumor-initiating stem or progenitor cells.9, 10, 11 We further compared the RNA expression profiles of A2B5-sorted glioma cells with those of normal glial precursors, so as to identify differentially expressed genes and pathways by which to selectively target resident glioma TPCs. By doing so, we identified a discrete cohort of genes specifically dysregulated in glioma TPCs, among which the thrombin receptor F2R/PAR1 was one of the most prominently and selectively overexpressed.7
The protease-activated receptor (PAR) family that includes PAR1 comprises four distinct G-protein coupled receptors, which share a unique mechanism of activation that relies on the proteolytic cleavage of their N-terminal ectodomains by serine proteases.12 PAR1, the prototype of this receptor family, is the principal thrombin-activated receptor in most cell types.13 In addition to its role in coagulation and thrombogenesis,12 PAR1 is expressed by a variety of cancers. PAR1 has been shown to promote the proliferation and survival of tumor cells, as well as tumor angiogenesis and metastasis through endothelial cell activation, in a variety of epithelial malignancies.14, 15, 16, 17 Among neuroectodermal cancers, PAR1 has been recently proposed as a highly expressed therapeutic target in melanoma.18, 19 PAR-1 is similarly overexpressed by GBM,13, 20, 21, 22 and its level of expression has been reported to correlate with tumor grade, such that higher levels of PAR1 predict low Karnovsky performance scores and poor prognosis.20, 23
Extending these observations, we reported that PAR1 was differentially overexpressed by glioma-derived TPCs relative to normal glial progenitor cells. Importantly, its overexpression was observed at the earliest stages of gliomagenesis, and was maintained as such at all stages of glioma development, from WHO-II through WHO-IV. On that basis, we investigated the necessity of PAR1 to glioma TPC expansion and tumor development. We found that lentiviral shRNAi knockdown (KD) of PAR1 significantly inhibited the growth and self-renewal of GBM-derived TPCs in vitro, and then noted that pharmacological inhibition of PAR1 similarly inhibited the growth and migration of glioma TPCs in culture. Importantly, we then noted that PAR1 silencing potently suppressed the malignant growth of glioma TPCs in vivo as well, after orthotopic transplantation into the brains of immunodeficient mice, and significantly prolonged the survival of these xenografted mice. Together, these data identify PAR1 as a potential target for the treatment of malignant gliomas.
Results
PAR1 is overexpressed in A2B5-defined glioma tumor precursor cells (TPCs)
To identify new pathways by which to target tumor-initiating cells in malignant gliomas, we previously compared the gene expression profiles of glioma TPCs to their normal homologs, using microarray analysis of A2B5-defined cells isolated from gliomas of different stages.7 This strategy was based on the immuno-isolation of normal GPCs from the adult human brain by monoclonal antibody A2B5, which recognizes neural and glial progenitor cell-selective gangliosides.8 By using this method, we identified a cohort of genes in A2B5+ TPCs that were differentially expressed relative to A2B5-sorted GPCs derived from normal adult brain tissue; among these was F2R, encoding for the thrombin receptor PAR1, which was potently upregulated by A2B5+ TPCs at every stage of glioma development (Table 1). Quantitative PCR confirmed a substantial upregulation of F2R mRNA in TPCs derived from both LG and HG gliomas, as derived from both astrocytomas and GBM, all relative to normal GPCs and unsorted normal dissociates (Table 1; Figure 1a). In addition, PAR1 mRNA was significantly overexpressed in glioma TPC lines, which we established from both unsorted (GICL 1-6) and A2B5-sorted cells (GICL 7-9), all derived from primary GBMs, and all cultured in serum-free media supplemented with FGF2, EGF and PDGF as described7 (Figure 1b). Under these conditions, the A2B5-expressing cells selectively expanded, such that the proportion of A2B5+ TPCs increased with time and passage in vitro.

PAR1 is overexpressed in glioma tumor progenitor cells (TPCs) (a), Expression of PAR1 mRNA using real-time PCR, in freshly sorted glioma-derived A2B5+ TPCs (n=12) relative to normal A2B5+ glial progenitor cells (GPCs) (n=4) and unsorted cells (UNS) (n=4), reveals that PAR1 mRNA was significantly upregulated at all stages of glioma development. (b–c) Relative quantification of PAR1 gene (b) and protein expression (c) using RT-PCR (b) and flow cytometry (c) detection in GBM-derived glioma-initiating cell lines (GICLs) established from unsorted or A2B5+ cells (*) maintained in serum-free media (SFM) supplemented with FGF, EGF (20 ng/ml) and PDGF (10 ng/ml) for less than 10 passages; and commercially available adherent GBM cells U87 and U251 (c) cultured in 10% serum culture conditions. Comparable quantities of cDNA were ensured by amplification of GAPDH (b). (d–f) Flow cytometry analysis of PAR1, A2B5 and CD133 expression in GBM-derived GICLs. Representative scatter plot of GICL8 stained with A2B5 (bottom right), PAR1 (upper left), both (upper right), or their corresponding isotype controls (bottom left) (D). Error bars indicate Means±s.e.m.
Flow cytometry confirmed that PAR1 protein was overexpressed by glioma TPCs derived from different GBMs, or from the widely used human glioma cell lines U87 and U251 (Figure 1c; Supplementary Table S1). Furthermore, the great majority of PAR1+ TPCs co-expressed either or both A2B5 (94.6±1.2%) and the neural stem cell protein prominin 1, recognized as CD133 (79.9±5.4%) (Figures 1d and f; Supplementary Table S1). Together, these data suggest that PAR1 is highly and differentially overexpressed by glioma TPCs relative to normal glial progenitor cells, and that these PAR1 overexpressors typically co-express both A2B5 and CD133, consistent with the predominant tumor-initiating stem cell phenotype of the PAR1+ cell population.
PAR1 overexpression correlates with the classical subtype of GBM
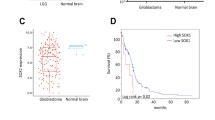
GBMs have been classified into four molecular subtypes—neural, mesenchymal, classical and proneural—each associated with distinct genetic alterations, gene expression patterns, phenotype and prognosis.24 The proneural (PN) subgroup has been preferentially associated with a glioma-CpG island methylator phenotype (G-CIMP), which is characterized by mutations in isocitrate dehydrogenase 1 and correlates with relatively good prognosis. In contrast, patients whose tumors express a mesenchymal (MES) or classical (CL) signature typically exhibit a more rapid disease course, coupled with therapeutic resistance.24, 25 To further explore the clinical significance of F2R in gliomas, we correlated its expression with GBM subtype, as well as with common copy number variants and mutations identified in The Cancer Genomic Atlas (TCGA). F2R was enriched across GBM subtypes relative to normal GPCs, but most prominently so in GBMs of the CL subtype (P<0.01) (Figure 2a). In contrast, no significant correlation of F2R expression to G-CIMP status or isocitrate dehydrogenase 1 mutation was noted (Figure 2b). Across all TCGA classes, we noted high levels of F2R expression in anaplastic astrocytoma relative to both grade II oligodendroglioma and oligoastrocytoma (Figure 2c), indicating that while F2R overexpression appeared with LG gliomas, its expression increased with malignant progression. Among GBMs, we then found a significant correlation between F2R expression and EGFR amplification, CDKN2A and PTEN deletion, all hallmarks of the CL subtype (Figure 2d).

Analysis of PAR1 expression in relation with TCGA-defined glioblastoma subtypes and copy number variations (a–b), PAR1 gene expression was enriched in the CL subtype of GBM defined by the TCGA (n=483) relative to normal brain tissue (n=10) (a) but was not correlated with the glioma-Cpg Island Methylator Phenotype (G-CIMP) subtype. (b, c) Expression value of PAR1 gene expression in low-grade gliomas (n=468) derived from the TCGA showing a significant enrichment of PAR1 in anaplastic AST, relative to grade II OLG and OA. Black lines in each group indicate mean±s.e.m. 1 way ANOVA, P<0.0001; *P<0.05; **P<0.01; ***P<0.001 after Tukey’s multiple test comparison. (d) PAR1 expression was significantly correlated with EGFR, PTEN and CDKN2A copy number variations. AST, astrocytoma; CL, classical; OLG,: oligodendroglioma; OA, oligoastrocytoma; II and III: WHO grades II and III, respectively; MES, mesenchymal; NL, neural; PN: proneural.
PAR1 silencing inhibits the growth, self-renewal and survival of A2B5+ TPCs in vitro
The robust overexpression of PAR1 by A2B5-defined TPCs prompted us to examine its contribution to glioma expansion. To determine if glioma TPCs were dependent upon PAR1 signaling, we first tested the effect of lentiviral shRNAi-induced KD of PAR1 expression on the growth of glioma TPCs in vitro. Quantitative PCR with reverse transcription and flow cytometry were used to respectively assess PAR1 mRNA and Par-1 protein expression in two lines of A2B5+ TPCs, each derived from A2B5+ cells sorted from fresh GBMs. These lines are designated in this paper as glioma-initiating cell lines, GICL-8 and GICL-9, each of which we validated as able to produce HG GBMs upon orthotopic xenograft into the brains of immunodeficient hosts. Six days after transduction with these lines with either of two PAR1 KD lentiviruses, each generated using distinct non-overlapping short hairpin RNA (shRNA) sequences, we compared the extents of RNA and protein KD to cells transduced with a scrambled control (SCR). The PAR1-KD lentiviruses significantly and substantially reduced the expression of both PAR1 mRNA (Figures 3a and b) and protein (Figures 3c and d), relative to SCR control lentivirus (Figure 3e), in each line.

Lentiviral induced PAR1 knockdown (KD) validation. Validation of PAR1 gene (a, b, e) and protein (c–e) induced silencing using quantitative RT-PCR (a, b) and flow cytometry (c, d) A2B5+ derived glioma-initiating cell lines (GICLs), 6 days after transduction with two different PAR1 KD lentiviruses, compared with glioma cells transduced with a SCR. Gene expression levels normalized to GAPDH. One-way ANOVA with repeated measures; *P<0.05; **P<0.01; ***P<0.001 after Tukey post-hoc comparisons. Means±s.e.m.
To determine if PAR1 influenced glioma development, we then examined the effect of PAR1 silencing on the growth of A2B5-sorted GBM cells. We found that within 6 days of transduction, lentiviral shRNAi KD of PAR1 significantly reduced the number of GBM-derived A2B5+ TPCs in both GICL8 and GICL9, relative to scrambled (SCR) shRNAi-transduced and non-transduced control (CT) cells (Figures 4a and c) (Supplementary Table S2). On that basis, we next investigated the effects of PAR1-induced KD on cell proliferation and cell cycle progression, by analyzing EdU incorporation in association with propidium iodide (PI) staining. We found that A2B5+ TPCs subjected to PAR1 KD all manifested fewer cells in S-phase relative to SCR and control cells, for both GICL8 and GICL9 (Figures 4d and e) (Supplementary Table S2).

PAR1 silencing inhibits the growth and self-renewal of glioma TPCs. (a) Representative photomicrograph illustrating the number of A2B5+ TPCs derived from GICL-8 6 days after transduction with either PAR1-KD or control lentiviruses. Scale bar, 100 μm. (b–g) Effects of PAR1 silencing on the in vitro expansion (b, c), proliferation (d, e), survival (f, g) and clonal sphere formation (h, i) of A2B5+ TPCs derived from two glioma-initiating cell lines (GICL8 and GICL9), 6 days after transduction with different PAR1 knockdown (KD) lentiviruses, compared with scrambled lentivirus (SCR) and control (CT) untransduced cells. (b, c) Lentiviral KD of PAR1 significantly reduced the number of A2B5+ GICL relative to both SCR shRNAi-transduced and non-transduced CT cells. (d, e) EdU incorporation in association with propidium iodide (PI) staining revealed that GICLs subjected to PAR1 KD manifested fewer cells in S phase relative to SCR and CT cells. (f, g) PAR1 KD significantly increased the percentage of apoptotic cells as determined by flow cytometry analysis of Annexin V, relative to SCR and CT cells. KD, Knockdown. Means±s.e.m. One-way ANOVA with repeated measures (P<0.0001) with Tukey post-hoc comparisons. *P <0.05; **P <0.01; ***P <0.001. Post-hoc comparisons between PAR1 KD cells and SCR and non-transduced control (CT) cells are illustrated by orange and black stars, respectively; three independent experiments for each cell line. (h, i) GICLs were plated into a 96-well plates for limiting dilution sphere formation assay, and counted 14 days later; wells were then scored for the presence or absence of sphere growth. The log fraction of the negative wells (non-responding) was plotted as a function of cell density per well.
We next asked if PAR1 suppression might be associated with increased cell death, and found that PAR1 KD TPCs exhibited a significantly increased incidence of Annexin V-defined apoptotic death, compared with both SCR and control cells (Figures 4f and g) (Supplementary Table S2). In addition, PAR1 KD also impaired the self-renewal of GBM-derived A2B5+ TPCs, as evidenced by a dramatic decrease in tumor sphere formation in limiting dilution assays of glioma TPCs transduced with PAR1 shRNA lentiviruses vs CT and SCR-transduced cells (Figures 4h and i; Supplementary Table S3).
Similar results were obtained after PAR1 KD in U87 glioma cells (Supplementary Figure S1), in which PAR1 silencing substantially decreased the number of glioma cells relative to both SCR and untransduced control cells, as evident using either of the two PAR1 KD lentiviral vectors (Supplementary Figures S1A, B and D). PAR1 KD also substantially decreased the percentage of glioma cells in S-phase, relative to SCR and CT cells (Supplementary Figures S1C and D), suggesting the contribution of PAR1 expression and signaling to cell cycle progression.
PAR1 silencing suppressed the in vivo expansion of glioma TPCs
We next sought to evaluate the effect of PAR1 inhibition on the tumorigenic competence of both U87 glioma cells and A2B5+ TPCs in vivo. PAR1-shRNA transduced U87 cells, as well as untransduced U87 control (CT) cells and those transduced with SCR lentivirus, were implanted intracranially into the brains of immunodeficient NOG mice, 6 days after lentiviral PAR1 shRNAi transduction (8 x 104 cells per animal, n=3 for each group). PAR1 silencing substantially suppressed the tumorigenicity of U87 cells, compared with both CT and SCR-infected cells, as seen both macroscopically (Supplementary Figure S2A) and histologically (Supplementary Figure S2B), at 4 weeks after transplant. Stereological analyses of the xenografts confirmed a significant and dramatic decrease in antero-posterior tumor extension and tumor volume in mice transplanted with U87 cells transduced with PAR1 KD1 and KD2 lentiviruses, relative to both the SCR-infected cells and uninfected control cells (Supplementary Figures S2C and E). We next examined the effect of PAR1 inhibition on the tumorigenicity of GBM-derived A2B5+ TPCs transplanted into the brains of immunodeficient mice, 6 days after lentiviral transduction with either of our two PAR1 lentiviruses (2.8 × 104 cells per animal, n=4–6 mice per group). We found that PAR1 silencing potently decreased both tumor extension (Figures 5a–c) and volume (Figures 5d and e), when assessed 6 weeks after transplant of A2B5+ glioma TPCs derived from both GICL8 and GICL9, relative to SCR-infected cells (Supplementary Table S4). We then assessed the effect of PAR1 KD on the proliferation of glioma TPCs by analyzing the percentage of donor-derived TPCs co-expressing the mitotic marker Ki67. The A2B5+ TPCs derived from GICL8 and GICL9, whether transduced with PAR1 KD1 or KD2 lentiviruses, exhibited significantly lower Ki67-defined mitotic indices than those transduced with SCR control viruses (Figures 5f and g; Supplementary Table S4).

PAR1 silencing suppresses the in vivo growth of TPC-derived tumors and prolongs survival Effects of PAR1 silencing on the in vivo expansion of A2B5+ GICLs derived from two different GBM (28 000 cells per animal, n=5–6 mice per group), 4 weeks after transduction with PAR1 knockdown (KD) lentiviruses, compared with scrambled lentivirus (SCR) and control (CT) untransduced cells. (a) Hematoxylin-eosin stained sections of xenografts following intracranial implantation of A2B5+ TPCs. (b–g) Graphs representing the stereological analysis of the tumor extension, measured along the antero-posterior axis of xenograft mice brain (b, c); tumor volume (d, e); and proliferation of glioma TPCs as shown by the number of xenografted cells stained with the anti-human nuclei antigen (HNA) co-expressing the mitotic marker Ki67 (f, g), demonstrating a prominent inhibitory effect of PAR1 silencing on the tumorigenicity and mitotic activity of glioma A2B5+ TPCs relative to SCR and CT cells. Means±s.e.m. P-values calculated using one-way ANOVA (P<0.0001) followed by Tukey post-hoc comparisons with *P <0.05; **P<0.01; ***P<0.001. Post-hoc comparisons between cells transduced with PAR1 KD shRNAi, compared with those transduced with SCR shRNAi untransduced CT cells are illustrated by orange and black stars, respectively. (h–i) Kaplan–Meier curves show an increase in median survival of mice bearing intracranial glioma TPCs transduced with PAR1 shRNA relative to SCR and CT mice. Log-rank analysis, (P<0.0001) followed by pairwise comparison between all groups with **P<0.01 and ***P<0.001.
Pharmacological inhibition of the PAR1/thrombin system suppressed the expansion and migration of A2B5+ glioma TPCs in vitro
We next asked whether pharmacological inhibition of PAR1 could slow the growth of glioma A2B5+ TPCs. The pleotropic effects of PAR1 activation, in particular those associated with thrombogenesis, have prompted the development of a number of selective PAR1 antagonists.16 Among these, the pyrroloquinazoline SCH-79797, first developed as an anti-angiogenic agent,26, 27 induced a significant, dose-dependent inhibition of in vitro expansion by A2B5+ TPCs (P<0.0001), compared with matched cultures treated with vehicle alone (dimethylsulfoxide, DMSO) (Figures 6a and b). We repeated this experiment using the himbacine-derived PAR1 antagonist SCH-530348 (vorapaxar), and found that it too significantly inhibited the growth of glioma TPCs, also in a dose-dependent manner (P<0.0001). In particular, at concentrations⩾5 μm, SCH-530348 potently reduced the growth of A2B5+ TPCs derived from both GICL8 and GICL9 (Figures 6c and d). We next asked if PAR1 inhibition might have a direct effect on the migration of TPCs. To that end, A2B5+ TPCs were seeded at 4000 cells per well into laminin-pretreated (3 μg/cm2) 96-well plates, and a day later switched into media containing escalating concentrations of SCH79797 (0.01–0.1 μm), SCH530348 (1–10 μm), or control vehicle (0.1% DMSO) media. The cells were exposed to these agents for 24 h, then imaged by time-lapse microscopy. Glioma TPCs derived from both GICL8 and GICL9 displayed dose-dependent decreases in their rates of migration in response to both SCH79797 (P<0.0001), starting at 0.01 μm (Figures 6e and f), and SCH530348, starting at ⩾1 μm for GICL9 (P<0.0001), and ⩾10 μm for GICL8 (P=0.0004) (Figures 6g and h). Thus, pharmacological inhibition of PAR1 by each of two structurally distinct agents substantially suppressed both the in vitro expansion and migration of two distinct lines of A2B5+ glioma TPCs.

Pharmacological inhibition of PAR1 impedes glioma TPC expansion and migration in vitro. Effects of the specific PAR1 inhibitors SCH79797 and SCH530348 on the in vitro expansion (a–d) and migration (e–h) of A2B5+ GICL derived from GBMs. Dose-dependent growth of GBM-derived TPCs was measured by counting the number of cells 4 days after administration of SCH79797 (a, b) and SCH530348 (c, d) relative to highest concentration of vehicle control (DMSO). P-values were calculated using one-way ANOVA with repeated measures followed by Tukey post-hoc comparisons with *P<0.05; **P<0.01; ***P<0.001. Results were obtained from three independent experiments for each cell line. Migration was measured by assessing the average cell speed of A2B5+ GICLs in a dose-dependent manner, 24 h after administration of SCH79797 (e, f) and SCH530348 (g, h), relative to 1% DMSO vehicle control solution. P-values were calculated using Kruskal–Wallis test followed by Dunn’s multiple comparison test post-hoc comparisons with **P<0.01; ***P<0.001. Means±s.e.m.
To investigate the potential mechanism of PAR1 activation, we next examined the effect of pharmacologically inhibiting thrombin using dabigatran etexilate. Dabigatran is an Food and Drug Administration-approved oral anticoagulant that acts as a direct inhibitor of thrombin,28, 29 and which has been shown to inhibit the invasiveness of breast carcinoma cells in mice.30 To examine whether dabigatran might exert similar antineoplastic effects on glial neoplasms, cultured A2B5+ glioma TPCs were treated with escalating doses of dabigatran, then counted 4 days after drug administration. The dabigatran-treated cells exhibited significantly less in vitro expansion (Supplementary Figures S3A and B) and migration (Supplementary Figures S3C and D) than did untreated and vehicle-exposed A2B5+ TPCs, and did so in a dose-dependent manner. These data suggested that at least some of the effects of PAR1 suppression on tumor growth may be due to thrombin antagonism.
PAR1 silencing prolonged the survival of mice implanted with glioma TPCs
Given the strong inhibition of in vivo tumor growth by glioma GPCs transduced with PAR1 shRNAi, and the corresponding decrements in mitotic expansion and migration by glioma TPCs in response to both PAR1 KD and pharmacological inhibitors, we next asked if PAR1 suppression might improve the survival of mice following intracranial implantation of glioma TPCs. For this study of the effects of PAR1 inhibition on net survival, we used lentiviral shRNAi KD rather than pharmacological agents, since none of the currently available pharmacological inhibitors of PAR1 have reliable blood–brain barrier penetration, and the blood–brain barrier permeability of a variety of agents into gliomas and GBMs is highly variable. To therefore validate PAR1 as an appropriate target for therapeutic inhibition, while setting aside the issues of blood–brain barrier permeability of candidate PAR1 antagonists, we used a genetic strategy for PAR1 inhibition. In particular, we transduced A2B5+ glioma TPCs derived from both lines GICL8 and GICL9 with lentiviral PAR1 shRNAi before their implantation into adult immunodeficient NOG mice, and then allowed these mice, or their SCR-transduced controls, to survive until moribund. We found that the mice engrafted with PAR1 KD TPCs derived from both TPC lines, GICL8 and GICL9, lived significantly longer than their counterparts transplanted with control TPCs that were either untreated (CT), or transduced with scrambled shRNA (Figures 5h and i). Thus, PAR1 inhibition yielded a prolongation of survival that accorded with the inhibition of glioma expansion noted both in vitro and in vivo as a result of PAR1 KD.
Discussion
We had previously noted that PAR1 was differentially upregulated by A2B5-defined TPCs isolated from primary gliomas at every stage of glioma development, including the earliest stages of gliomagenesis. On that basis, in the present study we asked whether PAR1, its encoded protein and its dependent signal effectors might specifically contribute to the growth and malignant spread of the tumor-initiating progenitor cells of malignant gliomas. We found that this was indeed the case, in that both genetic and pharmacological inhibition of PAR1 suppressed the expansion and malignant spread of A2B5-defined glioma TPCs.
PAR1 is but one of four members of the PAR family (PARs 1–4), a group of G-protein coupled receptors that share a unique mechanism of activation, via the proteolytic cleavage of their N-terminal extracellular domains rather than ligand binding. This cleavage event triggers multiple signaling pathways, and a panoply of cell type-specific responses.31, 32 PAR1 is the prototype of this family, and is the principal thrombin-activated receptor in most cell types; it is expressed by a variety of human cancers.13, 17, 33 Thrombin is a serine protease that activates several factors in the clotting cascade, and produces the enzymatic cleavage of fibrinogen to fibrin.15 Patients with GBM are frequently coagulopathic, with nearly 30% suffering from venous thromboembolism; GBM patients comprise one of the highest risk populations for pathological thrombosis in all of medical and surgical practice.34 Indeed, intravascular thrombosis is observed in over 90% of histological samples of GBM.35
On that basis, it has been proposed that intra-tumor thrombosis and its associated local hypoxia may stimulate the production of factors contributing to malignant progression, in tandem with the remodeling of the tumor microenvironment and cancer stem cell compartment.20, 34, 36, 37 In addition, a prominent role of tissue factor (TF) pathway has been suggested, through its role in thrombogenesis and coagulation cascade signaling through PAR1 activation.20, 34, 36, 37 Accordingly, PAR1 activation has been associated with the growth and metastasis of tumor cells, as well as with accentuated angiogenesis and drug resistance in epithelial malignancies.17, 38, 39 PAR1 levels have been negatively correlated with survival in breast, lung, gallbladder, melanoma and gastric cancers.40, 41, 42, 43, 44 In gliomas, PAR1 expression and malignant association has been demonstrated in both primary tumors and cell lines.13, 20, 21, 22, 23
Yet despite the wealth of data associating PAR1 expression with malignant potential across a variety of somatic cancers, few studies have evaluated the specific role of PAR1 in tumor-initiating cells,36, 45 and none have done so in glial malignancies. We found that A2B5-defined glioma-initiating cells indeed differentially overexpress PAR1, and that the levels of PAR1 are directly related to tumor expansion, both in vitro and in vivo. We then demonstrated that both the specific KD of PAR1 by lentiviral shRNAi, and by the selective PAR-1 antagonists SCH-79797 and SCH-530348, substantially impeded the growth and self-renewal of GBM-derived TPCs in vitro. Most importantly, we noted that PAR1 KD resulted in significantly diminished tumor growth in vivo, while significantly increasing the survival of glioma TPC-transplanted animals. Together, these data suggested a critical, phenotypically specific role for PAR1 in the biology of glioma tumor-initiating progenitor cells, and further indicated that PAR1 inhibition might impede the progression or maintenance of malignant gliomas.
PAR1 is a G-protein coupled receptor, which may signal through a number of pathway intermediates.16, 17, 38 Apart from thrombin, other coagulation enzymes as well as tumor-derived proteases, that include the anti-coagulant-activated protein C, plasmin and the matrix metalloprotease-1 can also activate PAR1 and elicit PAR1-dependent signaling in a thrombin-independent manner.16, 46 PARs can also activate alternative parallel pathways, and signal through the transactivation of other receptors, most notably the sphingosine-1-phosphate receptor, the activation of which has been implicated in neural stem cell expansion.31, 47, 48 Indeed, PAR1 activation has been found to promote the growth of both colon and renal cell carcinomas by transactivating EGFR,49, 50 a mechanism that might prove significant in EGFR-overexpressing glial malignancies. In that regard, we noted that F2R/PAR1 was enriched in the classical (CL) subtype of GBM, and correlated with both EGFR amplification and reduced PTEN/CDKN2A copy number, in accord with previous reports.51
The robust suppression of glioma growth occasioned by KD of PAR1 gene expression in tumor-initiating cells strongly suggests the viability of targeting PAR1-dependent signaling and intermediates thereof as a therapeutic strategy in glioma, specifically so in those for which the A2B5-defined phenotype is the dominant tumor-initiating phenotype. Furthermore, the anti-angiogenic effects of PAR1 antagonism, and the disruption of the tumor stem cell microenvironment resulting from abrogating PAR1-dependent interactions of the microvasculature with glioma progenitor cells,22 may serve to further potentiate the antineoplastic effects of PAR1 inhibition. That said, the attractiveness of PAR1 as a therapeutic target is tempered by its expression by normal astrocytic and neuronal populations alike,52 as well as by resident neural stem cells.53, 54 Although tumor cells, and especially the tumor-initiating fraction thereof, selectively overexpress the receptor, its lower level basal expression by other normal cells of the central nervous system introduces the possibility of dose-dependent off-target effects that will mandate cautious titration in any therapeutic efforts targeting PAR1 itself. In that regard, those PAR1-targeted small molecules now available for clinical use, such as the novel direct PAR1 antagonist vorapaxar, have limited brain–barrier penetrance, further limiting current options for targeting PAR1-overexpressing TPCs. Nonetheless, the strong antineoplastic effects noted by pharmacological PAR1 inhibition in vitro, and by PAR1 KD both in vitro and in vivo, suggest that the development of blood–brain barrier penetrant PAR1 antagonists may be of great therapeutic value. Together, these data identify PAR1 as a substantial contributor to the expansion and growth of glioma-derived tumor-initiating cells, and hence to the development of malignant gliomas; as such, the abrogation of PAR1 signaling may prove a promising strategy for the treatment of malignant glioma.
Materials and methods
Tissue samples
Human tissue samples were obtained from glioma patients diagnosed in accord with WHO guidelines. Non-tumor epileptic tissue resections were used as controls. All samples were obtained from patients who consented to tissue use, under protocols approved by both the University of Rochester and Johns Hopkins University IRBs.
Cell preparation and isolation
Tissues were dissociated using papain, cultured in cell suspension plates, in serum-free media defined as DMEM/F12 media supplemented with N1, 20 ng/ml bFGF, EGF and PDGF-AA. Tumor-derived GPCs were isolated by tissue dissociation followed by A2B5-based cell sorting and cultured using described protocols.7, 8
Establishment of glioma initiating cell cultures
After dissociation, unsorted or A2B5+ cells derived from primary GBMs were further enriched for TPCs by culturing the cells in serum-free media supplemented with FGF, EGF and PDGF, at a clonal density (100 000 cells/ml), as described.7 Under these conditions, resultant cultures were defined as glioma TPCs when demonstrated (i) extensive self-renewal and sphere generation; (ii) expression of cancer stem cells markers including A2B5 and CD133; and (iii) tumorigenicity after transplantation into the brain of immunodeficient mice.
PAR1 expression analysis
Microarray analysis of PAR1/F2R gene was done following previously described procedure7 using RMA, R and packages from Bioconductor.55, 56, 57
Validation of PAR1 gene expression was assessed by quantitative real-time PCR analysis, using a 96-gene Taqman low-density array coupled with Taqman assay (Invitrogen, Carlsbad, CA, USA);58 as well as PAR1/F2R-directed PCR with reverse transcription using F2R-specific primers. The expression of each gene was normalized with 18S as an endogenous control. Relative gene expression was analyzed using the ΔΔCt approach and significance was determined by the t-test statistic with a 5% false discovery rate threshold. PAR1 protein expression was evaluated by flow cytometry analysis (FACS Aria, BD Biosciences, San Jose, CA, USA) of PAR1 stained cells (Clone WEDE15, 1/100).
Orthotopic transplantation and analysis
To assess in vivo tumorigenicity, immunodeficient mice (5–10 week old) were injected with TPCs and killed either 4–6 weeks thereafter, or once moribund for animals used for survival analysis. All transplantation procedures were reviewed and approved by the Rochester University Committee on Animal Resources. At the time of sacrifice, xenografted brains were cut as 14-μm cryostat sections, and donor cells identified by immunolabeling for human nuclear antigen, and the mitotic marker Ki67. Adjacent sections were stained with hematoxylin and eosin for histological assessment. Numerical, mitotic and volumetric analyses were estimated using the Optical Fractionator Program of Stereo Investigator (MBF Bioscience, Williston, VT, USA). In all determinations of animal use, group assignment, numbers, experimental design, randomization, blinding and description of results in this study, we attempted to follow the ARRIVE reporting guidelines.59
Generation, validation and assessment of PAR1 knockdown lentiviruses
A set of five lentiviral shRNAi vectors with distinct target sequences was purchased from Open Biosystems. We first validated PAR1-induced silencing constructs by transfection of multiple glioma cell lines, with subsequent qPCR and flow cytometry detection of PAR1 mRNA and protein expression, respectively. Viral production, cell transduction, validation of KD, and assessment of effects thereof on glioma cell proliferation are all described in the Supplementary Methods.
PAR1- and thrombin-specific inhibitors
The selective PAR1 antagonists SCH 79797 and SCH 530348 were purchased, respectively from Tocris Bioscience (Ellisville, MO, USA) and Axon medchem (Axon 1755) and dissolved in DMSO as per the manufacturer’s instructions. The chemical compound dabigatran etexilate (BIBR-1048) was purchased from Cedarlane (Burlington, NC, USA) (S2154-5 mg) and dissolved in DMSO following manufacturer’s instructions.
Self-renewal, cell proliferation, survival and cell migration assays
Self-renewal was assessed 14 days after dissociation of gliomaspheres into single cells and sorted to 96-well plates by FACS using the Automated Cell Deposit Unit (ACDU) at 1-80 cells per well followed by Extreme Limiting Dilution Analysis.60 Cell cycle analysis was performed using EdU (10 μm) administration followed by Click-it based Edu flow cytometry analysis (Invitrogen) following manufacturer’s instructions and as described.7 Cell survival was assessed using the Alexa-647 Annexin V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA), as described.7 Cell migration was assessed after seeding 4000 cells per well in a 96-well plate using time-lapse microscopy. Twenty-four hours after plating, cells were incubated with escalating concentrations of SCH 79797, SCH 530348, Dabigatran etexilate or vehicle for 24 h before imaging and cell tracking analysis.
Bioinformatic analysis
Gene expression and copy number alterations, along with their corresponding clinical data sets were obtained from TCGA (accessed at https://genome-cancer.soe.ucsc.edu/proj/site/hgHeatmap) for GBM multiforme and lower grade glioma sample populations. GBM data sets were as follows: (1) microarray gene expression (Agilent4502A_07_2, Agilent, Santa Clara, CA, USA, N=483); and (2) copy number alterations (gistic2, N=560). Gene expression subtype classification and G-CIMP status were obtained from corresponding GBM TCGA clinical data sets. Data sets for lower grade glioma were derived from RNA-seq gene expression (IlluminaHiSeq, Illumina, San Diego, CA, USA, N=468). Histological type and grade were obtained from corresponding TCGA clinical data sets. Pearson’s R coefficients and corresponding P-values for correlations between expression of PAR1 and copy number alterations were calculated in R software (https://www.r-project.com). The function heatmap.3 (https://gist.github.com/nachocab/3853004) was used to generate heatmaps with multiple stacked column identifiers.
Statistical analysis
All statistical analyses were performed using the GraphPad Prism (GraphPad Software, La Jolla, CA, USA) or R softwares (Vienna, Austria). The specific test used for each experiment to determine significance (P<0.05) is indicated in the text of the results and/or figure legends. in vitro data are representative of results obtained in at least three independent experiments. In all determinations of animal use, group assignment, numbers, experimental design, and description of results in this study, we attempted to follow the ARRIVE reporting guidelines
References
Gibson E, Monje M . Effect of cancer therapy on neural stem cells: implications for cognitive function. Curr Opin Oncol 2012; 24: 672–678.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–996.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444: 756–760.
Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006; 5: 67.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al. Identification of human brain tumour initiating cells. Nature 2004; 432: 396–401.
Venere M, Fine HA, Dirks PB, Rich JN . Cancer stem cells in gliomas: identifying and understanding the apex cell in cancer's hierarchy. Glia 2011; 59: 1148–1154.
Auvergne RM, Sim FJ, Wang S, Chandler-Militello D, Burch J, FanekY Al et al. Transcriptional differences between normal and glioma-derived glial progenitor cells identify a core set of dysregulated genes. Cell reports 2013; 3: 2127–2141.
Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G 2nd, Jiang L et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med 2003; 9: 439–447.
Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery 2008; 62: 505–514.
Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R et al. Non-stem cell origin for oligodendroglioma. Cancer Cell 2010; 18: 669–682.
Tchoghandjian A, Baeza N, Colin C, Cayre M, Metellus P, Beclin C et al. A2B5 cells from human glioblastoma have cancer stem cell properties. Brain Pathol 2010; 20: 211–221.
Coughlin SR . Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost 2005; 3: 1800–1814.
Elste AP, Petersen I . Expression of proteinase-activated receptor 1-4 (PAR 1-4) in human cancer. J Mol Histol 2010; 41: 89–99.
Bar-Shavit R, Turm H, Salah Z, Maoz M, Cohen I, Weiss E et al. PAR1 plays a role in epithelial malignancies: transcriptional regulation and novel signaling pathway. IUBMB Life 2011; 63: 397–402.
Coughlin SR . Thrombin signalling and protease-activated receptors. Nature 2000; 407: 258–264.
Garcia-Lopez MT, Gutierrez-Rodriguez M, Herranz R . Thrombin-activated receptors: promising targets for cancer therapy? Curr Med Chem 2010; 17: 109–128.
Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD . Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov 2012; 11: 69–86.
Villares GJ, Zigler M, Bar-Eli M . The emerging role of the thrombin receptor (PAR-1) in melanoma metastasis—a possible therapeutic target. Oncotarget 2011; 2: 8–17.
Zigler M, Kamiya T, Brantley EC, Villares GJ, Bar-Eli M . PAR-1 and thrombin: the ties that bind the microenvironment to melanoma metastasis. Cancer Res 2011; 71: 6561–6566.
Carneiro-Lobo TC, Lima MT, Mariano-Oliveira A, Dutra-Oliveira A, Oba-Shinjo SM, Marie SK et al. Expression of tissue factor signaling pathway elements correlates with the production of vascular endothelial growth factor and interleukin-8 in human astrocytoma patients. Oncol Rep 2014; 31: 679–686.
Kaufmann R, Patt S, Schafberg H, Kalff R, Neupert G, Nowak G . Functional thrombin receptor PAR1 in primary cultures of human glioblastoma cells. Neuroreport 1998; 9: 709–712.
Kuhn SA, Martin M, Brodhun M, Kratzsch T, Hanisch UK, Haberl H . Overexpression of protease-activated receptor type 1 (PAR-1) in glioblastoma multiforme WHO IV cells and blood vessels revealed by NCAM-assisted glioblastoma border labeling. Neurol Res 2014; 36: 709–721.
Zhang Y, Zhan H, Xu W, Yuan Z, Lu P, Zhan L et al. Upregulation of matrix metalloproteinase-1 and proteinase-activated receptor-1 promotes the progression of human gliomas. Pathol Res Pract 2011; 207: 24–29.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17: 98–110.
Lin N, Yan W, Gao K, Wang Y, Zhang J, You Y . Prevalence and clinicopathologic characteristics of the molecular subtypes in malignant glioma: a multi-institutional analysis of 941 cases. PLoS One 2014; 9: e94871.
Tsopanoglou NE, Maragoudakis ME . Inhibition of angiogenesis by small-molecule antagonists of protease-activated receptor-1. Semin Thromb Hemost 2007; 33: 680–687.
Zania P, Kritikou S, Flordellis CS, Maragoudakis ME, Tsopanoglou NE . Blockade of angiogenesis by small molecule antagonists to protease-activated receptor-1: association with endothelial cell growth suppression and induction of apoptosis. J Pharmacol Exp Ther 2006; 318: 246–254.
Lee CJ, Ansell JE . Direct thrombin inhibitors. Br J Clin Pharmacol 2011; 72: 581–592.
Stangier J . Clinical pharmacokinetics and pharmacodynamics of the oral direct thrombin inhibitor dabigatran etexilate. Clin Pharmacokinet 2008; 47: 285–295.
DeFeo K, Hayes C, Chernick M, Ryn JV, Gilmour SK . Use of dabigatran etexilate to reduce breast cancer progression. Cancer Biol Ther 2010; 10: 1001–1008.
Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther 2011; 130: 248–282.
Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R . Proteinase-activated receptors. Pharmacol Rev 2001; 53: 245–282.
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A . PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005; 120: 303–313.
Perry JR . Thromboembolic disease in patients with high-grade glioma. Neuro-oncology 2012; 14: 73–80.
Brat DJ, Van Meir EG . Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest 2004; 84: 397–405.
Garnier D, Milsom C, Magnus N, Meehan B, Weitz J, Yu J et al. Role of the tissue factor pathway in the biology of tumor initiating cells. Thromb Res 2010; 125: S44–S50.
Milsom C, Magnus N, Meehan B, Al-Nedawi K, Garnier D, Rak J . Tissue factor and cancer stem cells: is there a linkage? Arterioscler Thromb Vasc Biol 2009; 29: 2005–2014.
Kaufmann R, Hollenberg MD . Proteinase-activated receptors (PARs) and calcium signaling in cancer. Adv Exp Med Biol 2012; 740: 979–1000.
Queiroz KC, Shi K, Duitman J, Aberson HL, Wilmink JW, van Noesel CJ et al. Protease-activated receptor-1 drives pancreatic cancer progression and chemoresistance. Int J Cancer 2014; 135: 2294–2304.
Du X, Wang S, Lu J, Cao Y, Song N, Yang T et al. Correlation between MMP1-PAR1 axis and clinical outcome of primary gallbladder carcinoma. Jpn J Clin Oncol 2011; 41: 1086–1093.
Eroglu A, Karabiyik A, Akar N . The association of protease activated receptor 1 gene -506 I/D polymorphism with disease-free survival in breast cancer patients. Ann Surg Oncol 2012; 19: 1365–1369.
Fujimoto D, Hirono Y, Goi T, Katayama K, Yamaguchi A . Prognostic value of protease-activated receptor-1 (PAR-1) and matrix metalloproteinase-1 (MMP-1) in gastric cancer. Anticancer Res 2008; 28: 847–854.
Ghio P, Cappia S, Selvaggi G, Novello S, Lausi P, Zecchina G et al. Prognostic role of protease-activated receptors 1 and 4 in resected stage IB non-small-cell lung cancer. Clin Lung Cancer 2006; 7: 395–400.
Massi D, Naldini A, Ardinghi C, Carraro F, Franchi A, Paglierani M et al. Expression of protease-activated receptors 1 and 2 in melanocytic nevi and malignant melanoma. Hum Pathol 2005; 36: 676–685.
Baumer N, Krause A, Kohler G, Lettermann S, Evers G, Hascher A et al. Proteinase-activated receptor 1 (PAR1) regulates leukemic stem cell functions. PLoS One 2014; 9: e94993.
Soh UJ, Dores MR, Chen B, Trejo J . Signal transduction by protease-activated receptors. Br J Pharmacol 2010; 160: 191–203.
Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R . Proteinase-activated receptors (PARs) - focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal 2013; 11: 86.
Zhao P, Metcalf M, Bunnett NW . Biased signaling of protease-activated receptors. Front Endocrinol 2014; 5: 67.
Arora P, Cuevas BD, Russo A, Johnson GL, Trejo J . Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 2008; 27: 4434–4445.
Bergmann S, Junker K, Henklein P, Hollenberg MD, Settmacher U, Kaufmann R . PAR-type thrombin receptors in renal carcinoma cells: PAR1-mediated EGFR activation promotes cell migration. Oncol Rep 2006; 15: 889–893.
Magnus N, Gerges N, Jabado N, Rak J . Coagulation-related gene expression profile in glioblastoma is defined by molecular disease subtype. J Thromb Haemost 2013; 11: 1197–1200.
Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR et al. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol 2004; 188: 94–103.
Feve M, Saliou JM, Zeniou M, Lennon S, Carapito C, Dong J et al. Comparative expression study of the endo-G protein coupled receptor (GPCR) repertoire in human glioblastoma cancer stem-like cells, U87-MG cells and non malignant cells of neural origin unveils new potential therapeutic targets. PLoS One 2014; 9: e91519.
Guo H, Zhao Z, Yang Q, Wang M, Bell RD, Wang S et al. An activated protein C analog stimulates neuronal production by human neural progenitor cells via a PAR1-PAR3-S1PR1-Akt pathway. J Neurosci 2013; 33: 6181–6190.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5: R80.
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP . Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003; 31: e15.
Talloen W, Clevert DA, Hochreiter S, Amaratunga D, Bijnens L, Kass S et al. I/NI-calls for the exclusion of non-informative genes: a highly effective filtering tool for microarray data. Bioinformatics 2007; 23: 2897–2902.
Smyth GK . Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3: 1–25.
Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG . Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010; 8: e1000412.
Hu Y, Smyth GK . ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009; 347: 70–78.
Acknowledgements
We thank Dr Mahlon Johnson for the histopathological validation of tumor stage, Dr Kevin Walter for arranging tissue donation and Shengtao Qu and Eric Franklin for technical assistance. This work was supported by the Dr Miriam and Sheldon G Adelson Medical Research Foundation, and by the New York State Stem Cell Research Program. Tissue collection, processing and studies on human tissue derived from Johns Hopkins University were supported by NIH R01NS070024.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Auvergne, R., Wu, C., Connell, A. et al. PAR1 inhibition suppresses the self-renewal and growth of A2B5-defined glioma progenitor cells and their derived gliomas in vivo. Oncogene 35, 3817–3828 (2016). https://doi.org/10.1038/onc.2015.452
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.452
- Springer Nature Limited
This article is cited by
-
Epigenetic modifiers either individually or in specific combinations impair viability of patient-derived glioblastoma cell line while exhibit moderate effect on normal stem cells growth
Investigational New Drugs (2023)
-
The intertwined fates of inflammation and coagulation in glioma
Mammalian Genome (2018)
-
Protease-activated receptor-1 inhibits proliferation but enhances leukemia stem cell activity in acute myeloid leukemia
Oncogene (2017)