
Abstract
Atypical teratoid/rhabdoid (AT/RT) tumor is a rare, highly malignant tumor of the central nervous system (CNS) most commonly found in children less than 5 years of age. Although the vast majority of cases are diagnosed in young children, there have been isolated case reports in adults. Since its histological appearance can be confused with other tumors, especially in adults, separating AT/RT from other neoplasms may be difficult. In many instances, a reliable diagnosis is not possible without demonstrating the lack of nuclear INI1 protein expression by immunohistochemical methods. The patients (three males and one female) ranged in age from 23 to 42 years (mean age, 32 years). Radiographically, two tumors were localized in the right fronto-parietal region, one was frontal and the other was found in the left temporal lobe. Varying degrees of hydrocephalus and heterogeneous enhancement were present on MRI. In all cases, diagnosis during intraoperative consultation and preliminary diagnosis was different from the final diagnosis after immunohistochemical analysis. Immunohistochemical staining showed that the tumor cells were positive for vimentin and reacted variably for keratin, epithelial membrane antigen (EMA), synaptophysin, neurofilament protein, CD34, and smooth muscle actin (SMA). All were negative for GFAP, S-100, desmin and CD99. Three of the four cases lacked nuclear expression of INI1. One patient is alive with no evidence of disease 17 years after the diagnosis. In adult examples of AT/RT, the diagnosis requires a high index of suspicion, with early tissue diagnosis and a low threshold for investigation with INI1 immunohistochemistry to differentiate this entity from other morphologically similar tumors. Although the prognosis is dismal in pediatric population, long term survival is possible in adult AT/RT cases after surgery and adjuvant radiotherapy and chemotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Described in 1978 by Beckwith and Palmer [1], rhabdoid tumor has become a well-accepted clinicopathological entity. Since the original description of an aggressive renal tumor in very young children [2], tumors with rhabdoid features have been reported in many extra-renal sites. In 1985, Rorke et al. [3] first recognized this tumor in the brain under the term AT/RT to emphasize the variable combination of rhabdoid cells, epithelial cells, primitive neuro-ectodermal as well as mesenchymal components. Most of these tumors are situated in the posterior fossa, or less commonly supratentorially, and in an exceptional instance in the spinal cord [4]. They show a propensity to spread through the subarachnoid space, which is often noted at the time of diagnosis. Although AT/RT is most often diagnosed in children less than 5 years, in rare instances, it may occur in adults [5–13]. The pathological characteristic to display multiple lines of differentiation in varying proportions can cause confusion of AT/RT with other neoplasms, especially in adults.
AT/RTs are associated with deletion of chromosome 22q and inactivation of INI1/hSNF5 (HIV-integrase interactor 1 (INI1)/human homolog of Saccharomyces Cerevisiae sucrose-nonfermenting 5 (hSNF5), referred for simplicity as INI1) tumor suppressor gene on chromosome 22q11.2 [12, 14, 15]. Mutation or deletion of both copies of the INI1/hSNF5/gene is observed in approximately 70% of primary tumors [16]. An additional 20–25% of tumors are estimated to have reduced expression at the RNA or protein level, indicative of a loss-of-function event. Recent development of the INI1 immunohistochemical stain has been helpful in identifying tumors with chromosome 22 deletions [17]. Therefore, the absence of INI1 nuclear expression by the tumor cells was used in this study as one of the hallmark criteria to identify AT/RT, although one of the cases retained the expression of this protein.
The aim of this study is to call attention to the occurrence of AT/RT in adult patients. Four additional adult cases are presented and the clinicopathologic features are discussed in relation to other cases described in the adult population.
Materials and methods
Clinical data
Two patients (Cases 1 and 2) diagnosed at Georgetown University Medical Center (GUMC) are included in this report. The other two cases (Cases 3 and 4) were received in consultation by one of the authors (EJR) at the Armed Forces Institute of Pathology (AFIP). When available, the clinical presentation, neuroradiologic appearance and operative findings were abstracted from the patient records or patient families. Preoperative computed tomographic (CT) and/or magnetic resonance imaging (MRI) scans was available for one case, Case 1.
Histology and immunohistochemistry
All four surgical specimens were formalin-fixed, routinely processed, sectioned at 5 μm, and stained by the hematoxylin and eosin (H&E) method. The H&E slides of each case, including the two cases received in consultation, were reviewed and assessed for features including the general architecture, degree of cellularity, necrosis and mitoses.
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections using the Envision+ (Dako, Carpinteria, CA)—horseradish peroxidase system after the tissue was deparaffinized and rehydrated according to standard protocol. The following antibodies were used: polyclonal antibodies against S-100, and synaptophysin (purchased from Dako, Carpinteria, CA), and monoclonal antibodies against epithelial membrane antigen (EMA), neurofilament proteins (Nf), MIB-1, glial fibrillary acidic protein (GFAP), vimentin, smooth muscle actin (SMA), muscle specific actin (MSA), desmin, CD34, CD31 and Factor 8 (from Dako, Carpinteria, CA), CD99 (O-13) (from Signet; Dedham, MA), BAF 47 (INI1) (from BD Biosciences Pharmingen, San Jose, CA) and keratin (a cocktail of AE1/AE3, CAM 5.2, and 35βH11) (from Ventana Medical Systems, Tucson, AZ). In every case, formalin-fixed tissue was subjected to heat-induced antigen retrieval.
Case descriptions
Case 1
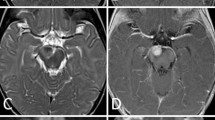
This 23-year-old-male presented with a history of two episodes of seizure activity occurring on the same day. In addition, he complained of a continuous headache for 2 weeks prior to the onset of seizures. On admission, MRI revealed a 3.6 × 3.5 × 3.2 cm heterogeneously enhancing mass with surrounding edema within the posterior left temporal lobe (Fig. 1). Craniotomy was performed and the mass was removed. The specimen was sent to surgical pathology, where a portion was used for frozen section diagnosis and the remaining tissue was submitted for permanent sections. The frozen section diagnosis was high grade malignant tumor with necrosis consistent with glioblastoma multiforme. However, a diagnosis of AT/RT was made after extensive immunohistochemical analysis. The patient had a stable post-operative course and was discharged after a week with a scheduled follow up appointment. Currently the patient is being followed at another institution, where he received adjuvant radiotherapy and chemotherapy after the surgery and is in remission 30 months after the diagnosis.

T2 weighted axial MRI shows a left posterior temporal mass showing with heterogenous signal intensity and surrounding vasogenic edema
Case 2
Twenty-five-year old female with 1 year history of headaches and who was being treated for migraine presented with bilateral gaze palsy. A CT scan showed a 5 cm right frontal brain tumor. The patient underwent a craniotomy and the tumor was completely excised. A diagnosis of rhabdoid tumor was made. Because of the diagnosis, no adjuvant therapy was given initially. However, the tumor recurred 2 years later and she received gamma knife radiosurgery. Over the next 5 years, she experienced five more episodes of recurrence in the same tumor bed and additionally in the left frontal region in the last episode. The recurrent tumors were resected completely. In all these tumors the histology showed similar features. She received adjuvant radiotherapy and later chemotherapy after the last recurrence at another institution. No recurrence has occurred in the last 10 years and she is alive 17 years after the diagnosis.
Case 3
A previously healthy 42-year-old-male presented to a tertiary care provider with dysphasia and right sided weakness. On admission he was found to have a 1.5 × 1.0 × 1.0 cm mass within the right fronto-parietal region. Because of the location of the tumor, a complete resection was not obtained. The specimen was sent to surgical pathology where the diagnosis of gemistocytic astrocytoma with the differential diagnosis of AT/RT was made. After surgery, the patient received adjunct radiotherapy and chemotherapy. The patient is alive 18 months after the diagnosis.
Case 4
This 37-year-old-male presented with a history of new-onset seizures that prompted referral to a tertiary care provider. Imaging studies revealed a right hemisphere mass. The contributor’s diagnosis (a recent case from abroad) was glioblastoma. AT/RT was considered as a differential diagnosis. There is no further clinical data available.
Results
Clinical features
Table 1 summarizes the clinical features of the four cases. The single female and three male patients ranged in age from 23 years to 42 years. All four tumors were located in the cerebrum. The cases presented with seizure (case 1 and 4), headache and gaze palsy (case 2) and dysphasia and right-sided weakness (case 3).
Pathological features
At low magnification, sections from the first case revealed a mixed population of cells comprising variable numbers of rhabdoid cells, primitive-appearing small blue cells, and spindle shaped cells having sarcomatoid appearance but on higher magnification the cells in all areas had rhabdoid features with eosinophilic cytoplasm (Fig. 2a–d). The second case had spindle cells without a discrete pattern or trabecular growth pattern with epithelioid cells with eosinophilic cytoplasm as well as typical rhabdoid morphology. The other two cases were relatively homogenous with rhabdoid cells and spindle cells and vague nested growth pattern (Fig. 3a–c). In none of the cases, a fascicular growth pattern was observed to consider a gliosarcoma. A common feature of these tumors, the rhabdoid cell, was characterized by discrete cell borders and eccentric reniform nuclei, often with prominent nucleoli and eosinophilic cytoplasm. Some of these cells contained intracytoplasmic hyaline globular inclusions adjacent to the nucleus. In some cases, the neoplastic cells exhibited oligodendroglial-like features due to artifactual tumor cell vacuolation. Necrosis and mitotic figures were present in all cases.

Representative morphologic features from formalin fixed paraffin embedded sections (case 1). Lower magnification shows variegated appearance with small blue cell areas with geographic necrosis (arrow) or sarcomatous-looking and rhabdoid areas (a) and (b) (hematoxylin and eosin, ×40, each). Higher magnification of the sarcomatous looking areas (c) and rhabdoid cells (arrow) (d) (hematoxylin and eosin ×400, each)

Representative morphologic features from other cases. Higher magnification shows epithelioid cells with vague trabecular (case 2) (a), or nested growth pattern (case 3) (b), and rhabdoid cells with hyaline globular inclusions (arrow) (case 4) (c) (hematoxylin and eosin ×400, each)
Immunohistochemistry
Table 2 summarizes the immunohistochemical results. The rhabdoid cells consistently displayed immunoreactivity for vimentin (Fig. 4a). They stained variably for EMA, keratin, SMA (Fig. 4b), and less frequently for synaptophysin, MSA and CD34. The primitive-appearing small blue cells and sarcomatous areas in the first case stained similar to rhabdoid cells. The rhabdoid component and all other tumor cells were negative for CD99, S-100, GFAP, and desmin. In all our cases, the tumors showed a high proliferation index, with MIB1 up-to 80% in one example (Fig. 4c). Three cases showed lack of nuclear staining for BAF 47 (INI1) antibody but showed normal nuclear positivity in adjacent control tissues (Figs. 4d, 5a–b). Case 4 was stained for INI1, but it was negative for other specific markers including Factor 8 (Fig. 5c–d). Additional cytogenetic, mRNA or protein analyses were not performed.

By immunohistochemistry (case 1), the tumor cells are positive for keratin (a) and SMA (b). Mib-1 shows a high proliferative index (c). The tumor cells are negative for INI1 (arrow) (D) (×400, each)

By immunohistochemistry, the tumor cells are negative for INI1 (cases 2 and 3) (a and b), whereas tumor cells in case 4 is positive for INI1 (arrows) (c) and negative for Factor 8 (D) (×400, each)
Discussion
In 1992, Horn et al. was the first to recognize AT/RT, as a malignant intracranial rhabdoid tumor in an adult patient [5]. Later, 16 additional cases were described in adults as malignant rhabdoid tumor of CNS or AT/RT in the English literature [4, 6–8, 11–13, 18–22]. Raisanen et al. described three adult cases where the diagnosis was supported by detection of 22q11.2 deletion, INI1/hSNF5 (INI1) mutation and/or loss of INI1 protein expression [12]. Of note, two patients were female, ages 20 and 31 and one was male, age 45 with two tumors occurring in the sella or sellar region and one in the cerebellum, respectively. More recently, Zarovnaya described a 43-year old woman with AT/RT in the cervical spinal cord that recurred throughout the neuraxis secondary to leptomeningeal spread [4]. Originally diagnosed as a rhabdoid meningioma, in retrospective review, the tumor cells were found to exhibit monosomy 22 with loss of expression of the INI1 gene in 22q11.2 by molecular studies and absence of INI1 gene expression by immunohistochemical methods. Table 3 summarizes the adult cases reported in the English literature with a diagnosis of AT/RT or rhabdoid tumor.
The pathogenesis of AT/RTs is not very well understood, but a pluripotent cell with the ability to diverge into epithelial and mesenchymal cells has been suggested. In earlier reports, rhabdoid tumors were described as rhabdomyosarcomatoid tumors due to their morphologic resemblance to rhabdomyoblasts [1]. However, ultrastructural studies did not reveal myoblast differentiation.
In all four patients described in this report, the tumors contained sheets of typical rhabdoid cells, and morphologically were consistent with AT/RT. As noted, the rhabdoid cells have abundant eosinophilic cytoplasm with discrete cell border and eccentric nuclei, prominent nucleoli and hyaline globular inclusion adjacent to the nuclei similar to the observation by other investigators (Fig. 2). The hyaline globular inclusions are known to be aggregates of intermediate filaments identified as vimentin by immunohistochemistry. Similar to our cases, variable mitotic activity, necrosis, hemorrhage and cystic changes have also been described [23, 24] as seen in our cases. Owing to the mixed population of cells or divergent differentiation, AT/RTs show polyphenotypic immunoreactivity with different immunohistochemical markers. The rhabdoid cells usually express vimentin and EMA. Also, there is variable expression of GFAP, S-100 and cytokeratin. Myogenous differentiation, when present, is usually observed in the small cell component [19, 25]. In the present cases, the rhabdoid cells expressed vimentin consistently, whereas in some cases they showed variable expression of keratin, EMA, SMA, MSA, synaptophysin, and neurofilament. All four cases were negative for S-100, GFAP, desmin and CD99. The lack of expression of CD99 as well as S-100 and EMA excludes the possible differential diagnosis of primitive neuroectodermal tumor (PNET) and rhabdoid meningioma, respectively, which may morphologically resemble AT/RT.
Molecular genetic studies have shown that majority of AT/RTs show deletion and/or mutation involving the INI1/hSNF5 tumor suppressor gene on chromosome 22q11 [12, 14, 15]. Loss of INI1/hSNF5 gene has been shown to affect the actin cytoskeleton which may explain the rhabdoid morphology seen in AT/RTs [26]. The recently developed INI1 antibody has been introduced as a useful marker to show the lack of expression of INI1 protein in tumor cells of AT/RT [17, 27]. The deletion can also be detected by fluorescent in-situ hybridization (FISH) although, 20–30% of tumors maintain a normal dosage of chromosome 22q suggesting that the absence of deletion should not exclude the consideration of ATRT. This could explain the positive INI1 reactivity observed in one of our cases (Case 4), where the diagnosis of AT/RT was strongly considered by the morphologic features and also other possible differential diagnoses were excluded by immunohistochemistry. In the workshop on childhood AT/RT reviewed by Biegel et al. [28], the molecular and cytogenetic findings in a series of 76 rhabdoid tumors were detailed. The authors reported that AT/RTs demonstrate monosomy 22 at a higher frequency than renal or extrarenal rhabdoid tumors and that extrarenal rhabdoid tumors had a very high rate of homozygous deletions of the entire INI1 gene. The loss of one allele with concomitant mutation in the remaining copy of INI1 gene was reported in 42 of 76 rhabdoid tumors and compound heterozygous mutations or splicing mutations in 4 cases. Approximately 10% of the cases had no coding sequence mutations but had decreased expression levels of INI1 by RT-PCR or Western blot analysis. Approximately 15% of tumors had no alteration of the INI1 gene at the DNA, RNA or protein levels. Therefore, it is possible that patients with monosomy 22 at an early age may not develop AT/RT until mutation occurs in the remaining allele latter in life.
INI1/hSNF5 is a component of SWI/SNF chromatin remodeling complex functioning as a tumor suppressor by positively regulating transcription of a particular set of eukaryotic genes including c-myc involved in differentiation and apoptosis [28–31]. Kalpana et al. [32] first isolated INI1 protein, in-vitro, using the yeast two-hybrid system by its interaction with HIV-1 integrase and named it Integrase interactor 1. INI1 protein (also called BAF47) forms functional core of SWI/SNF complex along with BRM (Brahma) or BRG-1 (Brahma-related gene 1) and BAF(BRG-1-Associated Factor)155 and BAF170. These other components may also show mutation in association with INI1 protein loss. Wong et al. [33] have identified mutation in the core ATPase subunit of BRG-1, along with INI1 protein loss, in approximately 10% of cell lines studied. The impact of lack of BRG-1 in tumorigenesis such as retinoblastoma has previously been described. Knudsen et al. [34] have shown that, retinoblastoma tumor suppressor protein inactivation is required for tumor progression and that BRG-1 is required for RB to exert antimitogenic activity [35].
The localization and behavior in adults appears to differ somewhat from childhood cases. The most common site for this tumor in children is posterior fossa and the overall prognosis of AT/RTs is rather bleak [36]. In adults, the tumor is seen mostly in cerebrum and survival based on the current cases and literature review averaged 38.7 months (Tables 1 and 3). Of the 21 adult patients, 11 patients survived more than 12 months and one patient is still alive 17 years after the diagnosis after complete resection of tumors and adjuvant multimodal treatment. These results suggest that depending on localization, AT/RT may be less aggressive in adults. Recent advances using aggressive therapy might have also contributed to a better outcome [37]. One of the major concerns, leptomeningeal spread, which can be seen very early in the disease, appears to respond to intensive adjunctive chemo-and/or radiation therapy and this may change the natural history. The presence of necrosis and high proliferation index (Mib1) correlates with the aggressiveness of the tumor. The Mib 1 index in one of our cases was 80%. Allen et al. [38] in their case report have suggested that transformation to an aggressive tumor in the clinical course of a stable brain tumor may be associated to acquisition of INI1 gene mutation with corresponding rhabdoid morphologic change. Of note, Litman et al. [39] have reported the appearance of rhabdoid tumor in the lung and ileum 20 years after radiation treatment for Wilms’ tumor. Based on the location of the tumor within the radiation field, the authors suggested that, in some cases, AT/RT may be radiation induced.
Conclusions
It is important to consider ATRT in the differential diagnosis of aggressive intracranial neoplasms with ambiguous morphology. The IN1 antibody can be a useful tumor marker in such cases. The association of INI1 protein loss with other proteins in the SWI/SNF chromatin remodeling complex, such as BRG-1, is important to better understand the pathogenesis and aid the search for therapy. Identifying the triggering factors that lead to mutation or deletion of INI1/hSNF5 tumor suppressor gene later in life is also crucial to our understanding of this entity. Further investigations that address the prevalence of the lack of expression of INI1 protein in adult AT/RT and other primary and metastatic brain tumors are needed to assess the specificity and reliability of this technique. Finally, the identification of a specific genetic abnormality is beneficial in searching for gene directed therapy and improving the prognosis of these aggressive and fatal tumors.
References
Beckwith JB, Palmer NF (1978) Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer 41:1937–1948
Haas JE, Palmer NF, Weinberg AG, Beckwith JB (1981) Ultrastructure of malignant rhabdoid tumor of the kidney. A distinctive renal tumor of children. Hum Pathol 12:646–657
Rorke LB, Packer RJ, Biegel JA (1996) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85:56–65
Zarovnaya EL, Pallatroni HF, Hug EB, Ball PA, Cromwell LD, Pipas JM, Fadul CE, Meyer LP, Park JP, Biegel JA, Perry A, Rhodes CH (2007) Atypical teratoid/rhabdoid tumor of the spine in an adult: case report and review of the literature. J Neurooncol 84:49–55
Horn M, Schlote W, Lerch KD, Steudel WI, Harms D, Thomas E (1992) Malignant rhabdoid tumor: primary intracranial manifestation in an adult. Acta Neuropathol (Berl) 83:445–448
Fisher BJ, Siddiqui J, Macdonald D, Cairney AE, Ramsey D, Munoz D, Del Maestro R (1996) Malignant rhabdoid tumor of brain: an aggressive clinical entity. Can J Neurol Sci 23:257–263
Ashraf R, Bentley RC, Awan AN, McLendon RE, Ragozzino MW (1997) Implantation metastasis of primary malignant rhabdoid tumor of the brain in an adult (one case report). Med Pediatr Oncol 28:223–227
Arrazola J, Pedrosa I, Mendez R, Saldana C, Scheithauer BW, Martinez A (2000) Primary malignant rhabdoid tumour of the brain in an adult. Neuroradiology 42:363–367
Kawaguchi T, Kumabe T, Watanabe M, Tominaga T (2004) Atypical teratoid/rhabdoid tumour with leptomeningeal dissemination in an adult. Acta Neurochir (Wien) 146: 1033–1038 (discussion 1038)
Lutterbach J, Liegibel J, Koch D, Madlinger A, Frommhold H, Pagenstecher A (2001) Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol 52:49–56
Pimentel J, Silva R, Pimentel T (2003) Primary malignant rhabdoid tumors of the central nervous system: considerations about two cases of adulthood presentation. J Neurooncol 61:121–126
Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A (2005) Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol 15:23–28
Rezanko T, Tunakan M, Kahraman A, Sucu HK, Gelal F, Akkol I (2006) Primary rhabdoid tumor of the brain in an adult. Neuropathology 26:57–61
Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59:74–79
Packer RJ, Biegel JA, Blaney S, Finlay J, Geyer JR, Heideman R, Hilden J, Janss AJ, Kun L, Vezina G, Rorke LB, Smith M (2002) Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24:337–342
Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8: 3461–3467
Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28:644–650
Byram D (1999) Regarding Weiss et al., IJROBP 41:103–109; 1998. Int J Radiat Oncol Biol Phys 45:247
Sugita Y, Takahashi Y, Hayashi I, Morimatsu M, Okamoto K, Shigemori M (1999) Pineal malignant rhabdoid tumor with chondroid formation in an adult. Pathol Int 49:1114–1118
Bruch LA, Hill DA, Cai DX, Levy BK, Dehner LP, Perry A (2001) A role for fluorescence in situ hybridization detection of chromosome 22q dosage in distinguishing atypical teratoid/rhabdoid tumors from medulloblastoma/central primitive neuroectodermal tumors. Hum Pathol 32:156–162
Kachhara R, Retnam TM, Kumar S, Nair S, Bhattacharya RN, Krishnamoorthy T, Radhakrishnan VV (2003) Rhabdoid tumor of the thalamus. Neurol India 51:273–274
Erickson ML, Johnson R, Bannykh SI, de Lotbiniere A, Kim JH (2005) Malignant rhabdoid tumor in a pregnant adult female: literature review of central nervous system rhabdoid tumors. J Neurooncol 74:311–319
Bhattacharjee M, Hicks J, Dauser R, Strother D, Chintagumpala M, Horowitz M, Cooley L, Vogel H (1997) Primary malignant rhabdoid tumor of the central nervous system. Ultrastruct Pathol 21:361–368
Parmar H, Hawkins C, Bouffet E, Rutka J, Shroff M (2006) Imaging findings in primary intracranial atypical teratoid/rhabdoid tumors. Pediatr Radiol 36:126–132
Lee MC, Park SK, Lim JS, Jung S, Kim JH, Woo YJ, Lee JS, Kim HI, Jeong MJ, Choi HY (2002) Atypical teratoid/rhabdoid tumor of the central nervous system: clinico-pathological study. Neuropathology 22:252–260
Medjkane S, Novikov E, Versteege I, Delattre O (2004) The tumor suppressor hSNF5/INI1 modulates cell growth and actin cytoskeleton organization. Cancer Res 64:3406–3413
Perry A, Fuller CE, Judkins AR, Dehner LP, Biegel JA (2005) INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol 18:951–958
Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, Weissman B, Smith M (2002) The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62:323–328
Lee CH, Murphy MR, Lee JS, Chung JH (1999) Targeting a SWI/SNF-related chromatin remodeling complex to the beta-globin promoter in erythroid cells. Proc Natl Acad Sci USA 96:12311–12315
Muchardt C, Yaniv M (1999) The mammalian SWI/SNF complex and the control of cell growth. Semin Cell Dev Biol 10:189–195
Phelan ML, Sif S, Narlikar GJ, Kingston RE (1999) Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 3:247–253
Kalpana GV, Marmon S, Wang W, Crabtree GR, Goff SP (1994) Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 266:2002–2006
Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, Ghaffari S, Iliev D, Penn B, Woodland AM, Smith R, Salada G, Carillo A, Laity K, Gupte J, Swedlund B, Tavtigian SV, Teng DH, Lees E (2000) BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res 60:6171–6177
Knudsen ES, Buckmaster C, Chen TT, Feramisco JR, Wang JY (1998) Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev 12:2278–2292
Strobeck MW, Knudsen KE, Fribourg AF, DeCristofaro MF, Weissman BE, Imbalzano AN, Knudsen ES (2000) BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci USA 97:7748–7753
Parham DM, Weeks DA, Beckwith JB (1994) The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohistochemistry or electron microscopy. Am J Surg Pathol 18:1010–1029
Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Walter AW, Rorke LB, Biegel JA (2004) Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22:2877–2884
Allen JC, Judkins AR, Rosenblum MK, Biegel JA (2006) Atypical teratoid/rhabdoid tumor evolving from an optic pathway ganglioglioma: case study. Neuro Oncol 8:79–82
Litman DA, Bhuta S, Barsky SH (1993) Synchronous occurrence of malignant rhabdoid tumor two decades after Wilms’ tumor irradiation. Am J Surg Pathol 17:729–737
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Makuria, A.T., Rushing, E.J., McGrail, K.M. et al. Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol 88, 321–330 (2008). https://doi.org/10.1007/s11060-008-9571-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-008-9571-z