
Abstract
Differentiating oligodendroglioma from extraventricular neurocytoma by conventional light microscopy alone can present a diagnostic challenge. We report pathologic findings of an unusual spinal cord tumor from a 33-year-old male patient which showed hybrid features of oligodendroglioma and extraventricular neurocytoma. Magnetic resonance imaging (MRI) showed an enhancing intramedullary mass in the cervicothoracic region (C7 through T6). Histologic examination revealed a clear cell neoplasm containing ganglion-like cells and calcifications, prompting the differential diagnosis of oligodendroglioma and extraventricular neurocytoma. The immunohistochemical analysis disclosed neural differentiation of the neoplastic cells with strong synaptophysin and neurofilament staining consistent with extraventricular neurocytoma, as well as strong S-100 and glial fibrillary acidic protein (GFAP) expression. Molecular studies with fluorescent in situ hybridization (FISH) revealed chromosome 1p/(partial) 19q deletions, a finding commonly observed in oligodendroglioma. The proliferation index (using antibody MIB1) of the tumor was ∼30%. The morphologic findings and these results strengthen the hypothesis that these tumors may share a common progenitor cell, which has also been observed by others. Because there are differences in patient management and long-term prognosis, it is important to attempt to distinguish between oligodendroglioma and neurocytoma. This unusual case and similar rare reported cases support the need to reclassify tumors showing pathologic features common to both neurocytoma and oligodendroglioma as a unique entity, while the effort continues to identify the cell of origin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neoplasms composed of oligodendrocyte-like cells often pose a diagnostic challenge. In the intramedullary spinal cord compartment, the principal differential diagnosis includes oligodendroglioma and extraventricular neurocytoma. Although immunohistochemical and ultrastructural investigation can be helpful in such cases, overlapping features may prove difficult to distinguish between the two entities. More recently, the application of molecular diagnostic techniques to certain types of primary central nervous system (CNS) tumors has increased the diagnostic accuracy over conventional microscopy, but at the same time raises important nosological questions.
Central neurocytomas are rare, generally circumscribed, intraventricular CNS tumors first recognized by Hassoun et al. [1]. Clinically, they often present as calcified supratentorial masses in young adults with symptoms of increased intracranial pressure. Microscopically, the neoplastic cells are monotonous with clear cytoplasm and small round nuclei, and often with a surrounding halo that resembles oligodendroglioma. In some cases, there are rosette-like structures consisting of irregular acellular zones with a fine, fibrillary matrix. Immunohistochemistry demonstrates evidence of neuronal differentiation with consistent synaptophysin positivity and variable staining for neurofilament proteins, especially prominent with the fibrillary matrix [2–4]. These findings are substantiated by ultrastructural features consisting of microtubules, neurofilaments, neurotic processes, secretory granules, and synaptic junctions [3, 5, 6]. Neurocytomas are thought to arise from mature neuronal cells but in vitro culture studies have shown that the progenitor cells are likely to be glioneural with the potential to differentiate into both glial and neuronal cells [7, 8]. Glial fibrillary acidic protein (GFAP) and S-100 expression is typically not observed in the tumor cells but in the surrounding reactive astrocytes [2]. More recently, extraventricular neurocytomas (EVN) have been reported as brain and spinal cord masses [9–19].
Oligodendrioglioma was first described by Bailey and Bucy in 1929 based on morphologic similarity of the neoplastic cells to non-neoplastic oligodendrogliocytes [20]. It is the third most common type of gliomas, comprising 4–7% of primary brain tumors [21]. The classic microscopic appearance is that of a neoplasm composed of clear cells surrounded by perinuclear halos and embedded in a scaffolding of “chicken wire” vessels. The tumor lacks expression of markers of mature oligodendrocytes; however, there may be focal expression of GFAP, especially in gliofibrillary oligodendrocytes and minigemistocytes [22]. Although the cell of origin of either oligodendroglioma or neurocytoma has not been identified, several investigators have suggested that these two tumors may have a common cell of origin [23, 24].
We report the pathologic findings of a spinal cord tumor from a 33-year-old male, which showed unusual combination of morphology, immunohistochemistry, and fluorescent in situ hybridization (FISH) results, underscoring the challenge of discriminating between oligodendroglioma and neurocytoma and suggesting a possible common origin.
Case history
A 33-year-old male patient with long standing history of neck pain, right hand numbness, and left ankle weakness presented to a tertiary care provider. On admission, there were no abnormal findings on general physical examination. Neurologic examination failed to reveal extremity weakness or sensory loss.
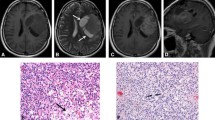
Magnetic resonance imaging (MRI) studies at the time showed abnormal T2 signal with an intramedullary mass in the cervico-thoracic spinal cord extending from C7 to T7. The mass lesion showed heterogeneous signal intensity and enhancement throughout the lesion (Fig. 1). A radiologic diagnosis of astrocytoma was considered. The patient underwent complete resection of the tumor, which was submitted to surgical pathology in its entirety.

T2 weighted sagittal MRI shows intramedullary mass in the thoracic spinal cord (C7–T7) with heterogenous signal intensity and enhancement throughout the lesion
The patient tolerated the procedure well. His postoperative hospital course was complicated by weakness of right iliopsoas and quadriceps dorsiflexion for which he received physical and occupational therapy. After 2 weeks of post-surgical hospital stay he was transferred to a rehabilitation center with a follow up visit in 1 month.
Material and methods
Light microscopy
A portion of the specimen was received for frozen section diagnosis. The remaining specimen was submitted entirely and processed routinely. Histological sections were prepared from 10% formalin-fixed, paraffin-embedded tissue, and stained with hematoxylin-eosin for routine light-microscopic examination.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections using an avidin-biotin-horseradish peroxidase system after the tissue was deparaffinized and rehydrated according to standard protocol. The following antibodies were used: polyclonal antibodies against S-100, chromogranin and synaptophysin (Dako, Carpinteria, CA, USA), and monoclonal antibodies against epithelial membrane antigen (EMA), neurofilament, neuron specific enolase (NSE), MIB-1, GFAP, vimentin (Dako), CD99 (O-13) (Signet; Dedham, MA, USA), and keratin (a cocktail of AE1/AE3, CAM 5.2, and 35βH11) (Ventana Medical Systems, Tucson, AZ, USA). In every case, formalin-fixed tissue was subjected to heat-induced antigen retrieval.
Fluorescence in situ hybridization (FISH) analysis
Chromosomal analysis was performed on deparaffinized formalin-fixed, paraffin-embedded tissue sections using commercially available Vysis dual-color probe sets (LSI 1p36/LSI 1q25 and LSI 19q13/LSI 19p13) as described in the manufacturer’s catalog (Vysis Inc., Downers Grove, IL, USA).
Results
H&E stained sections showed solid areas of uniform, polygonal cells with small round nuclei and delicate chromatin with clear perinuclear halos, clear cytoplasm accompanied by a capillary vascular arcade that compartmentalized the tumor cells, as well as foci of small or large cells with round nuclei and eosinophilic cytoplasm suggesting gangliocytic differentiation (Fig. 2a–c). Mitotic figures were rare and necrosis was not observed.

Representative morphologic features from formalin fixed paraffin embedded sections of the tumor. Lower magnification shows monomorphic cells with clear cytoplasm as well as areas rich in ganglion-like cells (a) (hematoxylin and eosin, ×20). Higher magnification of the clear cells (b) and the ganglion-like cells (c) (hematoxylin and eosin ×100, each)
Table 1 summarizes the immunohistochemical results. The tumor cells including with clear cytoplasm stained strongly for NSE, neurofilament, synaptophysin (Fig. 3a), GFAP (Fig. 3b), and S-100 (Fig. 3c). The cells resembling ganglion cells stained for chromogranin (Fig. 3d), NSE, S-100, neurofilament, and synaptophysin and were negative for GFAP. The clear cells were negative for vimentin, whereas the cells with eosinophilic cytoplasm and ganglion-like cells were positive. The proliferative activity of the tumor was estimated to be 30% using MIB1 antibody in the clear cells, whereas it was negative or very low in the remainder of the tumor. The neoplastic cells did not express keratin, CD99, or EMA.

By immunohistochemistry, the clear cells are positive for synaptophysin (a), GFAP (b), and S-100 (c), (×100, each). The ganglion-like cells are positive for chromogranin (d) (×400)
Fluorescent in situ hybridization analysis revealed fewer than two orange signals for 1p36 indicating deletion of 1p36 region on chromosome 1 in ∼80% of the tumor cells (Fig. 4a), whereas no deletion of 1q25 region was present (green signal, Fig. 4b). Also, in ∼10–20% of the tumor cells fewer than two orange signals for chromosome 19q13 were also observed indicating deletion of chromosome 19q13 region, but not 19p13 region (data not shown).

FISH analysis demonstrates 1p36 deletion in the majority of the tumor cells (a) (×1,000), whereas no deletion of 1q25 was observed (b)
Discussion
This case highlights the limitations of the present diagnostic criteria for classifying primary clear cell neoplasms of the CNS. Since current nomenclature of brain tumors is based on morphology, ultrastructure, and immunohistochemistry rather than molecular genetic criteria, it may be considered incongruent to classify our case as an oligodendroglioma. At the same time, the prognostic role of chromosome 1p/19q deletion in oligodendrogliomas has generated great interest in targeted therapy which may be associated with chemosensitivity and higher survival [25–27]. In addition, our findings lend further support to a proposed common progenitor cell for these two tumors [23, 24].
According to in vitro animal studies, the O-2A oligodendrocyte precursor cell has the ability to generate oligodendrocytes or type 2 astrocytes in rats [28]. Similarly, human gliomas contain glial precursor cells that can give rise to cells of astrocyte or oligodendrocyte lineage [29]. Williams et al. [23] demonstrated the existence of a bi-potential precursor cell with the ability to differentiate to either oligodendrocytes or neurons in rats. Using immunohistochemical markers in an in vitro model, the authors identified mixed clones with neurons and oligodendrocytes. Similar findings were reported by Price and Thurlow [30], who demonstrated a progenitor cell in the rat cerebral cortex that could differentiate into both neurons and white matter glial cells. These findings are not surprising since cultured rat spinal cord neuroepithelial cells, which are distinct from neural crest stem cells, can differentiate into neurons, oligodendrocytes, and astrocytes [31]. O-2A cells have been shown to express platelet derived growth factor α receptor (PDGF α R) and NG2 chondroitin sulfate proteoglycan in vitro, which can be detected in tissue sections. Based on immunohistochemical analysis of human gliomas, Shoshan et al. [32] suggested that oligodendrogliomas might arise from NG2+/PDGF α R + progenitor cells. As cell division is important in tumorigenesis, proliferating progenitor cells are more susceptible to oncogenesis than post-mitotic cells supporting the hypothesis that NG2+/PDGF α R + cells could be the precursor of oligodendrogliomas. It is not clear if similar cells are also progenitors of neurocytomas, but because oligodendrocytes can be induced from adult human olfactory epithelial-derived precursors [33], it is possible that human oligodendriogliomas and neurocytomas are derived from a common precursor cell.
Few cases of neurocytoma have been reported in extraventricular sites including the spinal cord [9–19]. As in the present case, most spinal neurocytomas occur as intramedullary cervical or cervico-thoracic tumors. EVN are histologically similar to their centrally located counterparts, but often exhibit glial and ganglionic differentiation [11]. Accordingly, the typical immunohistochemical profile consists of synaptophysin and neurofilaments positivity with variable GFAP expression. Similar to our case, ganglion cells in EVN stain for chromogranin, which is not observed in central neurocytomas.
In general, central and extraventricular neurocytomas have a good prognosis [2, 11]. Complete resection of neurocytomas is the treatment of choice. The diagnosis of atypical neurocytoma has been assigned to those tumors showing necrosis, vascular proliferation, increased mitotic activity, and MIB1 labeling index of more that 2%. The MIB1 index has been shown to correlate better with survival and recurrence than cellular atypia alone [34]. Our case lacked cellular atypia and necrosis; however, the MIB1 proliferation index approached 30%, which is higher than reported cases of atypical neurocytoma [17, 18, 34].
Ho-Keung et al. [35] were the first to report that oligodendrogliomas exhibited staining for the neuronal markers synaptophysin and NSE. Subsequently, Wharton et al. [36] reported synaptophysin and neurofilament expression and neuron-like electrophysiologic properties in cultured oligodendroglioma cells. Although the neoplastic clear cells in our case showed inconsistent staining for neurofilaments and synaptophysin, this can be explained by loss of immunoreactivity with formalin fixation and embedding or true oligodenroglial differentiation. Morphologically, the identification of focal ganglion cell differentiation in our case confirmed by chromogranin immunostain and synaptophysin and neurofilament expression in clear cells supports the diagnosis of EVN rather than oligodendroglioma. However, the presence of S-100 and GFAP staining and lack of vimentin in clear cells is more in favor of oligodendroglioma. In in vitro experiments with neurocytomas, a shift from synaptophysin to GFAP expression has been observed with successive cell passages [7]. Also in rats transplacental ethyl-nirosourea administration induces oligodendrioglioma-like tumors with both neuronal and glial markers [37]. These observations favor a common bi-potential progenitor cell of origin for neurocytomas and oligodendrogliomas.
Given the lack of specific immunohistochemical, histological or ultrastructural markers for oligodendrogliomas, greater emphasis has been placed on molecular genetic criteria, that is, allelic deletions on chromosome 1p and 19q to make the diagnosis [25, 26]. Studies have shown that chromosome 1p loss occurs in 40–92% of oligodendrogliomas, whereas 19q loss occurs in 50–80% of cases [26]. Loss of chromosome 1p/19q is not commonly present in neurocytic tumors [38]; however, this has been observed in EVN [15, 24]. Some investigators found 1p/19q deletions in tumors showing immunohistochemical and ultrastructural features of neuronal differentiation similar to our case, which prompted reclassification of the tumors as oligodendroglioma [38].
In summary, this unusual case illustrates the need to reclassify tumors showing pathologic features of both neurocytoma and oligodendroglioma as a unique entity, while the effort to identify the cell of origin continues. Further study of larger numbers of cases of the biologic behavior and the possible expression of cell surface molecules such as NG2 and PDGF α R by tumors showing differentiation of both oligodendroglioma and neurocytoma may help in directing therapy and improving prognosis.
References
Hassoun J, Gambarelli D, Grisoli F, Pellet W, Salamon G, Pellissier JF, Toga M (1982) Central neurocytoma. An electron-microscopic study of two cases. Acta Neuropathol (Berl) 56:151–156
Figarella-Branger D, Pellissier JF, Daumas-Duport C, Delisle MB, Pasquier B, Parent M, Gambarelli D, Rougon G, Hassoun J (1992) Central neurocytomas. Critical evaluation of a small-cell neuronal tumor. Am J Surg Pathol 16:97–109
von Deimling A, Janzer R, Kleihues P, Wiestler OD (1990) Patterns of differentiation in central neurocytoma. An immunohistochemical study of eleven biopsies. Acta Neuropathol (Berl) 79:473–479
You H, Kim YI, Im SY, Suh-Kim H, Paek SH, Park SH, Kim DG, Jung HW (2005) Immunohistochemical study of central neurocytoma, subependymoma, and subependymal giant cell astrocytoma. J Neurooncol 74:1–8
Kim DG, Chi JG, Park SH, Chang KH, Lee SH, Jung HW, Kim HJ, Cho BK, Choi KS, Han DH (1992) Intraventricular neurocytoma: clinicopathological analysis of seven cases. J Neurosurg 76:759–765
Cenacchi G, Giangaspero F, Cerasoli S, Manetto V, Martinelli GN (1996) Ultrastructural characterization of oligodendroglial-like cells in central nervous system tumors. Ultrastruct Pathol 20:537–547
Westphal M, Meissner H, Matschke J, Herrmann HD (1998) Tissue culture of human neurocytomas induces the expression of glial fibrilary acidic protein. J Neurocytol 27:805–816
Ishiuchi S, Nakazato Y, Iino M, Ozawa S, Tamura M, Ohye C (1998) In vitro neuronal and glial production and differentiation of human central neurocytoma cells. J Neurosci Res 51:526–535
Tatter SB, Borges LF, Louis DN (1994) Central neurocytomas of the cervical spinal cord. Report of two cases. J Neurosurg 81:288–293
Coca S, Moreno M, Martos JA, Rodriguez J, Barcena A, Vaquero J (1994) Neurocytoma of spinal cord. Acta Neuropathol (Berl) 87:537–540
Giangaspero F, Cenacchi G, Losi L, Cerasoli S, Bisceglia M, Burger PC (1997) Extraventricular neoplasms with neurocytoma features. A clinicopathological study of 11 cases. Am J Surg Pathol 21:206–212
Stapleton SR, David KM, Harkness WF, Harding BN (1997) Central neurocytoma of the cervical spinal cord. J Neurol Neurosurg Psychiatry 63:119
Funato H, Inoshita N, Okeda R, Yamamoto S, Aoyagi M (1997) Cystic ganglioneurocytoma outside the ventricular region. Acta Neuropathol (Berl) 94:95–98
Brat DJ, Scheithauer BW, Eberhart CG, Burger PC (2001) Extraventricular neurocytomas: pathologic features and clinical outcome. Am J Surg Pathol 25:1252–1260
Mrak RE, Yasargil MG, Mohapatra G, Earel J Jr, Louis DN (2004) Atypical extraventricular neurocytoma with oligodendroglioma-like spread and an unusual pattern of chromosome 1p and 19q loss. Hum Pathol 35:1156–1159
Mut M, Guler-Tezel G, Lopes MB, Bilginer B, Ziyal I, Ozcan OE (2005) Challenging diagnosis: oligodendroglioma versus extraventricular neurocytoma. Clin Neuropathol 24:225–229
Sharma S, Sarkar C, Gaikwad S, Suri A, Sharma MC (2005) Primary neurocytoma of the spinal cord: a case report and review of literature. J Neurooncol 74:47–52
Moriguchi S, Yamashita A, Marutsuka K, Yoneyama T, Nakano S, Wakisaka S, Nabeshima K, Asada Y (2006) Atypical extraventricular neurocytoma. Pathol Int 56:25–29
Buccoliero AM, Caldarella A, Ammannati F, Mennonna P, Taddei A, Taddei GL (2002) Extraventricular neurocytoma: morphological and immunohistochemical considerations on differential diagnosis. Pathol Res Pract 198:627–633 discussion 635–638
Bailey P, Bucy P (1929) Oligodendrogliomas of the brain. J Path Bact 32:735–751
Chowdhary S, Chamberlain MC (2006) Oligodendroglial tumors. Expert Rev Neurother 6:519–532
Herpers MJ, Budka H (1984) Glial fibrillary acidic protein (GFAP) in oligodendroglial tumors: gliofibrillary oligodendroglioma and transitional oligoastrocytoma as subtypes of oligodendroglioma. Acta Neuropathol (Berl) 64:265–272
Williams BP, Read J, Price J (1991) The generation of neurons and oligodendrocytes from a common precursor cell. Neuron 7:685–693
Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM (2002) Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol 61:947–955
Bello MJ, Vaquero J, de Campos JM, Kusak ME, Sarasa JL, Saez-Castresana J, Pestana A, Rey JA (1994) Molecular analysis of chromosome 1 abnormalities in human gliomas reveals frequent loss of 1p in oligodendroglial tumors. Int J Cancer 57:172–175
Perry A, Fuller CE, Banerjee R, Brat DJ, Scheithauer BW (2003) Ancillary FISH analysis for 1p and 19q status: preliminary observations in 287 gliomas and oligodendroglioma mimics. Front Biosci 8:a1–a9
Fallon KB, Palmer CA, Roth KA, Nabors LB, Wang W, Carpenter M, Banerjee R, Forsyth P, Rich K, Perry A (2004) Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol 63:314–322
Raff MC, Miller RH, Noble M (1983) A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 303:390–396
Colin C, Baeza N, Tong S, Bouvier C, Quilichini B, Durbec P, Figarella-Branger D (2006) In vitro identification and functional characterization of glial precursor cells in human gliomas. Neuropathol Appl Neurobiol 32:189–202
Price J, Thurlow L (1988) Cell lineage in the rat cerebral cortex: a study using retroviral-mediated gene transfer. Development 104:473–482
Kalyani A, Hobson K, Rao MS (1997) Neuroepithelial stem cells from the embryonic spinal cord: isolation, characterization, and clonal analysis. Dev Biol 186:202–223
Shoshan Y, Nishiyama A, Chang A, Mork S, Barnett GH, Cowell JK, Trapp BD, Staugaitis SM (1999) Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci USA 96:10361–10366
Zhang X, Cai J, Klueber KM, Guo Z, Lu C, Qiu M, Roisen FJ (2005) Induction of oligodendrocytes from adult human olfactory epithelial-derived progenitors by transcription factors. Stem Cells 23:442–453
Mackenzie IR (1999) Central neurocytoma: histologic atypia, proliferation potential, and clinical outcome. Cancer 85:1606–1610
Ng HK, Ko HCW, Tse CCH (1994) Immunohistochemical and ultrastructural studies of oligodendriogliomas revealed features of neuronal differentiation. Int J Surg Pathol 2:47–56
Wharton SB, Chan KK, Hamilton FA, Anderson JR (1998) Expression of neuronal markers in oligodendrogliomas: an immunohistochemical study. Neuropathol Appl Neurobiol 24:302–308
Vaquero J, Coca S, Moreno M, Oya S, Arias A, Zurita M, Morales C (1992) Expression of neuronal and glial markers in so-called oligodendroglial tumors induced by transplacental administration of ethyl-nitrosourea in the rat. Histol Histopathol 7:647–651
Fujisawa H, Marukawa K, Hasegawa M, Tohma Y, Hayashi Y, Uchiyama N, Tachibana O, Yamashita J (2002) Genetic differences between neurocytoma and dysembryoplastic neuroepithelial tumor and oligodendroglial tumors. J Neurosurg 97:1350–1355
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Makuria, A.T., Henderson, F.C., Rushing, E.J. et al. Oligodendroglioma with neurocytic differentiation versus atypical extraventricular neurocytoma: a case report of unusual pathologic findings of a spinal cord tumor. J Neurooncol 82, 199–205 (2007). https://doi.org/10.1007/s11060-006-9268-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-006-9268-0