
Abstract
Magnetic nanoparticles (NPs) of magnetite Fe3O4 obtained by coprecipitation (COP), thermal decomposition (DT), and commercial sample (CM) have been degraded in similar conditions to physiological medium at pH 4.7 and in simulated body fluid (SBF) at pH 7.4. The formation of the nanoparticles was confirmed by FTIR spectroscopy, X-ray diffraction (XRD), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). In view of medical and environmental applications, the stability of the particles was measured with dynamic light scattering. The degradation processes were followed with atomic absorption spectroscopy (EAA) and TEM. Magnetic measurements were carried out using vibrating sample magnetometry (VSM). Our results revealed that the structural and magnetic properties of the remaining nanoparticles after the degradation process were significantly different to those of the initial suspension. The degradation kinetics is affected by the pH, the coating, and the average particle size of the nanoparticles.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Magnetic nanoparticles (NPs) have received great attention due to their biomedical applications such as drug delivery (Häfeli et al. 2009; Klostergaard and Sweeney 2012), contrast agents for magnetic resonance imaging (MRI) (Arbab et al. 2005; Corot et al. 2006; Na et al. 2009), photothermal agents for solid tumor therapy (Wu et al. 2016), and heating mediators for magnetic hyperthermia in cancer treatment (Pankhurst et al. 2009; Gazeau et al. 2008), referring to the introduction of ferromagnetic or superparamagnetic particles into the tumor tissues (Ang et al. 2007). The potential benefits of NPs are considerable because of their unique magnetic properties, low toxicity, and biodegradability (Mahmoudi et al. 2011; Markides et al. 2012; Smith and Gambhir 2017). However, there is a distinct need to identify the capacity of the NPs for disintegration into bioeliminable products and the potential of cellular damage associated with toxicity caused by these materials. Although human tissues contain a lot of endogenous iron, NPs containing iron oxide may have higher toxicity than iron in a molecular form. This has been associated with effects such as inflammation, altered mitochondrial function, and alteration of the functions of the major components of the cell (Nel et al. 2006; Oberdörster et al. 2007). Although the toxicity of iron oxide in NPs has been reported (Häfeli et al. 2009; Jeng and Swanson 2006; Veranth et al. 2007; Schwegmann et al. 2010; Gu et al. 2012; Malvindi et al. 2014), the mechanisms, progressions, and factors governing nanoparticle degradation are lacking. Synthesis conditions, size, magnetic interactions, coating, and pH of the media may influence the release of iron ions (Levy et al. 2010). Hence, a study of mechanisms of NP degradation is important to better understand their toxicity and their long-term effects as well as the implementation of these materials in medical practice. The chemical conditions of the degradation of dextran-coated magnetic nanoparticles have been reported (Arbab et al. 2005). These authors demonstrated the requirement of an acidic environment (pH 4.5) and the presence of chelates such as citrate with a high affinity for Fe(III). Considering that during cellular endocytosis, nanoparticles are exposed to a continuous decrease of pH, from that of the extracellular medium (7–7.5), to that of intracellular endosomes (6–5.5), and to the acidic microenvironment of lysosomes (4.5–4.8) (Ohkuma and Poole 1978). Hence, in the present work, iron oxide nanoparticles obtained by different synthesis methods, with various particle sizes were exposed at different media at pH 7.4 and 4.5. A study of the evolution of the structural and magnetic properties and the degradation mechanisms of these NPs is reported.
Materials and methods
Magnetic nanoparticle synthesis
The synthesis was carried out by two different methods: coprecipitation (COP) and thermal decomposition (DT). For comparison, commercial magnetite NPs were also tested. All the chemical reagents used in this work were of analytical grade and used without further purification.
Coprecipitation method
For the synthesis by the coprecipitation method (COP), a solution of iron(II) sulfate heptahydrated (FeSO4:7H2O), NaOH, and KNO3, was prepared. The solution was subjected to magnetic stirring for 30 min to ensure nucleation and growth of magnetite particles. Afterwards, the solution was aged at 60 °C for 90 min in ultrasonic water bath and cooled at room temperature. The solid phase was magnetically separated and washed with deionized water repeatedly, in order to ensure the elimination of residual salts. Particles were then dried at 60 °C under vacuum.
Thermal decomposition method
For the thermal decomposition method (DT), FeO(OH) (4 mmol), oleic acid (10 mmol), and 5 g of docosane were mixed under inert gas atmosphere (Ar) and vigorous stirring, heated to 340 °C, and refluxed at this temperature for 1 h. Subsequently, the precipitate obtained was dissolved in pentane/diethyl ether and 30 ml ethanol was added to the mixture. Particles were then dried at 60 °C in vacuum.
Commercial sample
Commercial magnetite nanoparticles (CM) were acquired from Alfa Aesar (Karlsruhe, Germany), 97% purity.
Characterization
The crystal structure of the Fe3O4 NPs was identified by X-ray powder diffraction (XRD) performed in a Bruker D8-Focus X-ray diffractometer, equipped with parallel beam geometry and Cu Kα radiation (λ = 1.5406 Ǻ), with a voltage of 40 kV, current of 30 mA, in the 2θ ranged 5–90 degrees, and 0.020 steps. Infrared spectra were recorded using a Perkin Elmer Spectrum 100 FTIR spectrometer between 4000 and 250 cm−1.
Average particle size analysis
Nanoparticle average particle size was determined from SEM and TEM. SEM analysis was performed in a FEI Inspect F50 scanning electron microscope, working with an acceleration voltage of 8–10 keV, attached with an energy dispersive X-ray microanalysis, EDAX 8500 (silicon drift detector (SSD)). TEM analysis was carried out in a FEI spirit microscope (at 120 kV). Sample preparation for TEM observation was carried out by dispersion of the particles in a solution of ethanol/water. A drop of the suspension was placed onto a carbon-coated copper grid and allowed to dry at room temperature for several minutes. Average particle size and distribution were evaluated by measuring the largest internal dimension of at least 150 particles, using image J, an image processing software to determine the size distribution of the NPs (apparent diameters from 2D measurements).
Zeta potential measurements
Colloidal stability and surface charge of the samples were measure using light scattering with a dynamic laser Malvern Instruments Zetasizer, model Nano ZS.
Magnetic characterization
Hysteresis loops with a maximum field of 1 T were measured in a vibrating sample magnetometer, Varian Techtron 155, at room temperature.
Degradation process
In order to induce degradation, 200 mg/ml NPs were immersed in three different media at 37 °C: (a) an aqueous buffer at pH 4.7 containing citrate ions (20 mM) as iron chelator to mimic lysosomal environment (Arbab et al. 2005); (b) in simulated body fluid (SBF) at pH 7.4; and (c) in distilled water at pH 6.7 as a control sample. The citrate buffer was prepared by mixing appropriate volumes of 20 mM aqueous solutions of citric acid (C6H8O7, Fluka, 99.5%) and sodium citrate tribasic (Na3C6H5O7:5H2O, Fluka, 99.5%) to achieve the final desired pH. The simulated body fluid (SBF) was prepared with the following concentrations: NaCl (6.55 g), NaHCO3 (2.27 g), KCl (0.373 g), Na2HPO4:2H2O (0.178 g), MgCl2:6H2O (0.305 g), 37 wt% HCL (5 ml), CaCl2 (0.278 g), NaSO4 (0.071 g), and NH2C(CH2OH)3 (Tris buffer, 6.055 g). The reactants were dissolved one by one into 500-ml Millipore water at 37 °C under continuous magnetic stirring. Millipore water was added to increase the total volume to 1 l, and the pH was balanced to 7.4 using 1 M HCl. The kinetics of the degradation process was evaluated continuously up to 30 days. The degradation media with the NPs were split into several tubes and placed in an incubator. Samples of each solution were taken at 1, 2, 3, 6, 24, 48, 72, 120, 240, 480, and 720 h, and the amount of free ions was measured by atomic absorption spectroscopy (EAA). At each time point, the Fe3O4 NPs were separated from the rest of solution through centrifugation at 13,000 rpm for 1 h in order to characterize them by SEM-TEM and magnetic measurements.
Atomic absorption spectroscopy
Iron determination was carried out in a flame atomic absorption spectrophotometer VARIAN Techtron 1200. An iron hollow cathode lamp was used at an operating current of 2 mA and a wavelength and spectral bandwidth of 238.2 and 0.2 nm, respectively. Limits of detection (LOD) were less than 0.005 g/l. Fe standard series solutions (1.00, 2.00, 3.00, and 4.00 g/ml) were used, and the Fe released was determined by a calibration curve in parts per million (ppm). The correlation coefficient was 0.998.
Results and discussion
X-ray diffraction
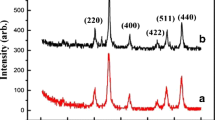
XRD patterns shown in Fig. 1 are characteristic of a single cubic spinel phase corresponding to a magnetite (JCPDS 39–1346). The sample prepared by thermal decomposition (DT) showed a broader diffraction line than that of the diffraction patterns of COP and CM samples, indicating the small crystal size of these particles.

X-ray powder diffraction patterns of the samples prepared by coprecipitation (COP) thermal decomposition (DT), and commercial sample (CM)
Fourier transform infrared
The FTIR spectra of the samples DT, COP, and CM are presented in Fig. 2. The characteristic band of the intrinsic Fe-O stretching of the spinel structure is observed at 580 cm−1 for all the samples and corresponds to vibrations of the metal at the tetrahedral site. In the range of 400–450 cm−1, are the Fe-O stretching vibrations of the octahedral site (Nel et al. 2006). The wide stretching vibration at 3400 cm−1 and a smaller one in 1629 cm−1 is probably due to the bending vibration H-O-H of physisorbed water onto the nanoparticle surface. For the DT sample, additionally, two characteristic bands are observed at 2926 and 2855 cm−1, corresponding to asymmetric and symmetric stretching of C-H bonds of the aliphatic chain and at 1415 cm−1, the band assigned to C-O-C stretching is observed which is a carbohydrate fingerprint; these absorptions suggest that the NPs prepared by thermal decomposition are coated with the oleic acid.

FTIR spectra of the samples prepared by (a) thermal decomposition (DT), (b) coprecipitation (COP), and (c) of the commercial sample (CM)
SEM and TEM
The morphology and average particle size of different NPs were analyzed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Figure 3 shows the evolution of morphology and particle size of the different NPs after the maximum period of the degradation process in buffer tribasic citrate at pH 4.7. The differences in size and morphology presented between the different NPs are evident. The commercial sample (CM) showed a very heterogeneous morphology with a very wide particle size distribution (20–800 nm, average particle size 60 nm). After 30 days of degradation, the average particle size for this sample was significantly reduced to 5 nm with a very narrow size distribution. The particles prepared by coprecipitation (COP) originally have an average particle size of 40 nm with a homogenous pseudo-faceted morphology. They undergo almost complete degradation after 10 days in this medium; a very small amount of material was recovered. After this period, a drastic change in size and morphology was observed and the particles showed sharp eroded edges (Fig. 3d), suggesting that diffusion takes place throughout the crystal facets. A similar morphology was observed for the sample obtained using the thermal decomposition method (DT) after 30 days degradation. Originally, this sample had a spherical morphology (Fig. 3e) with an average particle size distribution of 15 nm. The very good dispersion of these NPs is attributed to the coating with oleic acid remaining from the synthesis process and confirmed by FTIR. After 30 days of the degradation process, these NPs showed an irregular shape and a very narrow particle size distribution with an average size of 5 nm. (Lartigue et al. 2013) carried out a HRTEM in situ study of the morphological degradation of cubic oxide NPs with different coatings in a buffer medium with citrate acid at pH 4.7. The NPs, originally cubic, gradually erode and totally dissolve after 3 h, even if they were coated with amphiphilic polymer or with PEG. However, when the particles formed aggregates, the polymer coating acted as a protective layer slowing down degradation. A similar behavior was observed in the present work in the DT particles coated with oleic acid. The coating protects the NPs from the external medium, but the rate of degradation observed in our work was much slower than that reported by (Lartigue et al. 2013).

Morphology and average particle size evolution after degradation process in buffer tribasic citrate at pH 4.7. a Original CM NPs. b CM NPs after 30 days degradation. c Original COP NPs. d COP NPs after 10 days degradation. e Original DT NPs. f DT NPs after 30 days degradation. The third row shows the histograms of particle size distribution before and after the degradation process for each of the NPs
Additionally, all the NPs were also immersed in SBF solution at pH 7.4 and in distilled water at pH 6.7 for periods up to 30 days. None of these NPs is affected by these media and no particular changes in size or morphology were observed, implying that the pH is a critical factor in the degradation process.
Degradation process
The time-dependent iron release of the NPs was followed for 30 days at 37 °C starting with 100 ppm of each sample in 250 ml of buffer tribasic citrate at pH 4.7 and in SBF at pH 7.4 in order to achieve similar conditions to physiological medium. A change in color of the suspensions was observed during the process of degradation carried out at pH 4.7 from the original brown magnetite to the characteristic yellow color of free iron ions in the solution. A similar trend was observed for all the different samples treated at this pH.
Figure 4 shows the content of Fe in solution and the magnetite degradation during the degradation process up to 30 days. It has been reported that the kinetics of degradation follows different mechanisms for each of the NPs and varies with pH, particle size, and the surface coating (Levy et al. 2010). In the present work, a first-order reaction was observed for all the NPs following the equation y = a (1-exp−kt), (k is the first-order constant rate) with different exponential rate constants for each of them. Table 1 shows the different exponential rate constants and mean degradation rates for the different materials.

a Time and pH dependence of the different samples at pH 4.7 in buffer tribasic citrate and in SBF at pH 7.4. Time dependence of iron release for the samples b COP, c DT, and d CM
The COP sample presents the highest average degradation rate in the first 24 h, and almost complete degradation was reached after only 10 days. This sample has a pseudo-cubical morphology but with well-defined facets and its surface is exposed to the environment without any coating on it. As a consequence, the sides erode fast in the acidic medium in the first 24 h, with a reduction in particle size. After 48 h, there is a slight decrease in the degradation rate, probably due to a very thin oxide coating developed during the contact with the medium. However, after 10 days, the sample almost vanished and very little material remained in the solution.
For the DT NPs, different equations for different time intervals were required. The kinetics is of first order but with different exponential rate constants for each period. The exponential decay equation is of the form y = A o(exp(−k∆tp))n, where ∆tp corresponds to a specific time period and n is an integer corresponding to the number of time periods. For these NPs, the first few hours of the degradation process is relatively fast, however after 24 h, a drastic change in the mechanism of degradation is observed up to 480 h and then the degradation rate increases again. This may be probably due to the fact that some particles were not covered with the oleic acid and these degraded fast in the first 24 h. But most of the NPs that were coated with the oleic acid and a slow degradation were obtained (0.05 ppm/h) up to 480 h. After this period, it is possible that the oleic acid coating decomposes, leaving the NPs naked and exposed to the medium, so the degradation rate increases to 0.1 ppm/h. After 720 h, there are still some particles in solution with an average particle size of 5 nm.
The CM NPs showed three degradation rates. These particles present a very large particle size distribution, as was observed by TEM (Fig. 3). The slow degradation rate observed in the first 24 h could be attributed to the presence of a fraction of very small particles that dissolved easily but liberate small amount of ions to the media, and also probably a thin oxide coating is covering the surface of these NPs, which controls the initial degradation rate. From 24 to 720 h, the degradation rates are slow (0.09 ppm/h from 24 to 480 h and 0.07 up to 720 h), associated to the very wide particle size distribution present, therefore larger particles take longer to degrade.
The DT and CM NPs were not completely degraded even after 30 days at pH 4.7 in a citrate buffer, reaching 80 and 70% degradation after this period. The mean degradation rate for the complete degradation period for COP, DT, and CM materials is very similar (0.13, 0.11, and 0.11, respectively), as expected since all of them are magnetite NPs. Therefore, these results demonstrate that the degradation rate at pH 4.7 is mainly controlled by particle size and coating.
No measurable release was observed in SBF at pH 7.4 (star symbol). These results suggest that the NPs in SBF are relatively stable in neutral conditions.
Magnetic measurements
Figure 5 shows the field-dependent magnetization curves obtained for the samples at 300 K, before degradation (Fig. 5a) and after degradation up to 48 h for the CM and COP particles (Fig 5b, c, respectively). The magnetic parameters such as saturation magnetization (Ms), coercivity (Hc), and remnant magnetization (M R) are given in Table 2. The hysteresis loops display non-zero coercivity (Hc) and remnant magnetization (M R), confirming that these particles are largely blocked at room temperature. This is consistent with the small magnetic NP dimensions determined from XRD and TEM analysis. The high Ms reached by the COP sample (83.83 emu/g) is attributed to the high crystallinity (Fig. 1) and a uniform morphology (Fig. 3c). The CM sample has a very wide size distribution and probably a very thin oxide layer affecting the magnetization of the material reaching 72.91 emu/g. Magnetic measurements, such as magnetization versus applied field, accurately reflect any modification in the magnetic core size due to degradation. Besides, magnetic measurements also provide relevant quantitative data to probe the concentration of magnetic nanoparticles, independent of the dosage of released free iron species (Levy et al. 2010). As illustrated in Table 2, the magnetic properties of the samples studied dramatically decreased with time of degradation. The magnetic properties of nanosized materials are known to change drastically upon a tiny variation in size or due to surface effects (Gutiérrez et al. 2015). In Fig. 3, it was demonstrated that the morphology and average particle size distribution of the NPs were significantly modified during the degradation process at pH 4.7. Therefore, the changes in the magnetization observed with degradation time are mainly attributed to reduction in particle size.

Magnetization versus applied field at room temperature of original COP, CM, and DT NPs (a), CM NPs after 0, 1, and 48 h degradation (b), and COP NPs after 0, 2, and 48 h degradation (c)
Zeta potential measurements
Colloidal stability and surface charge of the samples as a function of time were determined using dynamic light scattering in water, and the results are presented in the Table 3. The surface charges of the samples DT, COP, and CM at 1 h are −42.7, −42.4, and −40.7 mV, respectively. These results are in agreement with those reported in the literature (Klostergaard and Sweeney 2012; Schwegmann et al. 2010) for hematite nanoparticles at pH 4. From the results of Table 3, it can be inferred that after 1 h of degradation, the samples are stable in solution, aggregation is low, and repulsion forces are dominant. After 48 h, the DT and CM samples are stable with −44.1 and −43.5 mV, respectively. However, the COP sample is destabilized with a surface net charge reduction of −31.6 mV, probably due to increased aggregation of nanoparticles as we can see in Fig. 3. For the samples subjected to the process of degradation for 120 h (5 days), a reduction in the net surface charge is observed for the COP and DT samples.
The release kinetics of the NPs depends on the nature of the surface coating, pH, particle aggregation, and the average particle size. It has been reported (Arbab et al. 2005) that the charged nanoparticles exhibit different stabilities in physiological media directly influenced by the surface coatings. In our case, a faster degradation process has been observed for the COP NPs, which have a uniform morphology and their surface is exposed directly to the environment in comparison with that for DT NPs coated with oleic acid, confirming the importance of the nanoparticle coating on the degradation process, as has been reported before (Levy et al. 2010; Lartigue et al. 2013). These results suggest that the NPs coated with oleic acid are relatively more stable in acidic condition than the NPs without coating. Several factors may influence the stability of the coating and subsequently the accessibility of soluble chelators to the iron oxide crystal, namely, the molecular weight of the coating, the linker ensuring the anchorage of the coating to the crystal, the respective affinity for iron species of the soluble chelators, and the surface ligands of the particles (Levy et al. 2010). A high degradation rate is observed for the COP sample, while the degradation process for DT NPs is slower and more controlled due to the NPs coating. The fact that the NPs change their structural and magnetic properties upon the degradation process at pH 4.7 has a significant impact on the understanding of biotransformation and subsequent toxicity in the body. This has a great importance for NPs to be used as contrast agents or as mediators in hyperthermia therapy where the application relies on the nanosize-related magnetic properties.
Conclusion
The degradation of magnetite nanoparticles in similar conditions to physiological medium has been followed by TEM and EAA. The important role of pH, particle size, and coating on the degradation kinetics has been demonstrated. Also, a reduction of the average particle size during the degradation process has been found affecting the magnetic properties of the NPs. The results confirm that it is possible to modify the kinetics of degradation of magnetite nanoparticles by varying the nature of the coating and the average particle size of the nanoparticles.
References
Ang KL, Venkatraman S, Ramanujan RV (2007) Magnetic PNIPA hydrogels for hyperthermia applications in cancer therapy. Mater Sci Eng C27:347–351. doi:10.1016/j.msec.2006.05.027
Arbab AS, Wilson LB, Ashari P, Jordan EK, Lewis BK, Frank JA (2005) A model of lysosomal metabolism of dextran coated superparamagnetic iron oxide (SPIO) nanoparticles: implications for cellular magnetic resonance imaging. NMR Biomed 18(6):383–389. doi:10.1002/nbm.970
Corot C, Robert P, Idee JM, Port M (2006) Recent advances in iron oxide nanocrystal technology for medical imaging. Adv Drug Deliv Rev 58:1471–1504. doi:10.1016/j.addr.2006.09.013
Gazeau F, Levy M, Wilhelm C (2008) Optimizing magnetic nanoparticle design for nanothermotherapy. Nanomedicine 3(6):831–844. doi:10.2217/17435889.3.6.831
Gu L, Fang RH, Sailor MJ, Park J-H (2012) In vivo clearance and toxicity of monodisperse iron oxide nanocrystals. ACS Nano 6(6):4947–4954. doi:10.1021/nn300456z
Gutiérrez L, Romero S, Da Silva GB, Costo R, Vargas MD, Ronconi CM, Serna CJ, Veintemillas-Verdaguer S, Morales M (2015) Degradation of magnetic nanoparticles mimicking lysosomal conditions followed by AC susceptibility. Biomed Eng-Biomed Tech 60(5):417–425. doi:10.1515/bmt-2015-0043
Häfeli UO, Riffle JS, Harris-Shekhawat L, Carmichael-Baranauskas A, Mark F, Dailey JPC, Bardenstein D (2009) Cell uptake and in vitro toxicity of magnetic nanoparticles suitable for drug delivery. Mol Pharm 6:1417–1428. doi:10.1021/mp900083m
Jeng HA, Swanson J (2006) Toxicity of metal oxide nanoparticles in mammalian cells. J Environ Sci Health A Tox Hazard Subst Environ Eng 41(12):2699–2711. doi:10.1080/10934520600966177
Klostergaard J, Sweeney CE (2012) Magnetic nanovectors for drug delivery. Nanomedicine 8(1):S37–S50. doi:10.1016/j.maturitas.2012.01.019
Lartigue L, Alloyeau D, Kolosnjaj-Tabi J, Javed Y, Guardia P, Riedinger A, Pechoux C, Pellegrino T, Wilhelm C, Gazeau F (2013) Biodegradation of iron oxide nanocubes: high-resolution in situ monitoring. ACS Nano 7(5):3939–3952. doi:10.1021/nn305719y
Levy M, Lagarde F, Maraloiu VA, Blanchin MG, Gendron F, Wilhelm C, Gazeau F (2010) Degradability of superparamagnetic nanoparticles in a model of intracellular environment: follow-up of magnetic, structural and chemical properties. Nanotechnology 21(39):395103. doi:10.1088/0957-4484/21/39/395103
Mahmoudi M, Laurent S, Shokrgozar MA, Hosseinkhani M (2011) Toxicity evaluations of superparamagnetic iron oxide nanoparticles: cell “vision” versus physicochemical properties of nanoparticles. ACS Nano 5(9):7263–7276. doi:10.1021/nn2021088
Malvindi MA, De Matteis V, Galeone A, Brunetti V, Anyfantis GC, Athanassiou A, Cingolani R, Pompa PP (2014) Toxicity assessment of silica coated iron oxide nanoparticles and biocompatibility improvement by surface engineering. PLoS One 9(1):e85835. doi:10.1371/journal.pone.0085835
Markides H, Rotherham M, El Haj AJ (2012) Biocompatibility and toxicity of magnetic nanoparticles in regenerative medicine. J Nanomat 2012:614094. doi:10.1155/2012/614094
Na HB, Song IC, Hyeon T (2009) Inorganic nanoparticles for MRI contrast agents. Adv Mater 21(21):2133–2148. doi:10.1002/adma.200802366
Nel A, Xia T, Madler L, Li N (2006) Toxic potential of materials at the nanolevel. Science 311(5761):622–627. doi:10.1126/science.1114397
Oberdörster G, Stone V, Donaldson KT (2007) Toxicology of nanoparticles: a historical perspective. Nanotoxicology 1(1):2–25. doi:10.1080/17435390701314761
Ohkuma S, Poole B (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci 75:3327–3330
Pankhurst QA, Thanh NKT, Jones SK, Dobson J (2009) Progress in applications of magnetic nanoparticles in biomedicine. J Phys D Appl Phys 42(22):224001. doi:10.1088/0022-3727/42/22/224001
Schwegmann HJ, Feitz AJ, Frimmel FH (2010) Influence of the zeta potential on the sorption and toxicity of iron oxide nanoparticles on S. cerevisiae and E. coli. J. Colloid. Interf. Sci. 347:43–48. doi:10.1016/j.jcis.2010.02.028
Smith BR, Gambhir SS (2017) Nanomaterials for in vivo imaging. Chem Rev 117(3):901–986. doi:10.1021/acs.chemrev.6b00073
Veranth JM, Kaser EG, Veranth MM, Koch M, Yost GS (2007) Cytokine responses of human lung cells (BEAS-2B) treated with micron-sized and nanoparticles of metal oxides compared to soil dusts. Part Fibre Toxicol 4:2. doi:10.1186/1743-8977-4-2
Wu M, Guo Q, Xu F, Liu S, Lu X, Wang J, Gao H, Luo P (2016) Engineering phosphopeptide-decorated magnetic nanoparticles as efficient photothermal agents for solid tumor therapy. J Colloid Interf Sci 476:158–166. doi:10.1016/j.jcis.2016.05.023
Acknowledgements
We thank the Electron Microscopy Unit in the Chemistry Center of the IVIC for the TEM observation. The authors acknowledge Lic. Lisbeth Lozada and Joseba Echebarrieta for their support in sample preparation and SEM observation. The author (GG) acknowledges the financial support to FONACIT, grant number 20012001241 and to the Prometeo Fellowship provided by the Secretaría Nacional de Educación Superior, Ciencia, Tecnología e Innovación (SENESCYT) of Ecuador.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Briceño, S., Hernandez, A.C., Sojo, J. et al. Degradation of magnetite nanoparticles in biomimetic media. J Nanopart Res 19, 140 (2017). https://doi.org/10.1007/s11051-017-3800-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-017-3800-3