
Abstract
Molecular dynamics simulation has been implemented for doping effect on melting temperature, heat capacity, self-diffusion coefficient of gold–copper bimetallic nanostructure with 55 total gold and copper atom numbers and its bulk alloy. Trend of melting temperature for gold–copper bimetallic nanocluster is not same as melting temperature copper–gold bulk alloy. Molecular dynamics simulation of our result regarding bulk melting temperature is consistence with available experimental data. Molecular dynamics simulation shows that melting temperature of gold–copper bimetallic nanocluster increases with copper atom fraction. Semi-empirical potential model and quantum Sutton–Chen potential models do not change melting temperature trend with copper doping of gold–copper bimetallic nanocluster. Self-diffusion coefficient of copper atom is greater than gold atom in gold–copper bimetallic nanocluster. Semi-empirical potential within the tight-binding second moment approximation as new application potential model for melting temperature of gold–copper bulk structure shows better result in comparison with EAM, Sutton–Chen potential, and quantum Sutton–Chen potential models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Melting of small particle is one interesting topic of early day of modern cluster science (Couchman and Jesser 1977). The first of theoretical research regarding melting temperature was on macroscopic framework (Ross and Andres 1981). From a theoretical point of view, melting of nanoalloys has been studied by classical thermodynamic methods, (Valle et al. 2001; Liang et al. 2003; Shirinyan and Gusak 2004; Jesser et al. 2004; Shirinyan and Pasichnyy 2005; Shirinyan et al. 2006) while several systems have been studied by computer simulations, which are very well suited to analyze the melting process in detail. The simulations have shown that nanoalloys may undergo complex structural transformations before complete melting (Jellinek and Krissinel 1999; Van Hoof and Hou 2005). Many theoretical jobs have been done for understanding how solid to liquid phase changes in far below bulk limit (Berry and Wales 1989; Maillet et al. 1996; Bulgac and Kusnezov 1992; Kunz and Berry 1993; Wales and Berry 1994; Kim and Tomanek 1994; Luo et al. 1987; Doye and Wales 1998).
Nanoclusters are aggregation of atoms or molecules of nanometric size, containing a number of constituent particles ranging from 10 to 106 (Baletto and Ferrando 2005). There are unusual chemical and physical properties in metallic nanocluster in comparison with those metal bulk properties. Due to extended application of metallic nanoclusters in heterogeneous catalysts, electronic devices and microelectronic, investigation of structural and dynamical properties of them are interested topics of previous literature (Ferrando et al. 2008; Huang et al. 2003, Negreiros et al. 2012).
Applications of metal nanoclusters to heterogeneous catalysis have shown that good selectivity can be achieved toward specific reactions by controlling the catalyst surface composition (Bracey et al. 2009). Gold nanoparticle catalysts are used mainly in selective oxidation reactions and a broad range of organic transformations besides possessing in hydrogenation activity (Della Pina et al. 2008). Copper nanoparticles are known to be active for the oxidation of alcohols to aldehydes (Yoo and Li 2007). Therefore, by mixing the two metals, Au–Cu bimetallic nanoclusters offer a way to finely tune catalytic properties of these two metals. Nevertheless, understanding their structural–thermal stability relationship is a difficult task. Molecular dynamic simulation is an ideal tool for investigating of thermal physical properties such small cluster. Among different bimetallic alloy nanoparticle catalysts, AuCu catalysts have recently attracted interests for catalytic oxidation of CO, benzyl alcohol, and propene (Liu et al. 2008; Zhang 2012; Zhang et al. 2008) and for partial oxidation of methanol to produce hydrogen fuels. Previous literature shows that genetic algorithm has been used to get optimum structure of gold–copper bimetallic nanocluster up to 56 atoms with Gupta potential model (Darby et al. 2002). Global optimization techniques and molecular dynamics simulations are extended to investigate the structure and the chemical order of AuCu bimetallic nanocluster clusters, with different compositions (Toai et al. 2008). Structural morphology of some bimetallic nanocluster such as copper–silver, cobalt–gold, nickel–rhodium has been investigated by Panizon et al. Small impurities convert morphology transitions from crystalline to icosahedrons (Ih) structures that can release some strain at the core of Ih matrices (Panizon et al. 2014). Single impurity metal has significant effect on melting temperature of metallic nanoparticle (Mottet et al. 2005). Molecular dynamics simulation shows that for melting of bimetallic nanocluster Ag–Ni, Ag–Co, melting phenomena occur from silver surface atom at shell of cluster however Ni and Co atoms remain solid in the mentioned bimetallic nanocluster (Kuntova et al. 2008). Calculation local pressure shows that impurity has significant effect on local streets in metallic nanoparticle (Laasonen et al. 2013).
Experimental results show that the Ih phase structure can be stabilized with the addition of some impurities. Previous literature shows that Ag55 and Cu55 have the Ih structure whereas Au55 cluster has a low-symmetry C 1 structure (Huang et al. 2008). Previous study shows that a single Cu atom makes C 1 structure in Au55 cluster which converts to the Ih structure. As a result, melting temperature of Au55 with the presence of copper as a doping increases significantly (Gao et al. 2010; Häkkinen et al. 2004, Xing et al. 2005; Cheng et al. 2006). It is well known that the melting temperature of Au55 (380 K) is much lower than that of Cu1Au54 (530 K) (Sankaranarayanan et al. 2005). A Cu–Au cluster does not experience a smooth transition from a pure Cu-like behavior to a pure Au cluster with a decrease in the concentration of Cu. The thermal behavior of Cu–Au is mainly influenced by the presence of Cu atoms in the bimetallic structure (Rodriguez-López et al. 2003). Studies of phase conformations and thermal behavior of AuCu binary clusters by classical molecular dynamics simulations showed that the cluster size, the concentration in the alloy, and the annealing temperature have a dominant effect on this morphology reconstruction process (Yin et al. 2012). For example, when the starting morphology is of a cuboctahedral type, by changing the concentration of copper from 50 to 10 %, an optimum stability of an icosahedral-type morphology is found at a bimetallic concentration of gold 75 % and copper 25 %, in fair agreement with experimental reports (Taherkhani et al. 2014). In small of nanostructure surface-to-volume ratio is large, and then surface effect has significant contribution on physical quantities such as transport properties, melting point, and thermal energy (Huang and Balbuena 2002; Akbarzadeh and Taherkhani 2013; Taherkhani et al. 2012).
The knowledge of diffusion behavior is of interest for production of metallic materials and for their practical use in the near phase transition. In this base, investigation is performed on the Pt–Pd (Sankaranarayanan et al. 2005), Cu–Ni (Huang and Balbuena 2002), Pt (Akbarzadeh and Parsafar 2009), Au–Pd (Mejía-Rosales et al. 2006), and Fe–Al (Mehrer et al. 1997) systems. In all cases, self-diffusion coefficient increases with temperature. Self-diffusion coefficient decreases with the pressure variable (Holzapfel et al. 2009; Lott et al. 2002).
Semi-empirical potential model has been used for obtaining optimum structural of gold nanocluster with size 38, 55, and 75 as well as density functional theory (Garzón et al. 1998). A simple analytical embedded-atom method (EAM) model has been extended for seven fcc metals (Al, Ag, Au, Cu, Ni, Pd, and Pt) and their binary alloys (Cai and Ye 1996). Dissolution of one nanocrystal can be investigated via an ab initio molecular dynamics method (Holmberg et al. 2014).
In present study, we show that the presence of the impurity at the pure cluster affects on melting temperature for gold–copper bimetallic nanostructure as well as bulk structure. In this work, we study the structure details and dynamic behavior of Au–Cu nanoclusters for the gold–copper bimetallic nanocluster. We explore variation of the heat capacity, self-diffusion coefficient, and structural energy with temperature and doping variables. Comparison of melting temperature of bulk gold–copper alloy has been done with different potential models.
MD simulation details
In classical MD, each particle is treated as a point mass, and the motion is governed by Newton’s second law. After solving Newton equations and getting velocity and position of all particles versus time, the thermodynamic properties of system can be obtained from time averages over the ensembles of particles.
Molecular dynamics simulations were done in the constant temperature ensemble (NVT) using the Verlet algorithm for the integration of Newton’s equations of motion, with a time step of one femto second resulting in long simulation runs of 500 picoseconds (ps). Ensemble averaging has been done after 400 ps. The Nose–Hoover thermostat is used for controlling the temperature of molecular dynamics simulation with DLPOLY package (Smith and Todorov 2006). The quantum Sutton–Chen (QSC) many-body inter-atomic potential leads to an accurate description of many properties of metallic system with icosahedrons (Ih) geometrical symmetries and their alloys. In this work, we have applied the QSC inter-atomic potential to model the interaction in the Au–Cu alloy clusters. A cut-off ratio for the range of the interaction of 7.0 Å was used in all the cases. For obtaining optimum structure of all component in nano and bulk structure, first heating of system has been done above melting temperature, and then during the annealing technique cooling, the system have been done with rate 3 × 1010 K/s slowly (Kim et al. 2008)
Potential function
Molecular dynamics simulations using DLPOLY were used to gain insights into the melting process at the atomistic level. Molecular dynamics simulation by using DLPOLY package via different semi-empirical potential models has been done on melting temperature of gold–copper bulk alloy. All the thermodynamic and transport properties were obtained as time averages over the particle positions and velocities. The local electronic density is included to account for the many-body terms (Sankaranarayanan et al. 2005). Based on the Sutton–Chen potential, the potential energy of the finite system is given by
where r ij is the distance between atoms i and j, c a positive dimensionless parameter, and ɛ is a parameter with the dimensions of energy. Parameter v(r ij ) is a pair potential for including the repulsion of Pauli’s exclusion principle,
The local density accounting for cohesion associated with any atom i is given by
where ρ i is a local electron density accounting for cohesion associated with atom i, and a is a length parameter scaling all spacing (leading to dimensionless V and ρ); n and m are both positive integer parameters with condition n > m.
Sutton and Chen restricted values of m to be greater than 6 and fitted it to give close agreement with the bulk modulus and the elastic constants. The Sutton–Chen potential predicts properties involving defects, surfaces, and interfaces poorly. The quantum Sutton–Chen potential includes quantum corrections and takes into account the zero-point energy allowing better prediction of temperature-dependent properties. The QSC potential function was found to be better suited to melting and phase transformation studies of bulk Cu–Ni (Qi et al. 1999), Cu–Au (Qi et al. 2001), and Pd–Ni (Özdemir et al. 2004) alloys. The QSC parameters for Au and Cu are listed in Table 1 (Qi et al. 2001). Semi-empirical potential derived within the tight-binding second moment approximation by Cleri and Rosato was applied to describe interactions for bulk systems (Cleri and Rosato 1993).The parameters of mentioned empirical potential can be defined with exact work Cleri and Rosato (1993).
Self-diffusion coefficient
The self-diffusion coefficients for the two atomic species were calculated from the slope of the mean-square displacement (Taherkhani et al. 2010) T(MSD). The self-diffusion coefficient:
where Δτ is the simulation time and r i refers to the vector position of the molecule at time t. The ensemble average is taken over all molecules in the system and over several time origins. A linear least squares regression is performed on the MSD curve to give the slope from which the diffusivity at any temperature can be found (Huang and Balbuena 2002). Information regarding diffusion mechanism and its estimation value can be investigated via scanning tunneling microscopy experimentally (Huijben et al. 2006a, b).
Result and discussion
Solid–liquid phase transition in bulk Au–Cu alloy
Copper doping has been done on melting temperature of gold–copper Cu x Au55−x bimetallic nanostructure. Sutton–Chen (SC) (Kart et al. 2005), quantum Sutton–Chen (QSC) many-body (Kart et al. 2005), and EAM (Lv and Chen 2011) potentials have been used for investigation of solid–liquid phase transition of gold–copper bulk alloy previously. For comparison and validation of different potential models, we have chosen semi-empirical potential model for investigation of melting temperature of copper–gold alloy in bulk structure. Many techniques such as distortion parameter (Akbarzadeh et al. 2013, 2014), thermal conductivity (Taherkhani and Rezania 2012), self-diffusion coefficient (Ala-Nissila et al. 2002), and heat capacity (Taherkhani et al. 2012, 2014) can be used for recognition of solid–liquid phase transition (Akbarzadeh et al. 2013). Chemical-ordering effect is investigated on phase transition of solid–liquid copper–gold bulk structure. Heat capacity is used for investigation of melting temperature of gold–copper bulk structure. Heat capacity can be calculated from fluctuation of internal energy as a following equation:
where C V, \(\langle \delta E^{ 2} \rangle_{\text{NVT}}\), k B, and T are heat capacity, fluctuation of internal energy in NVT ensemble, Boltzmann constant, and temperature, respectively. Caloric curve, derivative average configurational energy with respective to the temperature, has been used for getting heat capacity for present case of study.
Peak in heat capacity graph is proportional to the melting temperature for pure and gold and its alloy in bulk structure.
Result of melting temperature with semi-empirical potential is represented at Table 2. According to Table 2, there is following order for melting temperature:
Simulation result for bulk melting temperature with semi-empirical potential model shows good agreement with available experimental data. On the basis of Table 2, melting temperature with semi-empirical potential shows a good agreement with available experimental data in comparison with other potential model such as quantum Sutton–Chen, EAM, and Sutton–Chen potential models.
On the basis of Table 2, results for copper and gold in bulk, solid–liquid phase transition are 1338 and 1325 K, respectively. Our results regarding melting point for bulk copper and gold metal are consistence with the experimental result 1356 and 1337 K, respectively.
Solid–liquid phase transition in bimetallic nanocluster Au–Cu
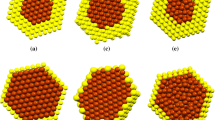
Molecular dynamics simulation has been used for melting temperature of bimetallic Au–Cu system with total atom 55. The most direct way to analyze the thermal behavior is considering the heat capacity versus temperature. For this purpose, we calculated the heat at different temperatures; the melting temperature (T m) of the system would be located in the point where heat capacity presents a jump in temperature. Result of heat capacity for different chemical compositions namely Cu6Au49, Cu43Au12, Cu18Au37, Cu28Au27, Cu41Au14, Cu49Au6, and Cu55 gold–copper bimetallic nanoclusters is presented at Fig. 1. The melting point of bimetallic Au–Cu 55 atom nanoclusters is examined with using the quantum Sutton–Chen many-body potential function. Impurity effect on the melting point for gold–copper bimetallic nanoclusters is considered. Result of melting temperature as a function of doping is presented as a Fig. 2. According to Fig. 2, melting temperature of gold–copper bimetallic nanocluster increases with copper atom fraction. Result of melting temperature via molecular dynamics is compared to the Monte Carlo (MC) simulation with semi-empirical potential (Cheng et al. 2006). Our simulation result confirms MC result regarding melting temperature with impurity copper doping in gold–copper bimetallic nanocluster. In both methods of simulations, melting temperature increases with copper doping in gold–copper bimetallic nanocluster. Result of melting temperature shows that a different application of potential model does not change trend of melting temperature of gold–copper bimetallic nanocluster with copper doping. Snapshot including brown atom for Cu yellow atom for Au about Cu12Au43, Cu6Au49, and Cu55 structures before and after melting temperature is presented at Fig. 3a–f, respectively. On the basis of Fig. 3, structure of nanocluster is destroyed completely after melting temperature, and it is completely set in solid phase before melting temperature. Our result shows that melting phenomena are started with the surface gold atom at the shell of gold–copper bimetallic nanocluster; however, copper atoms stay solid in core of the cluster. Trend of melting temperature for bulk gold–copper is not same as melting temperature trend for gold–copper bimetallic nanocluster. Surface energy effect is an important phenomenon that observes in nanosystem not in bulk structure. Previous literature shows that surface energy of gold nanoparticle is less than copper nanoparticle (Taherkhani et al. 2014). As a result, most of gold atom moves on surface. Strength of metallic bond for copper is greater than gold, as well. From what has been discussed, it can be concluded that meting temperature of gold–copper bimetallic increases with more copper atom fraction due to surface effect. On the basis of Fig. 2, melting temperature for gold–copper bimetallic nanocluster is lower than melting temperature of bulk structure significantly. Average pressure tensor upon the insertion of Cu impurities in Au icosahedra has been calculated to see whether the stabilization of the icosahedral structure is due to strain relaxation. Based on the following equations for average pressure tensor calculation (Laasonen et al. 2013),

Heat capacity for Cu6Au49, Cu43Au12, Cu18Au37, Cu28Au27, Cu41Au14, Cu49Au6, and Cu55 as a function of temperature which is shown at a–g, respectively

Melting temperature versus copper fraction with MD and MC methods

Snapshot structure for Cu12Au43, Cu6Au49, and Cu55 before and after melting temperature with a–f. (brown atom, Cu; yellow atom, Au)
Local pressure P i for each atom i can be defined as a following equation:
Average pressure for the nano cluster can be defined as
P −, σ i , \(r_{i,j}^{a}\), \(r_{i,j}^{b}\), V i , and N are average pressure, stress tensor, cartesian components of the vector r i,j with (a, b = x, y, z), atomic volume, and number of atom in nanocluster, respectively (Laasonen et al. 2013). Our calculations regarding average tensor pressure for Cu6Au49 and Cu27Au28 of gold–copper bimetallic nanocluster are 0.0266 and −0.0822 GPa, respectively. Average pressure analysis shows that for Cu6Au49, pressure value is positive; however, for Cu27Au28, average pressure is negative. As a result, with copper doping, icosahedron gold–copper bimetallic nanocluster becomes stable due to lower stress, and more melting temperature is expected.
Self-diffusion coefficient result
Molecular dynamics simulation has been performed for calculation of self-diffusion coefficient. Self-diffusion coefficient is calculated from the slope of the mean-square displacement, Eq. 4. Result of self-diffusion coefficient gold, copper versus temperature is shown in Fig. 4 for Cu12Au43, Cu18Au37, and Cu41Au14 bimetallic nanoclusters. On the basis of Fig. 4 both self-diffusion coefficient values for copper and gold increase as a function of temperature. Self-diffusion result for each gold–copper composition shows that there is a jump for both self-diffusions of gold and copper atom near the its melting temperature. On the basis of Fig. 4 for all compositions of gold–copper bimetallic nanocluster namely Cu12Au43, Cu18Au37, and Cu41Au14, self-diffusion of copper is more than gold atom. Self-diffusion of gold and copper atom in gold–copper bimetallic nanocluster is greater that the bulk value (Butrymowicz et al. 1974) due to surface effect in nanosystem.

Self-diffusion coefficient versus temperature for Cu12Au43, Cu18Au37, and Cu41Au14 with a–c, respectively
Conclusion
Molecular dynamics simulation has been done for investigation disorder effect on melting temperature, self-diffusion coefficient, and heat capacity in copper–gold bimetallic nanocluster. Molecular dynamics simulations regarding melting temperature of gold–copper bimetallic nanocluster with total 55 atoms show that melting temperature increases with copper atom fraction due to lower surface energy and metallic bond energy of gold atom. Molecular dynamics simulation with Quantum Sutton–Chen potential model estimates melting temperature of gold–copper bimetallic nanocluster around 600–800 K temperature range which is lower than its bulk structure significantly. Molecular dynamics simulation shows that self-diffusion coefficient increases with temperature and there is jump for self-diffusion coefficient, which confirms solid-to-liquid phase transitions. On the basis of molecular dynamics simulation, self-diffusion coefficient of copper is greater than silver atom, and self-diffusion value for copper and gold atom is greater than those in bulk structure. Heat capacity has been used for determination of melting temperature of gold–copper bimetallic nanocluster. Molecular dynamics simulation with semi-empirical potential within the tight-binding second moment approximation has been used as a new application potential model for melting temperature of gold–copper bulk structure. There is following order for melting temperature of gold–copper alloy in bulk structure:
Semi-empirical potential model for melting temperature of bulk structure shows good agreement with available experimental data in comparison with EAM, quantum Sutton–Chen, and Sutton–Chen potential models.
References
Akbarzadeh H, Parsafar GA (2009) A molecular-dynamics study of thermal and physical properties of platinum nanoclusters. Fluid Phase Equilib 280:16
Akbarzadeh H, Taherkhani F (2013) Cluster size dependence of surface energy of Ni nanocluster: a molecular dynamics study. Chem Phys Lett 558:57
Akbarzadeh H, Yaghoubi H, Shamkhali AN, Taherkhani F (2013) Effects of gas adsorption on the graphite-supported Ag nanoclusters: a molecular dynamics study. J Phys Chem C 117:26287–26294
Akbarzadeh H, Yaghoubi H, Shamkhali AN, Taherkhani F (2014) CO adsorption on Ag nanoclusters supported on carbon nanotube: a molecular dynamics study. J Phys Chem C 118:9187–9195
Ala-Nissila T, Ferrando R, Ying S (2002) Collective and single particle diffusion on surfaces. Adv Phys 51:949–1078
Baletto F, Ferrando R (2005) Structural properties of nanoclusters: Energetic, thermodynamic, and kinetic effects. Rev Mod Phys 77:1
Berry RS, Wales DJ (1989) Freezing, melting, spinodals, and clusters. Phys Rev Lett 63:156
Bracey CL, Ellis PR, Hutchings GJ (2009) Application of copper–gold alloys in catalysis: current status and future perspectives. Chem Soc Rev 38:2231–2243
Bulgac A, Kusnezov D (1992) Phase transitions in Na7–Na9 microclusters. Phys Rev B 45:988
Butrymowicz DB, Manning JR, Read E (1974) Diffusion in copper and copper alloys part V. Diffusion in systems involving elements of group VA. Phys Chem Ref Data 3:572
Cai J, Ye YY (1996) Simple analytical embedded-atom-potential model including a long-range force for fcc metals and their alloys. Phys Rev B 54:8398
Cheng D, Huang S, Wang W (2006) Thermal behavior of core–shell and three-shell layered clusters: melting of Cu1Au54 and Cu12Au43. Phys Rev B 74:064117
Cleri F, Rosato V (1993) Tight-binding potentials for transition metals and alloys. Phys Rev B 48:22
Couchman PR, Jesser WA (1977) Thermodynamic theory of size dependence of melting temperature in metals. Nature (London) 269:81
Darby S, Mortimer-Jones TV, Johnston RL, Roberts C (2002) Theoretical study of Cu–Au nanoalloy clusters using a genetic algorithm. J Chem Phys 116:1536
Della Pina C, Falletta E, Prati L, Rossi M (2008) Selective oxidation using gold. Chem Soc Rev 37:2077–2095
Doye JPK, Wales DJ (1998) Thermodynamics of global optimization. Phys Rev Lett 80:357
Ferrando R, Jellinek J, Johnston RL (2008) Nanoalloys: from theory to applications of alloy clusters and nanoparticles. Chem Rev 108:845–910
Gao Y, Shao N, Pei Y, Zeng XC (2010) Icosahedral crown gold nanocluster Au43Cu12 with high catalytic activity. Nano Lett 10:1055–1062
Garzón IL, Michaelian K, Beltrán MR, Posada-Amarillas A, Ordejón E, Artacho P, Sánchez-Portal D, Soler JM (1998) Lowest energy structures of gold nanoclusters. Phys Rev Lett 81:1600
Häkkinen H, Moseler M, Kostko O, Morgner N, Hoffmann MA, Issendorff Bv (2004) Symmetry and electronic structure of noble-metal nanoparticles and the role of relativity. Phys Rev Lett 93:093401
Holmberg N, Chen JC, Foster AS, Laasonen K (2014) Dissolution of NaCl nanocrystals: an ab initio molecular dynamics study. Phys Chem Chem Phys 16:17437
Holzapfel C, Chakraborty S, Rubie D, Frost D (2009) Fe–Mg interdiffusion in wadsleyite: the role of pressure, temperature and composition and the magnitude of jump in diffusion rates at the 410 km discontinuity. Phys Earth Planet Inter 172:28–33
Huang S-P, Balbuena PB (2002) Melting of bimetallic Cu–Ni nanoclusters. J Phys Chem B 106:7225–7236
Huang S-P, Mainardi DS, Balbuena PB (2003) Structure and dynamics of graphite-supported bimetallic nanoclusters. Surf Sci 545:163–179
Huang W, Ji M, Dong C-D, Gu X, Wang L-M, Gong XG, Wang L-S (2008) Relativistic effects and the unique low-symmetry structures of gold nanoclusters. ACS Nano 2:897–904
Huijben J, van Houselt A, Zandvliet HJW, Poelsema B (2006a) Semiconductors II: surfaces, interfaces, microstructures, and related topics-influence of dimer buckling on dimer diffusion: a scanning tunneling microscopy study. Phys Rev B 73:73311
Huijben J, van Houselt A, Zandvliet HJW, Poelsema B (2006b) Influence of dimer buckling on dimer diffusion: a scanning tunneling microscopy study. Phys Rev B 73:073311
Jellinek J, Krissinel EB (1999) In: Jellinek J (ed) Theory of atomic and molecular clusters. Springer, Berlin, p 277
Jesser WA, Shneck RZ, Gile WW (2004) Solid–liquid equilibria in nanoparticles of Pb–Bi alloys. Phys Rev B 69:44121
Kart H, Tomak M, Çağin T (2005) Thermal and mechanical properties of Cu–Au intermetallic alloys. Modell Simul Mater Sci Eng 13:657
Kim SG, Tomanek D (1994) Melting the fullerenes: a molecular dynamics study. Phys Rev Lett 72:418
Kim DH, Kim HY, Kim HG, Ryu JH, Lee HM (2008) The solid-to-liquid transition region of an Ag–Pd bimetallic nanocluster. J Phys 20:035208
Kuntova Z, Rossi G, Ferrando R (2008) Melting of core–shell Ag–Ni and Ag–Co nanoclusters studied via molecular dynamics simulations. Phys Rev B 77:205431
Kunz RE, Berry RS (1993) Coexistence of multiple phases in finite systems. Phys Rev Lett 71:987
Laasonen K, Panizon E, Bochicchio D, Ferrando R (2013) Competition between icosahedral motifs in AgCu, AgNi, and AgCo nanoalloys: a combined atomistic–DFT Study. J Phys Chem C 117:26405
Liang LH, Liu D, Jiang Q (2003) Size-dependent continuous binary solution phase diagram. Nanotechnology 14:38
Liu X, Wang A, Wang X, Mou C-Y, Zhang T (2008) Au–Cu alloy nanoparticles confined in SBA-15 as a highly efficient catalyst for CO oxidation. Chem Commun 27:3187–3189
Lott K, Nirk T, Volobujeva O (2002) Chemical self-diffusion in undoped ZnS and in undoped CdSe. Cryst Eng 5:147–153
Luo J, Landmann U, Jortner J (1987) In: Jena P, Rao BK, Khanna S (eds) Physics and chemistry of small cluster, vol 158., NATO ASI series BPlenum, New York, p 155
Lv YJ, Chen M (2011) Thermophysical properties of undercooled alloys: an overview of the molecular simulation approaches. Int J Mol Sci 12:278–316
Maillet JB, Boutin A, Fuchs AH (1996) Numerical evidence of an embryonic orientational phase transition in small nitrogen clusters. Phys Rev Lett 76:336
Mehrer H, Eggersmann M, Gude A, Salamon M, Sepiol B (1997) Diffusion in intermetallic phases of the Fe–Al and Fe–Si systems. Mater Sci Eng A 239:889–898
Mejía-Rosales SJ, Fernández-Navarro C, Pérez-Tijerina E, Montejano-Carrizales JM, José-Yacamán M (2006) Two-stage melting of Au–Pd nanoparticles. J Phys Chem B 110(26):12884–12889
Mottet C, Rossi G, Baletto F, Ferrando R (2005) Single impurity effect on the melting of nanoclusters. Phys Rev Lett 95:035501
Negreiros F, Taherkhani F, Parsafar G, Fortunelli A (2012) Kinetics of chemical ordering in Ag–Pt nanoalloy via first-principal simulation. J. Chem. Phys. 137:194302
Özdemir S, Tomak M, Uludoğan M, Çağın T (2004) Liquid properties of Pd–Ni alloys. J Non-Cryst Solids 337:101–108
Panizon E, Bochicchio D, Rossi G, Ferrando R (2014) Tuning the structure of nanoparticles by small concentrations of impurities. Chem Mater 26:3354
Qi Y, Çağın T, Kimura Y, Goddard WA III (1999) Molecular-dynamics simulations of glass formation and crystallization in binary liquid metals: Cu–Ag and Cu–Ni. Phys Rev B 59:3527
Qi Y, Çağin T, Kimura Y, Goddard W III (2001) A. scosities of liquid metal alloys from nonequilibrium molecular dynamics. J Comput Aided Mater Des 8:233–243
Rodrıguez-López J, Montejano-Carrizales J, José-Yacamán M (2003) Molecular dynamics study of bimetallic nanoparticles: the case of Au x Cu y alloy clusters. Appl Surf Sci 219:56–63
Ross J, Andres RP (1981) Melting temperature of small cluster. Surf Sci 106:1
Sankaranarayanan SK, Bhethanabotla VR, Joseph B (2005) Molecular dynamics simulation study of the melting of Pd–Pt nanoclusters. Phys Rev B 71:195415
Shirinyan AS, Gusak AM (2004) Phase diagrams of decomposing nanoalloys. Philos Magn 84:79
Shirinyan AS, Pasichnyy MO (2005) Size-induced hysteresis in the process of nucleation and phase separation in a nanopowder. Nanotechnology 16:724
Shirinyan A, Wautelet M, Belogorodsky YJ (2006) Solubility diagram of the Cu–Ni nanosystem. J Phys 18:537
Smith W, Todorov ITA (2006) Short description of DL_POLY. Mol Simul 32:935–943
Taherkhani F, Rezania H (2012) Temperature and size dependency of thermal conductivity of aluminum nanocluster. J Nanopart Res 14:1–8
Taherkhani F, Negreiros FR, Parsafar G, Fortunelli A (2010) Simulation of vacancy diffusion in a silver nanocluster. Chem Phys Lett 498:312–316
Taherkhani F, Akbarzadeh H, Abroshan H, Fortunelli A (2012) Dependence of self-diffusion coefficient, surface energy, on size, temperature, and Debye temperature on size for aluminum nanoclusters. Fluid Phase Equilib 335:26
Taherkhani F, Akbarzadeh H, Rezania H (2014) Chemical ordering effect on melting temperature, surface energy of copper–gold bimetallic nanocluster. J Alloy Compd 617:746
Toai TJ, Rossi G, Ferrando R (2008) Global optimisation and growth simulation of AuCu clusters. Faraday Discuss 138:49–58
Valle´e R, Wautelet M, Dauchot JP, Hecq M (2001) Size and segregation effects on the phase diagrams of nanoparticles of binary systems. Nanotechnology 12:8
Van Hoof T, Hou M (2005) Structural and thermodynamic properties of Ag–Co nanoclusters. Phys Rev B 72:15434
Wales DJ, Berry RS (1994) Coexistence in finite systems. Phys Rev Lett 73:875
Xing X, Danell RM, Garzón IL, Michaelian K, Blom MN, Burns MM, Parks JH (2005) Size-dependent fivefold and icosahedral symmetry in silver clusters. Phys Rev B 72:081405
Yin J, Shan S, Yang L, Mott D, Malis O, Petkov V, Cai F, Shan Ng M, Luo J, Chen BH (2012) Gold–copper nanoparticles: nanostructural evolution and bifunctional catalytic sites. Chem Mater 24:4662–4674
Yoo W-J, Li C-J (2007) Copper-catalyzed oxidative esterification of alcohols with aldehydes activated by Lewis acids. Tetrahedron Lett 48:1033–1035
Zhang L (2012) A molecular dynamics study of thermal behavior of melting an Au54Cu1 cluster. Proc Eng 36:207–211
Zhang L, Zhang C-B, Qi Y (2008) Local structure changes of 54-, 55-, 56-atom copper clusters on heating. Phys Lett A 372:2874–2880
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Taherkhani, F., Akbarzadeh, H., Feyzi, M. et al. Disorder effect on heat capacity, self-diffusion coefficient, and choosing best potential model for melting temperature, in gold–copper bimetallic nanocluster with 55 atoms. J Nanopart Res 17, 29 (2015). https://doi.org/10.1007/s11051-015-2860-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-015-2860-5