
Abstract
Three different batches of pure nanoparticles (NPs) of nicergoline (NIC) were prepared by spray drying a water:ethanol solution by a new Nano Spray Dryer Büchi B-90. Spherical pure NPs were obtained, and several analytical techniques such as differential scanning calorimetry and X-ray powder diffractometry permitted to assess their amorphous character. A comparison of the solubility, intrinsic dissolution, and drug release of original particles and pure amorphous NPs were determined, revealing an interesting improvement of biopharmaceutical properties of amorphous NPs, due to both amorphous properties and nanosize dimensions. Since in a previous work, the high-thermodynamic stability of amorphous NIC was demonstrated, this study is addressed toward the formulation of NIC as pure amorphous NPs.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The number of poorly soluble synthetic drugs is steadily increasing. About 40 % of the drugs in the development pipelines (Speiser 1998) and approximately 60 % of the newly synthesized drugs are poorly soluble (Merisko-Liversidge 2002). This increasing number of poorly soluble drugs requires innovative formulation approaches in order to reach a sufficiently high bioavailability after oral administration or at least to make available intravenously injectable forms (Keck and Müller 2005).
A classical formulation approach for such poorly soluble drugs is micronization. This technique is used to reduce the coarse drug powder into particles with a mean size typically ranging between 2 and 5 μm. According to the Noyes–Whitney law (Noyes and Whitney 1897) indeed, the dissolution velocity is increased by enlarging the surface area of the drug powder.
Nowadays, many of the new drugs exhibit such a low solubility that micronization does not lead to a sufficiently high bioavailability. The next step taken is to move from micronization to “nanonization” producing drug nanocrystals.
Nanocrystals, consisting of pure drugs and minimum of surface active agents required for stabilization, are a carrier-free submicron colloidal drug delivery with a mean particle size in the nanometer range, typically between 10 and 100 nm (Junghanns and Müller 2008).
Not only drug nanocrystals further increase the dissolution velocity, but they also augment the saturation solubility. They could therefore more efficiently improve the oral absorption of the drugs and achieve a higher bioavailability compared to the microparticles (Gao et al. 2008).
Classical methods for the production of nanocrystals at both laboratory and industrial scale are precipitation, milling, and high-pressure homogenization methods (Shegokar and Müller 2010).
A new spray dryer, the Nano Spray Dryer B-90 (Büchi Instruments), was used in this study for the generation of particles of nano-dimensions. This is the only spray dryer system that permits to obtain particles with mean size in the nanometric range, narrow particle size and yields about 90 %. Thus, it is very different from conventional spray dryer which instead permit to recover particles in the micrometric range (Maa et al. 1999; Elversson et al. 2003).
In this innovative system, the droplet generation is based on a piezoelectric driven actuator, vibrating a thin, perforated, stainless steel membrane in a small spray cap. The drying gas enters in laminar flow from the top into the drying chamber and is heated up to the set inlet temperature. The piezodriven spray head generates ultra-fine droplets, which are gently dried into solid particles. The dried solid particles are electrostatically charged and collected at the collecting electrode. The drying gas exits the spray dryer, the outlet temperature is measured, and the gas is filtered.
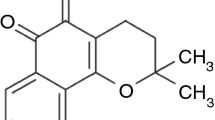
This new method was applied in this study to nicergoline (NIC). NIC (10α-methoxy-1,6-dimethylergoline-8β-methanol-5′-bromonicotinate) (C24H26BrN3O3), a semi-synthetic ergot derivative, is a potent blocking agent for α1-adrenoreceptors and thus has found effective clinical use in neurology (Heitz et al. 1986). Additionally, NIC demonstrated remarkable effects in lowering systemic blood pressure and dilating blood vessels, thus increasing peripheral blood flow (Brossi 1990). NIC has also been clinically used for improving metabolism in the brain, particularly for treatment of senile dementia (Venn 1980). The drug has been proven to have cerebral anti-ischemic action, also exhibiting platelet anti-aggregation and disaggregating actions (Goo et al. 1988; Pastoris et al. 1988). Several clinical studies have demonstrated that NIC is effective in lowering total peripheral resistance and normalizing blood pressure without producing reflex tachycardia, therefore the drug is useful in the early phases of acute myocardial infarction, due to the decrease of myocardial oxygen consumption (Triulzi et al. 1981).
Nicergoline is a poorly water soluble (0.002 mg/mL) active substance; an enhancement of solubility could potentially improve its bioavailability for oral use.
The objective of this study is to evaluate the feasibility of the generation of NIC nanoparticles (NPs) by the new nano-spray drying method and to characterize the NPs obtained by comparing their physicochemical properties with those of the original particles.
Depending on the production technology, nanocrystals can be either obtained in the crystalline or in the amorphous state, with the latter referred to as “nanocrystals in the amorphous state.” During the development of nanosized NIC particles, amorphous particles were obtained, but in this case the term “pure NPs” was preferred by the authors.
Materials and methods
Materials
Nicergoline (NIC) was purchased from China–Japan Shandong Hongfuda Pharmchem Co, Ltd. (Shandong, China) as white crystalline powder. NIC is referred to as native crystals (NCs) in the text. NIC is stable under normal conditions of use. Ultrapure water was produced by Gradient Milli-Q® (Millipore, Molsheim, France). Ethanol, 1-butanol, potassium acetate, potassium carbonate, magnesium nitrate, fructose, sodium chloride, potassium chloride, monobasic potassium phosphate, and sodium hydroxide were supplied by Sigma–Aldrich (Steinheim, Germany).
Preparation of NPs
Pure NPs were obtained by spraying a solution of 25 g/L (w/w) of NIC in a mixture of ethanol:ultrapure water 1:3 (50 g of NIC in a solution composed of 50 g of ethanol and 150 g of water). The spraying was performed by a Nano Spray Dryer B-90 (Büchi, Flawil, Switzerland) using a membrane with pores of 7 μm size, at an inlet temperature of 50 °C and a feeding rate of 90 L/min. Three different batches (A, B, and C) were produced in order to verify the reproducibility of the procedure.
Water uptake experiments
Approximately 100 mg of powders was carefully weighed and stored at 25 °C in desiccators containing saturated inorganic salt solutions giving various relative humidity (RH) percentages. The salts used were potassium acetate (22 % RH), potassium carbonate (43 % RH), magnesium nitrate (55 % RH), fructose (64 % RH), sodium chloride (75 % RH), and potassium chloride (86 % RH). Water uptake of powders stored at these conditions was followed by a discontinuous procedure, i.e., taking and weighing samples at regular intervals until equilibrium weight was reached (Kontny and Zografi 1995). The water content of the powders was determined considering the initial water content and the change in weight induced by storing the samples under each RH condition. The initial water content of powders was determined by drying them in a desiccator under vacuum in the presence of P2O5 as desiccant at 25 °C over night. Assays were carried out in triplicate. Powders were checked by X-ray powder diffractometry (XRPD) and thermal analysis before and after the water absorption test in order to assess any change in powder solid physical state.
Gas phase chromatography
The determination of ethanol residual solvent content in the NPs was performed by GPC according to the USP method (27-467 Organic Volatile Impurities) on an Agilent Shimadzu GC-14B chromatograph (Agilent Technologies, Waldbronn, Germany) fitted with a flame ionization detector. The packed column was a DB-624, 30 m × 3.0 mm; the carrier gas was helium (constant flow, 35 cm/s). Standard solutions were injected to validate the method for linearity, specificity, and repeatability. Assay solutions were obtained by a micro-distillation of 5 mL of 1-butanol containing 500 mg of the NIC to be analyzed. The general conditions were column temperature: isotherm at 210 °C; injection volume: 5 μL; and retention time: 4 min.
Karl Fisher titrimetry
The water content was determined according to the titrimetric method of Karl Fisher (Automat E 547, Metrohm, Herisau, Switzerland) after calibration with sodium tartrate (Sigma-Aldrich, Steinheim, Germany) and dissolution of crystals in methanol (Sigma-Aldrich, Steinheim, Germany). The Fischer reagent solutions were from Riedel-de-Haen (Ref: 36116 and 36117, Germany).
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was carried out by the simultaneous thermal analysis (STA) which enables to simultaneously analyze a sample for change in weight (TGA) and change in enthalpy flow (differential scanning calorimetry, DSC). In this text, the STA–TGA will be used to refer to TGA. The analysis was performed with a STA (STA 6000, Perkin–Elmer, Inc., Waltham, MA), under nitrogen atmosphere (20 mL/min) in 0.07 mL open aluminum oxide pans. STA was calibrated for temperature and heat flow with three standard metals (tin, indium, and zinc), taking into account their expected melting temperatures (505.08, 429.75, and 692.68 K, respectively), and for weight with an external Perkin–Elmer standard (Calibration Reference Weight P/NN520-0042, Material lot 91101 GB, Weight 55.98 mg, 01/23/08 VT). Calibration was repeatedly checked to assure deviation ≤±0.3 K.
DSC analysis
Differential scanning calorimetry analysis was performed on a Pyris 1 (Perkin–Elmer, Co. Norwalk, USA) equipped with a cooling device (Intracooler 2P, Cooling Accessory, Perkin–Elmer, Co. Norwalk, USA). A dry purge of nitrogen gas (20 mL/min) was used for all runs. DSC was calibrated for temperature and heat flow using a pure sample of indium and zinc standards. Sample mass was about 4–5 mg, and aluminum perforated pans of 50 μL were used.
X-ray powder diffractometry
X-ray powder diffractometry was used to check the amorphous state of the studied samples and to follow their physical stability. To this purpose, a Philips PW 1730 (Philips Electronic Instruments Corp., Mahwah, NJ, USA) as X-ray generator for Cu Kα radiation (λα1 = 1.54056 Å, λα2 = 1.54430 Å) was used. The experimental X-ray powder patterns were recorded on a Philips PH 8203. The goniometer supply was a Philips PW 1373, and the channel control was a Philips PW 1390. Data were collected in the discontinuous scan mode using a step size of 0.01° 2α. The scanned range was 2θ–40° (2θ).
Scanning electron microscopy
Nicergoline crystal morphology was determined using a scanning electron microscopy (SEM) (Stereoscan 360, Cambridge Instruments, Cambridge, United Kingdom). Samples were mounted on a metal stub with double-sided adhesive tape and then sputtered under vacuum with a gold layer of about 200 Å thickness using a metallizator (Balzer MED 010, Linchestein). The particle size of the original NIC powder was determined by measuring manually the mean Feret’s diameter of 500 particles.
Dynamic light scattering (DLS) analysis
The NP size was determined by DLS (Zetasizer Nano, S90, Malvern Instruments, Worcestershire, UK), by dispersing NPs in water.
Solubility measurements
An excess of drug powder was added to an empty glass vessel, stirred at 200 rpm, and placed in an incubator (Velp Scientifica, FTC 90E, Usmate, Italy). The amount of powder assured that, during the entire time interval for the experiment, an excess of powder was always present. Then, 200 mL of water or HCl 0.1 N or phosphate buffer pH 8.0 (USP 2011) was added. At regular intervals, 15 mL of the liquid phase was withdrawn after centrifugation (High Speed Refrigerated Centrifuge, Scilogex, Berlin, CT) and replaced with deionised water of the same temperature. After appropriate dilution, the concentration of the filtrate was determined by UV spectrophotometer at a wavelength of 288 nm (Cary 1E UV–VIS, Varian, Leinì, Italy). Assays were performed in triplicate. Solubility versus time profiles (over 120 min period) were determined at four different temperatures (10, 20, 30, and 37 °C). The undissolved material was immediately recovered after each solubility experiment and analyzed by XRPD in order to ascertain the characteristics of the solid material.
Intrinsic dissolution rate (IDR) study
Dissolution study was carried out by the rotating disk method (Banakar 1992). Thirteen millimeter diameter tablets were obtained by compressing 300 mg of powder in a Perkin–Elmer hydraulic press for IR spectroscopy KBr disks, at a force of 15 kN for 10 min. This process yielded tablets with a surface area of 132.73 mm2 that would not disintegrate during the test. Tablets were inserted into a stainless steel holder, so that only one face was exposed to the dissolution medium. The holder was then connected to the stirring motor of a dissolution apparatus (Erweka DT6, Gloucestershire, England), centrally immersed in a 1,000-mL beaker containing 900 mL of demineralized water or HCl 0.1 N at 37 °C and rotated at 50 rpm. Suitable aliquots were withdrawn with a regenerated cellulose filter syringe at specified times and assayed for drug content spectrophotometrically at a wavelength of 288 nm. A correction was calculated for cumulative dilution caused by replacement of the sample with an equal volume of original medium. Each test was repeated six times. Low-standard deviations were obtained, indicating the good reproducibility of this technique. The IDR were calculated from the slope of the straight line of cumulative drug release.
Particle dissolution
Particle dissolution was carried out in a 200-mL beaker containing 100 mL of demineralized water or HCl 0.1 N at 37 °C and rotated at 50 rpm. Sink conditions were assured during the experiments. Suitable aliquots were withdrawn with a regenerated cellulose filter syringe at specified times and assayed for drug content spectrophotometrically at a wavelength of 288 nm. A correction was calculated for cumulative dilution caused by replacement of the sample with an equal volume of original medium. Each test was repeated six times. Low-standard deviations were obtained, indicating the good reproducibility of this technique.
Stability study
Stability study was carried out at different conditions of storage. NPs were stored at 5 ± 3 °C for the long-term study (12 months) and at 25 °C ± 2 °C with 60 ± 5 % RH for the short-term stability study (6 months). In any case, particles were stored in tightly closed glass containers.
Statistical analysis
Data were analyzed by one-way analysis of variance (ANOVA), using a Bonferroni test. The statistical analysis was conducted using an Origin® software (version 8.5) (Northampton, MA). Results are shown as mean ± SD (standard deviation) and considered significantly different when P < 0.05.
Results and discussion
Physicochemical characterization of NPs
Several essays were performed to assess the formation of NPs by the new nanospray dryer. Several parameters were modified such as the solvent, the inlet temperature, and the feeding rate. Selected conditions, described in “Preparation of NPs” section, were used to produce three batches A, B, and C showing a good reproducibility of the powder characteristics, as demonstrated by the results presented in next paragraphs. The process yield was satisfying and ranges for the three batches from 91.0, 93.2, and 90.7 for, respectively, batches A, B, and C (Table 1).
The formation of NPs was firstly confirmed by SEM (Fig. 1). NCs were rather irregular and characterized by the presence of sharp edges. NPs of any batch were all spherical, and the particle size distribution was quite narrow. Figure 1b shows NPs of batch A as an example. Size and shape of NPs of batches B and C are practically identical to those of batch A. The particle size of the NPs was determined by DLS, and results were compared to those of NCs (Fig. 2). The aqueous nanosuspension had a mean particle diameter of 0.79 μm, while the mean particle diameter of NCs was 9.24 μm.

SEM microphotographs of NIC native crystals ×1,500 (a) and NIC nanoparticles batch A ×6,000 (b)

Particle size distribution profiles of NIC particles. The particle size of native crystals was determined by manually measuring the mean Feret’s diameter of 500 particles, while particle size of nanoparticles was determined by dynamic light scattering
The spray drying process favored the transformation of NIC in an amorphous form, which was assessed by DSC (Fig. 3) and XRPD (Fig. 4). XRPD patterns of NCs showed a typical diffractogram of a crystalline material and distances corresponded to the triclinic form I which is the most stable polymorph of NIC (Malaj et al. 2011). The diffractogram of NPs is nonetheless typical of an amorphous material characterized by the complete absence of diffraction peaks. The DSC thermogram of NCs shows the melting endotherm of NIC (Table 1), while the melting peak is absent in the thermogram of NPs confirming the result of XRPD that NPs are in the amorphous state. In the DSC thermogram of NPs, an endotherm approximately between 40 and 80 °C is clearly evident, corresponding to the dehydration of the amorphous material, as also confirmed by STA–TGA. The weight loss for NPs associated to this endotherm was 8.29 ± 1.24 %, while no significant dehydration was observed for NCs (weight loss 0.12 ± 0.10 %). In view of the water absorption of the amorphous NPs, a water uptake experiment was carried out in order to evaluate the ability of the amorphous material to uptake water as a function of RH. During this experiment, NCs absorbed very low percentages of water (max 1.8 ± 1.5 % at the highest 86 % RH) because of the crystalline character of this product. Unlike NCs, NPs absorbed up to 30.56 ± 1.2 % at the highest 86 % RH, confirming that the amorphous character of the NPs favored the absorption of water (Hamaura and Newton 1999). Amorphous materials showed indeed high hygroscopicity, as water can be absorbed not only at the surface, but also into the bulk of the material (Zografi 1988; Ahlneck and Zografi 1990; Crowley and Zografi 2002), according to the physicochemical and structural properties of the drug. In addition to the high hygroscopicity of the amorphous material, it must be noted that the high-particle surface strongly increased the water uptake. Powders were checked by XRPD and DSC before and immediately after water absorption test, and no changes in the solid physical state were observed.

DSC thermograms of NIC native crystals (a) and NIC nanoparticles batch A (b)

Powder X-ray diffraction pattern for native crystalline nicergoline (a) and nicergoline nanoparticles batch A (b)
In a previous work (Martena et al. 2011), the authors described the formation of amorphous NIC by melting or crystallization in chloroform and substantiated the physicochemical stability of the amorphous form when stored at temperatures typical of the refrigerator. The ability of amorphous material to absorb water may interfere with the normal product shelf life as it was proven that the presence of water favors the crystallization of amorphous materials (Andronis et al. 1997; Schmitt et al. 1996; Shamblin and Zografi 1999). As a consequence, a stability testing according to the ICH (ICH Topic Q 1 A (R2), 2003) was performed on the amorphous NPs, and the procedure for “Drug substances intended for storage in a refrigerator” was followed. Such steps were taken in the light of a previous study, where amorphous NIC showed a good stability at 3 °C. The following storage conditions were used:
-
1.
Long term 5 ± 3 °C 12 months.
-
2.
Accelerated 25 ± 2 °C/60 ± 5 % RH 6 months.
The effect of high humidity (60 %) on the stability of amorphous NPs at accelerated conditions was evaluated by regularly checking changes in physicochemical characteristics using XRPD and DSC. It emerged that no changes in the physical properties were observed during the testing period.
Solubility measurements and drug release
The determination of experimental solubilities for amorphous pharmaceutical materials is extremely difficult because of the tendency for such material to rapidly revert to the crystalline state upon exposure to the solvent (Hancock and Parks 2000). Consequently, it is generally accepted that the equilibrium solubility for an amorphous material cannot be measured. Actually, in contact with dissolution medium, water is absorbed into the amorphous matrix and consequently the molecular mobility increases, favoring crystallization of the hydrated amorphous solid (Taylor and Shamblin 2009).
Considering these premises, solubility measurements were carried out according to the method previously described and taking into account changes in instantaneous solubility versus time. Results are given in Fig. 5, where solubilities in water, acidic medium, and phosphate buffer pH 8.0 were determined at 10, 20, 30, and 37 °C. Solubilities in acidic medium were always higher than those in water and phosphate buffer (this last being always intermediate), regardless of the temperature and the sample (NPs or NCs). As expected, the solubility profile for NCs was always constant: the amount of drug dissolved in the solvent rapidly increased only during the first few minutes of the analysis and subsequently reached a plateau that remained constant for the entire experimental interval. The solubility of amorphous NPs followed a different behavior being also influenced by the experimental temperature. The solubility rapidly increased in a few minutes reaching a maximum value, which can be considered as the maximum solubility at any temperature. Depending on the experimental temperature, the solubility decreased from this maximum value at different rates until reaching a limiting value. In detail, the lower the temperature, the lower the drop in solubility and the greater the difference in limiting values between NPs and NCs. This difference was less pronounced as the experimental temperature increased.

Experimental solubility profiles of NIC native particles (square symbol) and nanoparticles (round symbol) (batch A) in distilled water, HCl 0.1 N and phosphate buffer pH 8.0 at 10, 20, 30, and 37 °C
Finally, it must be pointed out that, in general, the equilibrium solubility was undoubtedly influenced by the physical state of the particles and not by their size. In the present study, the maximum solubility and the limiting value were not influenced by the particles size. The particle size influenced only the rate at which the maximum value was reached. Taking into account these considerations, it can be thus concluded that the amorphous NPs showed higher solubility than the crystalline particles.
Cumulative drug releases of NCs and NPs were determined in water and HCl 0.1 N in order to calculate the intrinsic dissolution. The analysis of variance revealed that cumulative drug releases were significantly different (significance level P < 0.05). Cumulative drug releases in HCl 0.1 N were always higher than those in water, in agreement with the results of drug solubility. Furthermore, NPs always exhibited the highest cumulative drug release in all the studied media. The slopes of the straight lines determined by fitting the data points of each release curve were used to calculate the IDRs. Table 2 shows that IDRs were always higher for NPs in both media. Since intrinsic dissolution is not influenced by the particle size, but rather by the physicochemical properties of the solid material, it is possible to state that, in comparison with the crystalline form of NCs, the higher intrinsic dissolution of NPs was due to their amorphous character.
Results of solubility and IDRs proved that NPs are more soluble and dissolve faster than NCs because of their amorphous form, but these two factors are not influenced by the particle size, because the solubility is independent from the particle size and the intrinsic dissolution is a characteristic of the solid but not of the particles.
In order to assess the influence of particle size on the dissolution rate, the dissolution of NCs and NPs was evaluated (Fig. 6). Cumulative drug release in water and HCl of NPs was always higher than that of NCs, and differences are statistically relevant (significance level P < 0.05). This result is influenced by the amorphous state of the NPs as well as by their nanometric size. Thus, it is possible to conclude that the concomitant positive effect of amorphous state and particle nanodimensions may potentially concur to noticeably improve the bioavailability of NIC.

Cumulative drug release of NIC NCs and nanoparticles (batch A) as a function of time in distilled water and HCl 0.1 N
Conclusions
This study demonstrated that it was possible to obtain pure NPs of NIC using a new nano-spray drying method. This method favored the formation of spherical NPs into their amorphous state in a reproducible way. Both the amorphous state and nanosize dimensions of particles were responsible for a pronounced improvement in NIC drug release. Studies of physicochemical stability of the amorphous form proved that the amorphous form was stable for at least a period of 1 year when stored at 3 °C. These results encourage a future development of new oral formulations of NPs with the aim to improve the drug dissolution rate. To this aim, NPs may be formulated as an aqueous nanosuspension or even as a tablet or capsule. In the latter case, the formulation must assure NIC could be easily released from the pharmaceutical dosage form as original NPs (Mauludin et al. 2009).
In the future, authors will try to support results of in vitro experiments with in vivo results.
References
Ahlneck C, Zografi G (1990) The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state. Int J Pharm 62:87–95
Andronis V, Yoshioka M, Zografi G (1997) Effects of sorbed water on the crystallization of indomethacin from the amorphous state. J Pharm Sci 86:346–351
Banakar UV (1992) In: Swarbrick J (ed) Pharmaceutical dissolution testing. Marcel Dekker, New York, pp 55–105
Brossi A (1990) The alkaloids. Chemistry and pharmacology, 38. Academic Press Inc, San Diego, p 142
Crowley KJ, Zografi G (2002) Water vapour absorption into amorphous hydrophobic drug/poly(vinylpyrrolidone) dispersions. J Pharm Sci 90:2150–2165
Elversson J, Millqvist-Fureby A, Alderborn G, Eloffson U (2003) Droplet and particle size relationship and shell thickness of inhalable lactose particles during spray drying. J Pharm Sci 92:900–910
Gao L, Zhang D, Chen M (2008) Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J Nanopart Res 10:845–862
Goo D, Palosi E, Szporny L (1988) Comparison of the effects of vinpocetine, vincamine, and nicergoline on the normal and hypoxia-damaged learning process in spontaneously hypertensive rats. Drug Dev Res 15:75–85
Hamaura T, Newton JM (1999) Interaction between water and poly(vinylpyrrolidone) containing polyethylene glycol. J Pharm Sci 88:1228–1232
Hancock BC, Parks M (2000) What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res 17:397–403
Heitz C, Descombes JJ, Miller RC, Stoclet JC (1986) α-Adrenoceptor antagonistic and calcium antagonistic effects of nicergoline in the rat isolated aorta. Eur J Pharmacol 123:279–285
ICH Topic Q 1 A (R2) (2003) Stability testing of new drug substances and products. Note for guidance on stability testing: stability testing of new drug substances and products. (CPMP/ICH/2736/99). EMEA, London, pp 1–20
Junghanns JAH, Müller RH (2008) Nanocrystal technology, drug delivery and clinical applications. Int J Nanomed 3:295–309
Kontny MJ, Zografi G (1995) Sorption of water by solids. In: Brittain HG (ed) Physical characterization of pharmaceutical solids. Marcel Dekker, New York, pp 387–418
Maa YF, Nguyen PA, Sweeney T, Shire SJ, Hsu CC (1999) Protein inhalation powders: spray drying vs spray freeze drying. Pharm Res 16:249–254
Malaj L, Censi R, Capsoni D, Pellegrino L, Bini M, Ferrari S, Gobetto R, Massarotti V, Di Martino P (2011) Characterization of nicergoline polymorphs crystallized in several organic solvents. J Pharm Sci 100:2610–2622
Martena V, Censi R, Hoti E, Malaj L, Di Martino P (2011) Physicochemical characterization of nicergoline and cabergoline in its amorphous state. J Therm Anal Calorim doi: 10.1007/s10973-011-1954-2
Mauludin R, Müller RH, Keck CM (2009) Development of an oral rutin nanocrystal formulation. Int J Pharm 370:202–209
Merisko-Liversidge E (2002) Nanocrystals: resolving pharmaceutical formulation issues associated with poorly water-soluble compounds. In: Marty JJ (Ed) Particles. Marcel Dekker, Orlando
Noyes A, Whitney WR (1897) The rate of solution of solid substances in their own solutions. J Am Chem Soc 19:930–934
Pastoris O, Vercesi L, Allorio F, Dossena M (1988) Effect of hypoxia, aging and pharmacological treatment on muscular metabolites and enzyme activities. Il Farmaco Ed Sci 43:627–642
Schmitt E, Davis CW, Long ST (1996) Moisture-dependent crystallization of amorphous lamotrigine mesylate. J Pharm Sci 85:1215–1219
Shamblin SL, Zografi G (1999) The effects of absorbed water on the properties of amorphous mixtures containing sucrose. Pharm Res 16:1119–1124
Shegokar R, Müller RH (2010) Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm 399:129–139
Speiser PP (1998) Poorly soluble drugs: a challenge in drug delivery. In: Benita S, Böhm B, Müller RH (eds) Emulsions and nanosuspensions for the formulation of poorly soluble drugs. Medpharm Scientific Publishers, Stuttgart, pp 15–28
Taylor LS, Shamblin SL (2009) Amorphous solids. In: Brittain HG (ed) Polymorphism. Informa Healthcare, New York, pp 594–596
Triulzi E, Devizzi S, Margonato A (1981) Use of nicergoline in acute myocardial infarction with diastolic hypertension. Il Farmaco (ed Prat) 36:449–455
Venn RD (1980) Review of clinical studies with ergots in gerontology. In: Goldstein M, Liberman A, Calne DB, Thorner MO (eds) Ergot compounds and brain function. Raven Press, New York, pp 363–377
Zografi G (1988) States of water associated with solids. Drug Dev Ind Pharm 14:1905–1926
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Martena, V., Censi, R., Hoti, E. et al. A new nanospray drying method for the preparation of nicergoline pure nanoparticles. J Nanopart Res 14, 934 (2012). https://doi.org/10.1007/s11051-012-0934-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-012-0934-1