
Abstract
We have investigated the crystal structure of nanosized iron-oxide by X-ray diffraction (XRD), extended X-ray absorption fine structure measurements at the iron K-edge as well as by transmission electron microscopy (TEM). Iron-oxide nanoparticles were produced by thermal treatment of horse spleen ferritin molecules. The structure of these particles was compared to α-Fe2O3 and γ-Fe2O3 nanopowder references. The thermal treatment of a submonolayer film of ferritin molecules results in pure γ-Fe2O3 nanoparticles, while for films above a certain thickness α-Fe2O3 and γ-Fe2O3 coexist, exhibiting two different crystallite sizes. TEM shows a characteristic particle diameter of ~7 nm for γ-Fe2O3 resulting from thermal treatment of monolayers, consistent with the crystallite size of the γ-phase as obtained from XRD measurements on multi-layered samples. XRD shows the α-Fe2O3 phase to be characterized by a crystallite size of ~34 nm.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nanosize transition metal oxide particles can be expected to show size-dependent optical, magnetic, electronic and chemical properties with possible applications in catalysis, as well as magnetic and optical devices. It has already been shown that catalytic and magnetic properties of nanoparticles can be quite different from bulk materials. For example, nanosize particles of the compound Fe2O3 exhibit superparamagnetism with a size-dependent blocking temperature (Janzen et al. 2003). At the same time they show enhanced catalytic properties compared to bulk material (Li et al. 2003). Surface effects play an important role for the properties of nanoparticles, since the surface to volume ratio increases with decreasing particle diameter. It is also well known that the structure strongly influences the properties of such particles. For example, γ-Fe2O3 (maghemite) is biocompatible and widely used in life science, especially due to its magnetic properties (Chirita and Grozescu 2009; Fang and Zhang 2009). The structural phase of Fe2O3 nanoparticles can depend on both, the particle size and preparation method. For example it is reported that for particle diameters of less than ~20 nm γ-Fe2O3 is stable but that larger particles exhibit the α-Fe2O3 (hematite) structure (Schimanke and Martin 2000; Janzen et al. 1999; Sadeghi et al. 2010). This is consistent with thermodynamic calculations which yield a critical particle diameter of 16 nm for the transformation between α- and γ-Fe2O3 at ambient conditions (Navrotsky et al. 2008). Other investigations found Fe2O3 nanoparticles with diameters of 12 nm and smaller to exhibit the α-phase (Jacob and Khadar 2010; Bermejo et al. 1997).
For thin films a thickness dependence of the crystal structure of Fe2O3 was already observed by Barbier et al. (2005). They found that the structure grows in the γ-phase up to a layer thickness of 3.5 nm. On further growth it transforms to α-Fe2O3. The transformation is completed at a thickness of 20–30 nm.
A similar behaviour is reported for the compound Al2O3 which crystallizes in an α-phase isostructural to α-Fe2O3 and a spinel-type γ-phase. This γ-phase is stable down to surface areas of 125 m2/g corresponding to spherical particles with a diameter of less than 13 nm at ambient conditions. Larger particles crystallize in the α-phase. This is explained by the lower surface energy of the γ-phase compared to the α-phase (McHale et al. 1997).
FerritinFootnote 1 can be used to prepare nanoparticles with a diameter below 8 nm.
In this study we make use of this property to investigate the structure of such nanoparticles arranged in monolayers and multilayers. For Fe2O3 nanoparticles with a size of ~7 nm we have shown that, depending on the preparation process, the electronic excitation gap of the surface states can be different from that of the bulk (Preisinger et al. 2005). This was attributed to surface reconstruction in the presence of a defect rich Fe2O3-phase.
To shed further light on the structural properties of Fe2O3 nanoparticles prepared from ferritin, we have performed a structural investigation utilizing X-ray diffraction (XRD), extended X-ray absorption fine structure (EXAFS) and transmission electron microscopy (TEM). From atomic force microscopy (AFM) and TEM we find that nanoparticles prepared from monolayers of ferritin exhibit a diameter of ~7 nm. Their structure is best described by the defect spinel-type γ-Fe2O3 phase. For multi-layered samples the experiments indicate in addition the existence of the corundum like α-Fe2O3 phase with a crystallite size of ~34 nm. The coexistence of both phases can be explained by the influence of the preparation route of iron-oxide nanoparticles on the resulting structure.
Experimental
The crystal structure of the samples has been investigated by EXAFS and XRD measurements which were carried out at the ANKA-XAS and ANKA-PDIFF beam-lines of ANKA Angstroemquelle Karlsruhe. The samples for the measurements at ANKA were prepared from commercially available horse spleen ferritin stored in a NaCl buffer solution (from SIGMA) with a concentration of 85 mg/mL ferritin. Up to 300 μL of the solution were dried on a glass substrate and subsequently annealed in air at ~675 K for 5 h. In the case of EXAFS measurements, the dried and annealed substance was then ground to a fine powder and mixed with spectrophotometric grade polyethylene powder (SIGMA) and pressed to pellets. For XRD measurements, the grounded powder was suspended as a thin layer on a glass substrate. These samples are referred to as multi-layered (MuL) samples in the following.
As reference samples for the EXAFS experiments we measured pressed pellets of powder of α-Fe2O3 and γ-Fe2O3, respectively, mixed with polyethylene powder. The crystallinity and phase purity of the respective iron-oxide phases were checked by X-ray diffraction. The iron-oxide powders were obtained from Alfa Aesar and had an average particle size of 20–50 nm for α-Fe2O3 and 20–30 nm for γ-Fe2O3. To analyze the EXAFS measurements we used the ATHENA (Ravel and Newville 2005) and IFEFFIT (Newville 2001) software. After normalization to the edge step, a smooth, atomic like background was subtracted, minimizing the short distance contribution to the Fourier-transformed (FT) spectra of the experimental data (Newville et al. 1993). The k 2 weighted spectra of the nanoparticles were fitted by a superposition of the α-Fe2O3 and γ-Fe2O3 reference spectra in the region between 3 and 9 Å−1 in k-space.
The XRD measurements were performed in Bragg–Brentano geometry with wavelength of 0.118313(3) nm. A single analyser channel (Ge111-crystal) and a NaI scintillation detector were used for the detection of the Bragg reflections. The zero shift and instrumental line broadening were determined by measuring a LaB6 NIST standard. The glass substrate carrying the iron-oxide sample was mounted on a low background Si (911) sample holder, rotating at 2 Hz at ambient temperature and pressure. Diffraction powder patterns were recorded in the range between 15° and 58° in a θ–2θ step scan with a step width of 0.01°. A Rietveld refinement of the measured pattern was carried out after background removal using the program FULLPROF (Rodríguez-Carvajal 1993). For the refinement, the starting structural parameters were taken from Sawada (1996), Shmakov et al. (1995), Rasmussen et al. (1996) and Nickels et al. (1949).
As a third method for structural investigations, high resolution transmission electron microscopy (HR-TEM) was used. In contrast to EXAFS and XRD, the crystal structure of single particles can be investigated in HR-TEM. In particular it is possible to distinguish between single- and polycrystalline nanoparticles as well as between different iron-oxide phases of single particles. Using Fourier analysis of the HR-TEM images, one can calculate a diffraction pattern. The Bravais lattice and the lattice constants of the structure are determined by indexing the patterns. For HR-TEM sample preparation the particles were dispersed in isopropanol and a few micro litres of this solution were dropped on carbon coated copper grids. The grids were then dried in air.
Information about the iron-oxide particle size of the investigated samples was obtained by TEM and AFM. For this purpose monolayers of the native ferritin molecules were prepared on a highly oriented pyrolytic graphite (HOPG) surface by the Langmuir–Schaefer technique (Johnson et al. 2000; Furuno et al. 1989) and annealed in air at ~675 K for 5 h. For TEM investigations, the top layers of the HOPG substrate were peeled off and mounted on a sample holder (75 mesh Cu grid). This preparation method enables AFM and TEM measurements on the same sample and therefore allows direct comparison of the extracted parameters. From TEM measurements on these samples only the lateral dimension (parallel to HOPG surface) of the particles can be determined. In contrast, AFM measurements allow an accurate determination of the particle height (perpendicular to HOPG surface), while the lateral resolution is limited due to tip broadening effects. By combining the two methods it is possible to determine both, lateral and vertical dimension of the particles with high accuracy. In addition, the chemical nature of the particles is confirmed by TEM energy dispersive X-ray (EDX) measurements.
The TEM images as well as the EDX mappings were obtained using a JEOL JEM 2100F microscope equipped with an EDAX detector and a CCD camera. All the AFM images were taken using a Veeco Dimension 3100 AFM in Tapping Mode™.
Results
Figures 1 and 2a show a TEM image and an AFM image, respectively, of an iron-oxide nanoparticle monolayer on a graphite substrate obtained after annealing of a monolayer of native ferritin for 5 h at ~675 K.

TEM image of a nanoparticle monolayer resulting from thermal treatment of a native ferritin. The white spot represents the average particle diameter of 6.7 nm

(Color online) AFM image (a) of monolayer of native ferritin after thermal treatment. The solid blue line indicates the area where the height profile (b) is taken from
Although the particles are in contact with each other they can still be identified as single particles with a narrow size distribution and a lateral particle diameter of 6.7 ± 1.1 nm.
The particle size from AFM measurements is determined from height profiles as shown in Fig. 2b. Numerous scans were taken across various particle clusters. The difference in height between graphite surface and particle cluster averaged over those scans was taken as the actual particle height and yielded a value of 6.5 ± 0.5 nm, in good agreement with the lateral dimensions determined by TEM measurements. The combination of the two methods suggests an almost equal extension of the particles in all three dimensions.
To confirm the chemical nature of the particles we have performed TEM-EDX mapping. Figure 3a shows the TEM image and Fig. 3b shows the corresponding EDX iron mapping of the nanoparticles on a thin graphite layer.

TEM image of native ferritin after thermal treatment (a) and the corresponding TEM-EDX mapping (b). Bright spots indicate the presence of iron which is in agreement with the position of the nanoparticles
The bright areas indicate the presence of iron. Areas of high oxygen and iron concentration coincide. These areas match with the position of the nanoparticles, while no significant amount of elements other than iron and oxygen were found in those areas. In contrast, in areas without particles no iron was detected. This confirms the expectations that the particles imaged consist of iron oxide. Information on the valence state of the iron was obtained by core-level photo electron spectroscopy (XPS) performed on multi-layered samples and has been published earlier (Preisinger et al. 2005). The energies of the photoemission lines as well as the characteristic shake-up satellite indicate that iron is in a pure 3+ state as in Fe2O3.
Figure 4 shows a HR-TEM image of a Fe2O3 nanoparticle and the Fourier-transformed image (inset). Most of the particles are oriented on the substrate in such a way that lattice planes can be resolved. The overwhelming number of particles are single crystalline. The structure of the particles is stable under the influence of the electron beam during the measurements for at least several minutes. From Fourier analysis, one obtains a cubic structure with lattice constants similar to the defect spinel type structures of γ-Fe2O3 and Fe3O4. A hematite-like structure (hcp) can be excluded. Since photoelectron emission measurements show the iron ions to be in a pure 3+ state (Preisinger et al. 2005) we conclude, that the nanoparticles exhibit the γ-Fe2O3 phase.

HR-TEM image of a single particle obtained by annealing of native ferritin on an amorphous carbon film. The inset shows the Fourier transform diffraction pattern of the particle. Spots at 2.41 and 2.09 Å match to the (222) and (004) lattice spacing of γ-Fe2O3; the viewing direction is [110]
Figure 5 shows a typical X-ray diffraction powder pattern for a multi-layered sample after annealing. In addition to the measurement (red circles) a Rietveld refinement (black solid line) and the difference between data and Rietveld refinement (blue line) are shown. The lines can be attributed to four different phases. The two strongest lines at 24.2° and 34.5° arise from NaCl which is a residue of the buffer solution in which the ferritin is stored. The second phase (strongest lines at 22.1° and 24.5°) is Na2SO4. Most likely its presence can be attributed to a reaction of Na ions from the buffer solution with sulphur of the ferritin protein shell. Two iron-oxide phases can be identified: α-Fe2O3 (strongest lines at 27.2° and 25.3°) and γ-Fe2O3 (strongest lines at 23.1° and 27.2°).

(Color online) XRD measurement of a multi-layered sample of native ferritin after thermal treatment (red circles) and Rietveld refinement (black solid line) composed of calculated diffraction patterns of α- and γ-Fe2O3, NaCl and Na2SO4. The blue line represents the difference between data and fit. The strongest reflections of γ- and α-Fe2O3 are indicated by the symbols hash and asterisk, respectively. Reflections without marker originate from NaCl and Na2SO4, respectively
The lattice constants of the refinement are given in Table 1. They are within 0.1% in agreement with bulk values reported in the literature.
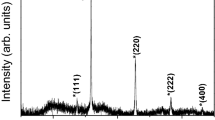
The width of the most intense lines of α-Fe2O3, γ-Fe2O3 and NaCl are compared in Fig. 6. The line width of the iron-oxide phases is broadened compared to NaCl reflections and Na2SO4 (not shown) reflections. The measured line width was corrected for instrumental broadening by measuring a LaB6 NIST standard. The corrected line widths of the γ-phase are broader by a factor of three compared to the line widths of the α-phase indicating a smaller crystallite size for γ-Fe2O3. The line width of the α-phase is only slightly wider than the line width of the α-Fe2O3 nanopowder reference sample exhibiting an average particle size of 20–50 nm. In comparison with the LaB6 reference, the lines of NaCl and Na2SO4 are only slightly broadened indicating a larger crystallite size than that of the iron-oxide phases. This is consistent with scanning electron microscopy measurements, which show NaCl particles with sizes between approximately 1–10 μm. Using the Scherrer formula (Scherrer 1918)
the mean diffraction length D can be calculated from the width β of a peak (in 2θ units) at angular position θ, the wavelength λ, and the Scherrer constant K, which is taken as unity since the integral width of the reflection is used (Stokes and Wilson 1942). This results in diffraction lengths of ~7.1 ± 1.6 nm for γ-Fe2O3 and ~34 ± 5 nm for α-Fe2O3. While the length scale of 7 nm agrees well with the particle size of γ-Fe2O3 as determined from TEM and AFM measurements on monolayers, particles with a diameter of ~34 nm have not been observed for mono-layered samples in microscopy. Since only an insignificant broadening of the lines of NaCl and Na2SO4 is observed, the calculated crystal size exceeds the validity of the Scherrer formula. Therefore, a crystallite size less than 100 nm can be excluded for both phases from XRD. The validity of the procedure to determine the particle size from the width of XRD reflection has been confirmed exploiting the reference samples. Here the analysis of the reflection width results in crystallite sizes of 53.2 ± 2.4 nm for α-Fe2O3 and 25 ± 1.8 nm for γ-Fe2O3, which is in good agreement with TEM measurements on α-Fe2O3 reference sample (53 ± 16 nm) and the suppliers specification of 20–30 nm for γ-Fe2O3, respectively.

Most intense diffraction lines of γ-Fe2O3, α-Fe2O3, and NaCl (top to bottom). Different width indicates different grain size
EXAFS measurements are sensitive to the local structure of the material. In particular EXAFS oscillations in the vicinity of the iron K-edge can give insight into iron-oxygen bond lengths as well as the oxygen coordination of iron atoms. The Fourier-transformed EXAFS spectra shown in Fig. 7 were taken from iron-oxide nanoparticles prepared from ferritin (sample MuL), as described above, as well as from α- and γ-Fe2O3 nanopowder reference samples, labelled α-ref and γ-ref, respectively. In the following we will limit the discussion to atomic shells in the spatial region 1.4 Å < R < 4.2 Å. In this region the spectrum of our nanoparticles consists of three peaks (P1, P2, and P3), which are almost symmetrically centred around R ~ 1.8 Å, R ~ 2.9 Å and R ~ 3.6 Å. Since both bulk α-Fe2O3 and bulk γ-Fe2O3 structures exhibit an oxygen coordination around the scattering iron ion in the range around 1.8 Å the first peak (P1) is attributed to these nearest oxygen neighbours. In accordance with expectations from the bulk structure the second peak (P2) is assigned to next nearest iron and oxygen neighbours. For R > 3 Å the EXAFS signal appears to be strongly damped.

(Color online) Magnitude of the Fourier-transformed EXAFS spectra of α-/γ-Fe2O3 reference samples and multi-layered sample. Vertical lines indicate differences in peak positions. For this figure, the scattering phase shift of the central Fe ion has been corrected, which leads to a shift of the Fourier-transformed spectra and the fitting ranges to the right by roughly 0.3 Å
Comparing the FT-spectra of sample MuL to the α-Fe2O3 and γ-Fe2O3 reference samples, we note that the position of peak P1 of the sample MuL is in agreement with the γ-Fe2O3 reference as is indicated in Fig. 7. Peak P2 of sample MuL appears to be located midway between the respective positions of peak P2 of the two reference samples. The position and magnitude of peak P3 of sample MuL rather agrees with the one observed for the spectra of α-Fe2O3. This peak is missing in the γ-Fe2O3 FT-spectrum or has merged with peak P2. In summary, a visual inspection of the peak positions of the recorded spectra yields a satisfactory agreement neither between sample MuL and α-Fe2O3 nor between sample MuL and γ-Fe2O3. Since XRD measurements show that both iron-oxide phases coexist in multi-layered samples, the signature of the presence of both phases is expected in EXAFS measurements as well. This can explain the features of the EXAFS spectrum of sample MuL. While peak P1 of this spectrum looks similar to that of the γ-Fe2O3 spectrum, peak P3 of sample MuL only has a counterpart in the spectrum of the α-Fe2O3-reference. The relatively broad peak P2 of sample MuL can be interpreted as a superposition of peak P2 of the α-Fe2O3 and γ-Fe2O3 phase, respectively. This poses the question why peak P1 of the sample MuL is dominated by the γ-phase and peak P3 by the α-phase. A possible explanation might be that for the smaller γ-Fe2O3 nanoparticles disorder in the surface near region plays a more important role than for the larger α-Fe2O3 particles. The difference of the surface to volume ratio of the two phases amounts to a factor of five. It appears to be a reasonable assumption that in the surface near region the contribution due to longer scattering paths dominating peak P3 is suppressed. Therefore, for short scattering paths the shape of the EXAFS spectrum is defined by γ-Fe2O3 while long scattering paths are dominated by the α-Fe2O3 contribution, under the condition that the total volume of γ-Fe2O3 phase is much larger than that of the α-phase. To check qualitatively whether the EXAFS oscillations can be interpreted as a convolution of the two Fe2O3 phases, we have fitted the k 2-weighted EXAFS oscillations with a superposition of the measured hematite and maghemite reference spectra.
Figure 8 shows the EXAFS oscillations of the α- and γ-Fe2O3 references (a) as well as the oscillations of the nanoparticle sample and the fit resulting from a linear combination of the two. The fit reproduces the frequency of the EXAFS oscillations, i.e. the distance of the neighbouring shells, although not the amplitudes. The fact that the amplitudes are not very well reproduced could be attributed to the different magnitude of disorder in the α- and γ-Fe2O3 particles, as discussed above. The fact that the EXAFS spectra can be reproduced by the linear combination of α- and γ-Fe2O3 references is in accordance with the coexistence of the two phases in XRD measurements.

a EXAFS oscillations of α-Fe2O3 (solid line) and γ-Fe2O3 (dashed line) references. b EXAFS oscillations of sample (solid line) and linear combination fit of references (dashed line)
Discussion
The most important results of the measurements presented are (1) under thermal treatment of monolayers of horse spleen ferritin γ-Fe2O3 nanoparticles with a narrow size distribution form, (2) for multi-layered samples a mixture of α- and γ-Fe2O3 is found. The size of the γ-Fe2O3 particles in the multi-layered sample is in agreement with those found for a monolayer, while the volume of the α-Fe2O3 crystallites is about 100 times larger, (3) the lattice parameters of the nanoparticles of both phases agree with those of the corresponding bulk material.
Microscopic images (TEM, AFM) of annealed ferritin monolayers show clusters of nanoparticles in which the nanoparticles are in close contact but have not merged. The clustering suggests that the interface energy of the system nanoparticle cluster–substrate is smaller than that of the corresponding number of isolated particles with the substrate. Since such a two dimensional arrangement of nanoparticles is stable no energy is gained by formation of larger (single crystalline) particles at the applied temperature or the process is kinetically inhibited. An explanation for the first case could be due to the particle substrate interaction. An influence of the interaction between particles and substrate on particle growth has been observed for ferrihydrite. Banfield et al. (2000) attributed a reduced coarsening of ferrihydrite to the particle–substrate interaction.
The fact that particles, which form a monolayer, are single phase and of cubic structure is confirmed by HR-TEM. The observed structure is in agreement with the two inverse spinel type structures of γ-Fe2O3 and Fe3O4. Due to their similar structure the two phases can hardly be distinguished by diffraction measurements. In earlier photoemission spectroscopy experiments performed on multi-layered samples, we have shown the iron ions to be in a 3+ state. In addition scanning tunnelling spectroscopy (STS) measurements show an excitation gap of ~2 eV in agreement with that of bulk γ-Fe2O3 (Preisinger et al. 2005), i.e. we can exclude the Fe3O4 phase and, therefore, suggest the particles to be γ-Fe2O3.
XRD and EXAFS measurements on multi-layered samples confirm the presence of γ-Fe2O3 in the sample. In addition, the α-Fe2O3 phase is found. This is the thermodynamically stable Fe2O3 bulk phase. The analysis of the line width of reflections of XRD measurements yields different diffraction volumes of the two iron-oxide phases. While the width of the γ-phase reflections indicates a diffraction length of ~7 nm, in agreement with the particle size found from TEM and AFM measurements, a diffraction length of ~34 nm is calculated for the α-phase. A Rietveld refinement of the XRD pattern results in bulk-like values of the cell parameters of α- and γ-Fe2O3, i.e. the lattice parameters do not depend on the size of the nanoparticles, at least down to a particle size of 7 nm.
We now want to address the occurrence of α-Fe2O3 in multi-layered samples. From thermodynamic considerations it is known that metastable bulk phases can be stabilized in the form of nanoparticles due to an energetically more favourable surface enthalpy. With increasing particle size, the relative contribution of the surface enthalpy to the total enthalpy decreases. In the case of Fe2O3 the critical particle diameter for the stability of γ-Fe2O3 compared to α-Fe2O3 is 16 nm at room temperature, i.e. the γ-phase is stable for particles with a diameter of 16 nm or less assuming spherical particles (Navrotsky et al. 2008).
Investigations of Fe2O3 thin film growth shows that films with a thickness of less than 3.5 nm favour the Fe2O3 γ-phase, while films exceeding this thickness exhibit a mixture of γ- and α-Fe2O3. The transformation is completed for a film thickness of 20–30 nm (Barbier et al. 2005), which is consistent with the thermodynamic considerations presented above. If we consider the nanoparticle layers as a thin film with a thickness corresponding to the particle diameter the occurrence of the α-Fe2O3 phase for multi-layered samples is consistent with the observation of Barbier et al. (2005).
From XRD measurements we find that the α-Fe2O3 phase occurring in addition to the γ-phase consists of crystallites of size of ~34 nm with unknown shape. Assuming a spherical particle shape this diameter corresponds to a surface to volume ratio resulting in stable α-Fe2O3 phase as stated by Navrotsky et al. (2008). The occurrence of α-Fe2O3 in multi-layered samples is also in agreement with experiments on annealed ferrihydrite. The structure of ferrihydrite is reported to be similar to that of a native ferritin core. The oxidation of multi-layered systems of synthetic six-line ferrihydrite at ~670 K for 5 h leads to pure α-Fe2O3 with a crystallite size of ~50 nm as measured by XRD (not shown). The transformation of ferrihydrite to α-Fe2O3 under these conditions has also been shown by Stanjek and Weidler (1992).
In the light of these experiments the question arises why the multi-layered samples exhibit a significant amount of γ-Fe2O3 after annealing. One possible explanation is the presence of impurities in the form of NaCl, Na2SO4 and carbon. Impurities can influence the transformation of ferrihydrite to α-Fe2O3. Schwertmann and Fechter (1984) found that the oxidation of ferrihydrite in air at 720 K in the presence of organic material yields γ-Fe2O3 rather than α-Fe2O3. For the presented synthesis route the protein shell acts as carbon source and can influence the transformation of the ferrihydrite core. The effect of further impurities like NaCl or Na2SO4 is not clear yet. It is known that in aqueous Fe2+ solution the oxidation process slows down in the presence of Cl− and SO4 2− (Tamura et al. 1976). Therefore, the formation of the mixed valent iron-oxide Fe3O4 would be favourable. The oxidation of Fe3O4 can result in the isostructural γ-Fe2O3 phase rather than that of α-Fe2O3.
Microscopic measurements do not indicate a spatial correlation between iron oxide and NaCl or Na2SO4. In addition no correlation between the mass ratio of α-Fe2O3 to γ-Fe2O3 and the amount of NaCl or Na2SO4 present in the samples could be evaluated from XRD measurements. From literature it is known that the adsorption of phosphates on the surface of nanoscale γ-Fe2O3 particles even stabilizes this phase (Tronc and Jolivet, 1986). Therefore, we think it is not likely that NaCl or Na2SO4 act as center for the germination process of α-Fe2O3. However, we cannot rule out this possibility completely.
Another possibility is that γ-Fe2O3 is formed from ferritin but that the annealing temperature and time is not sufficient to complete the phase transition from nanoscale γ-Fe2O3 to α-Fe2O3. Ye et al. (1998) find the transition process to start at ~710 K for 12 nm γ-Fe2O3 particles. With increasing temperature the α-phase contributes to XRD pattern while the contribution of γ-Fe2O3 is reduced till only α-Fe2O3 is present. Although the crystallite size of γ-Fe2O3 is constant for all temperatures, the size of the α-Fe2O3 particles increases with temperature up to 54 nm at 1,030 K. They also find that the γ to α transition temperature increases with decreasing particle size. The transition temperature is in agreement with the finding of Belin et al. (2007) who observe reflections of α-Fe2O3 in XRD pattern at 723 K for 9 nm γ-Fe2O3 particles. In this context they find an increase of the transition rate with increasing temperature. While the γ-Fe2O3 particles size remains constant, the size of the α-Fe2O3 crystallites grows faster with increasing temperature. In contrast, Schimanke and Martin (2000) report a γ to α transition already at ~570 K with rather slow transition rates for similar γ-Fe2O3 particle size.
The XRD pattern presented in this report were obtained at an oxidation temperature of ~670 K. Regarding Schimanke and Martin (2000) and Belin et al. (2007) the transition rate constant varies between 1.70 × 10−6 and 4.83 × 10−4 s−1 for γ-Fe2O3 with a particle diameter of ~9 nm as a function of temperature. This is not consistent with the result of the present investigation that diffraction volume ratio of α- to γ-Fe2O3 is constant for oxidation times of 5 and 24 h, i.e. no time dependence of the α-Fe2O3 crystallite size has been observed. This is an indication that the γ-phase is stable at 670 K and is confirmed by the following experiment.
We have synthesized γ-Fe2O3 nanoparticles with a particle diameter of ~10 nm by thermal decomposition of iron oleate at ~570 K. The annealing of multi-layers of those particles at ~670 K for 24 h does not result in the occurrence of any α-Fe2O3 phase in XRD, confirming that the transition form γ- to α-Fe2O3 occurs at temperatures above 670 K.
The thermal stability of the γ-phase at the applied temperature suggests that the α-Fe2O3 crystallites were not formed by a transition of γ-Fe2O3 particles in multi-layered ferritin samples to α-Fe2O3. Therefore, a direct formation of α-Fe2O3 from the starting solution is proposed. A possible explanation is that defective ferritin molecules with a partial or complete denatured protein shell and free iron ions act as seeds for the α-Fe2O3 phase. These can aggregate and form α-Fe2O3 particles as in the case of ferrihydrite. The fact that no α-Fe2O3 is found in the mono-layered samples can be attributed to the preparation process which is selective with respect to the integrity to the ferritin molecules.
Conclusion
We have performed XRD, EXAFS, and TEM measurements on iron-oxide nanoparticles synthesized from thermally treated native ferritin. While nanoparticle monolayers exhibit only the γ-Fe2O3 phase, XRD and EXAFS measurements indicate the coexistence of α-Fe2O3 and γ-Fe2O3 in multi-layered samples. The observation of γ-Fe2O3 nanoparticles in monolayers is consistent with the phase described in literature for iron-oxide particles synthesized on various routes. The occurrence of α-Fe2O3 is attributed to presence of defective ferritin molecules. These can form particles in a process similar to the transformation of ferrihydrite to α-Fe2O3.
Notes
To produce nanosized iron oxide particles we made use of the ability of ferritin to self-assemble and construct a core, which shows a structure similar to that of the mineral ferrihydrite (5Fe2O3·9H2O). Ferritin is the major cellular iron-storage protein. It is able to store iron as hydrated iron oxide in the internal cavity which is composed of 24 polypeptide subunits. The inner and outer diameters of the protein shell are about 8 and 12.5 nm, respectively. Hydrophilic and hydrophobic channels penetrate the protein shell and provide the means by which iron ions can be accumulated within or removed from the molecules. (Cornell and Schwertmann 2003; Massover 1993; Mann et al. 1989).
References
Banfield JF, Welch SA, Zhang H, Thomson Ebert T, Penn RL (2000) Aggregation-based crystal growth and microstructure development in natural iron oxyhydroxide biomineralization products. Science 289:751–754
Barbier A, Belkhou R, Ohresser P, Gautier-Soyer M, Bezencenet O, Mulazzi M, Guittet M-J, Moussy J-B (2005) Electronic and crystalline structure, morphology, and magnetism of nanometric Fe2O3 layers deposited on Pt(111) by atomic-oxygen-assisted molecular beam epitaxy. Phys Rev B 72:245423
Belin T, Millot N, Bovet N, Gailhanou M (2007) In situ and time resolved study of the γ/α-Fe2O3 transition in nanometric particles. J Sol State Chem 180:2377–2385
Bermejo E, Becue T, Lacour C, Quarton M (1997) Synthesis of nanoscaled iron particles from freeze-dried precursors. Powder Technol 94:29–34
Chirita M, Grozescu I (2009) Fe2O3—nanoparticles, physical properties and their photochemical and photoelectrochemical applications. Chem Bull POLITEHNICA Univ. (Timişoara) 54:1–8
Cornell RM, Schwertmann U (2003) The iron oxides: structure, properties, reactions, occurence and uses, 2nd edn. VCH, Weinheim
Fang C, Zhang M (2009) Multifunctional magnetic nanoparticles for medical imaging applications. J Mater Chem 19:6258–6266
Furuno T, Sasabe H, Ulmer KM (1989) Binding of ferritin molecules to a charged polypeptide layer of poly-1-benzyl-l-histidine. Thin Solid Films 180:23–30
Jacob J, Khadar MA (2010) VSM and Mössbauer study of nanostructured hematite. J Magn Magn Mater 322:614–621
Janzen C, Roth P, Rellinghaus B (1999) Characteristics of Fe2O3 nanoparticles from doped low-pressure H2/O2/Ar flames. J Nanopart Res 1:163–167
Janzen C, Knipping J, Rellinghaus B, Roth P (2003) Formation of silica-embedded iron-oxide nanoparticles in low-pressure flames. J Nanopart Res 5:589–596
Johnson CA, Yuan Y, Lenhoff AM (2000) Adsorbed layers of ferritin at solid and fluid interfaces studied by atomic force microscopy. J Colloid Interface Sci 223:261–272
Li P, Miser DE, Rabiei S, Yadav RT, Hajaligol MR (2003) The removal of carbon monoxide by iron oxide nanoparticles. Appl Catal B 43:151–162
Mann S, Webb J, Williams RJP (1989) Biomineralization: chemical and biochemical perspectives. VCH, Weinheim
Massover WH (1993) Ultrastructure of ferritin and apoferritin: a review. Micron 24:389–437
McHale JM, Auroux A, Perrotta AJ, Navrotsky A (1997) Surface energies and thermodynamic phase stability in nanocrystalline aluminas. Science 277:788–791
Navrotsky A, Mazeina L, Majzlan J (2008) Size-driven structural and thermodynamic complexity in iron oxides. Science 319:1635–1638
Newville M (2001) IFEFFIT: interactive XAFS analysis and FEFF fitting. J Synchrotron Rad 8:322–324
Newville M, Līviņš P, Yacoby Y, Rehr JJ, Stern EA (1993) Near-edge X-ray-absorption fine structure of Pb: a comparison of theory and experiment. Phys Rev B 47:14126–14131
Nickels JE, Fineman MA, Wallace WE (1949) X-ray diffraction studies of sodium chloride–sodium bromide solid solutions. J Phys Chem 53:625–628
Preisinger M, Krispin M, Rudolf T, Horn S, Strongin DR (2005) Electronic structure of nanoscale iron oxide particles measured by scanning tunneling and photoelectron spectroscopies. Phys Rev B 71:165409
Rasmussen SE, Jørgensen JE, Lundtoft B (1996) Structures and Phase Transitions of Na2SO4. J Appl Cryst 29:42–47
Ravel B, Newville M (2005) ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Rad 12:537–541
Rodríguez-Carvajal J (1993) Recent advances in magnetic structure determination by neutron powder diffraction. Physica B 192:55–69
Sadeghi M, Sarabadani P, Karami H (2010) Synthesis of maghemite nano-particles and its application as radionuclidic adsorbant to purify 109Cd radionuclide. J Radioanal Nucl Chem 283:297–303
Sawada H (1996) An electron density residual study of α-ferric oxide. Mater Res Bull 31:141–146
Scherrer P (1918) Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Göttinger Nachrichten 2:98–100
Schimanke G, Martin M (2000) In situ XRD study of the phase transition of nanocrystalline maghemite (γ-Fe2O3) to hematite (α-Fe2O3). Solid State Ionics 136–137:1235–1240
Schwertmann U, Fechter H (1984) The influence of aluminum on iron oxides: XI. Aluminum-substituted Maghemite in soils and its formation. Soil Sci Soc Am J 48:1462–1463
Shmakov AN, Kryukova GN, Tsybulya SV, Chuvilin AL, Solovyeva LP (1995) Vacancy ordering in γ-Fe2O3: synchrotron X-ray powder diffraction and high-resolution electron microscopy studies. J Appl Cryst 28:141–145
Stanjek H, Weidler PG (1992) The effect of dry heating on the chemistry, surface are and oxalate solubility of synthetic 2-line and 6-line ferrihydrites. Clay Minerals 27:397–412
Stokes AR, Wilson AJC (1942) A method of calculating the integral breadths of Debye–Scherrer lines. Math Proc Camb Phil Soc 38:313–322
Tamura H, Goto K, Nagayama M (1976) Effect of anions on the oxygenation of ferrous ion in neutral solutions. J Inorg Nucl Chem 38:113–117
Tronc E, Jolivet JP (1986) Surface effects on magnetically coupled iron oxide “γ-Fe2O3” colloids. Hyperfine Interact 28:525–528
Ye X, Lin D, Jiao Z, Zhang L (1998) The thermal stability of nanocrystalline maghemite Fe2O3. J Phys D Appl Phys 31:2739–2744
Acknowledgments
We acknowledge the ANKA Angstroemquelle Karlsruhe for the provision of beamtime and we would like to thank S. Mangold and S. Doyle for their valuable assistance using beamline ANKA-XAS and ANKA-PDIFF. We thank M. Klemm and P.S. Riseborough for fruitful discussion. This work was supported by the Deutsche Forschungsgemeinschaft in context of SFB 484 and PROALMEX Mexico-Germany binational collaboration project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Krispin, M., Ullrich, A. & Horn, S. Crystal structure of iron-oxide nanoparticles synthesized from ferritin. J Nanopart Res 14, 669 (2012). https://doi.org/10.1007/s11051-011-0669-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-011-0669-4