
Abstract
Plant tissue culture techniques have been extensively employed in commercial micropropagation to provide year-round production. Tissue culture regenerants are not always genotypically and phenotypically similar. Due to the changes in the tissue culture microenvironment, plant cells are exposed to additional stress which induces genetic and epigenetic instabilities in the regenerants. These changes lead to tissue culture-induced variations (TCIV) which are also known as somaclonal variations to categorically specify the inducing environment. TCIV includes molecular and phenotypic changes persuaded in the in vitro culture due to continuous sub-culturing and tissue culture-derived stress. Epigenetic variations such as altered DNA methylation pattern are induced due to the above-mentioned factors. Reportedly, alteration in DNA methylation pattern is much more frequent in the plant genome during the tissue culture process. DNA methylation plays an important role in gene expression and regulation of plant development. Variants originated in tissue culture process due to heritable methylation changes, can contribute to intra-species phenotypic variation. Several molecular techniques are available to detect DNA methylation at different stages of in vitro culture. Here, we review the aspects of TCIV with respect to DNA methylation and its effect on crop improvement programs. It is anticipated that a precise and comprehensive knowledge of molecular basis of in vitro-derived DNA methylation will help to design strategies to overcome the bottlenecks of micropropagation system and maintain the clonal fidelity of the regenerants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The advent of plant tissue culture techniques becomes one of the most important tools in modern plant science research, and adoption of these techniques in crop production may provide the answer for adequate food manufacturing for the community. These techniques are not only used in the crop breeding programs but also have commercial importance, including micropropagation at a large-scale, recombination DNA technology, germplasm conservation, and natural plant metabolite production [1]. Micropropagation techniques have great potential for crop improvement as it allows the production of pathogen-free genetically and physiologically similar plants in large numbers [2]. Although tissue culture regenerants are planned to be alike; this is not always the case.
Due to fluctuations in the in vitro microenvironment, plant cells go through additional stresses, which induce genetic and epigenetic variabilities in the regenerants leading to tissue culture-induced variations (TCIV). Somaclonal variation is a widely accepted term representing TCIV, it was first proposed in plants by Larkin, Scowcroft [3]. Somaclonal variation occurs universally in all cell and tissue cultures indifferently of the micropropagation system [3]. The occurrence of TCIV variation in the micropropagation system, is the reason behind the clones that are not true-to-type to donor plants. This type of variations causes serious problems for the researchers and plant propagators who require fidelity in their clones. Despite of that, variability associated with the in vitro system provides a pool of natural variants upon which selection pressure can be appointed to isolate the desirable regenerants in the form of clones [4]. TCIV can be demonstrated as mitotically or meiotically stable phenomenon [5], which may occasionally lead to the production of plant variants, useful in crop improvement programs [6, 7]. Mitotically stable variants create phenotypes which are physiologically different only among the primary regenerants and rarely transmitted to subsequent generations. This technique has been successfully used in case of ornamental plants and tree species where primary regenerants are the end products [8]. However, in a plant’s lifecycle environmental and genetic changes induce DNA methylation, which could eventually create epigenetic variations and affect up to several generations [9]. Occationally, epigenetic variations induce heritable changes in gene expression without altering the DNA sequences [10].
DNA methylation involves methylation in the DNA at the cytosine residues [11]. DNA methylation is allied with various molecular mechanisms such as regulation of genes, chromatin inactivation, genomic imprinting, and cell differentiation in plants [12]. Available evidence shows that altered cytosine methylation pattern is much more frequent in the plant genome in the in vitro system and leads to discrete phenotypic changes [10, 13]. Tissue culture-induced mutations such as, activation of transposable elements, chromosome breakage, changes in DNA sequence are hypothesized to occur as a reason of DNA methylation, which eventually leads to high-rate occurrence of phenotypic variation [8, 12]. DNA methylation is the most frequently found covalent base modification across the various taxa [14]. Specific DNA methylases generate several methylated bases during the state of post-replicative DNA modification, among which 5-methylcytosine (5-mC) is prevalent in higher plants and in mammals [14].
Various methods are available to detect changes in the genome-wide methylation pattern in tissue culture plants. Most of these methods are based on comprehensive knowledge of an organism’s genome sequence [15], such as bisulfite modification [16, 17] and chromatin immunoprecipitation (ChIP) [18, 19]. Methylation-sensitive amplification polymorphism (MSAP) is a modified amplified fragment length polymorphism (AFLP) technique [20] that does not require genome sequencing. High-performance separation techniques such as high-performance capillary electrophoresis (HPCE) [21] and high-performance liquid chromatography (HPLC) [22] are similarly used in detection of cytosine methylation. The review uncovers the studies showing alteration of DNA methylation patterns that are linked with the variation in the characters at single gene or genome level, by which a large number of high quality breeding material or improved agronomic traits can be attainable [23]. On these grounds, cytosine methylation can be potentially used as an important source of variation for crop improvement. This review provides detailed information on various aspects of TCIV and its implications on a crop improvement program. This review highlights the recent progress on this topic and fills in any existing gaps which have not been clarified yet.
Plant tissue culture and micropropagation systems
A few possible pathways are available in plant tissue culture and regenerants can be obtained either by axillary bud proliferation, adventitious shoot regeneration or via formation of somatic embryos [24]. Organogenesis, i.e. formation of either shoot or root and somatic embryogenesis (SE)—the formation of a bipolar structure containing the root and shoot meristem at the same time from a somatic cell [24]. This technique is used for clonal propagation of various economically important plants [25], including grape [26], pineapple [27], chili [28], strawberry [29], Siberian ginseng [30], Cymbidium orchids [31], raspberry [32], and blueberry [33]. Similar to organogenesis, somatic embryos can be obtained directly from the explants or indirectly with the interference of the callus formation at any stage of development [25]. Although direct SE has been reported from microspores, ovules, embryos, and seedlings [26, 31], leaf explants are also found to be responsive to SE [30, 33]. However, there is a great variability on leaves´ response to SE induction across plant taxa.
In vitro-induced variation: History and origin
The term TCIV or somaclonal variation has been widely used to denote the variations originated during the tissue culture process [3]. Many other names are also used to describe this variation such as gametoclonal, protoclonal, and meridional variation, depending on their tissue of origin [34]. Ideally, clonal propagation process involves only mitotic division to produce plantlets asexually. Thus, all the regenerants should be phenotypically and genotypically identical [35]. However, with the first observation of TCIV [36], it imposed a major problem in clonal propagation system [35]. The unmanageable occurrence of variations might be the reason for not including somaclonal variation regularly in crop improvement programs, as this could disturb the already present desirable traits in the crop plants. Despite that, several tissue culture-induced variants have been released as commercial varieties and cultivars [37]. Unlike spontaneous mutations, TCIV arise frequently in explants and the occurrence of spontaneous mutations is much more frequent during in vitro culture than in in vivo culture [38]. In tissue culture system, the genetic material is unprotected. Thus chances of exposure to chemicals present in the medium is higher due to which survival of the variants in non-inclusive environment increases with the rate of occurrence of mutation in tissue culture system than in field grown plants [38]. Even though, when the mutation rate is same in the somaclones and conventionally grown plants, detection of physiological variants is quite difficult in a crop grown in field because of larger production area [38]. The variations in somaclones emerge due to spontaneously induced major uncontrolled changes in cells, tissues, or organs which may occur genetically or epigenetically [34] (Fig. 1). For instance, in twenty-five regenerants from callus cultures of wild barley, no phenotypic differences were identified, although there were differences in the their rDNA spacer length and SDS PAGE profile of hordein [39]. However, often somaclones are morphologically different from the donor plants and are permanently different when the variations are genetic and heritable [8]. On the other hand, temporary changes occur due to the epigenetic or physiological factors which always do not inherit to the next generation [8, 35]. Chromosomal aberrations, sequence variations, point mutations, transposable element activation, and most importantly alterations in DNA methylation patterns are examples of some genetic and epigenetic variations which contribute to somaclonal variation [4].

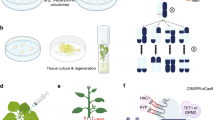
Schematic diagram of tissue culture-induced variation
DNA methylation: A chromatin modification
In plants and mammals, DNA methylation is conserved and specific patterns of genomic DNA methylation are extremely important for development [40]. It is well accepted that DNA retains information in a bigger context than the DNA sequence alone, while DNA sequences harbor genetic codes, and in some cases imprint these codes in a epigenetic fashion into various phenotypic traits [14]. DNA methylation is an evolutionarily ancient covalent modification (cytosine converts to 5-mC), often associated with gene silencing in eukaryotes [41]. Among all the heritable and major epigenetic changes DNA methylation is one of the best studied process [23]. DNA methylation level varies from 0 to 3% in insects, 2–7% in vertebrates, up to 10% in fish and amphibians, while plant genome shows a high rate of DNA cytosine methylation of over 30% [16, 41]. In plants, DNA methylation usually occurs in CpG islands in all symmetric (CG or CHG, where H = A, T or G), and asymmetric (CHH) contexts unlike animal where CG methylation prevails [23]. Among these, three types of cytosine methylation occurs in plants, frequency of CG sequences getting methylated is highest followed by CHG and CHH sequences [23]. For instance, in Arabidopsis genome-wide cytosine methylation 24% was reported from CG context, while 6.7 and 1.7% were from CHG and CHH contexts [16]. Another study on whole genome bisulphite sequencing (WGBS) on mulberry under drought stress tolerance deciphered that methylated cytosines were most prevalent in CG sites (44.28%) followed by CHG (28.50%) and CHH (27.22%) sites [17].
To ensure if DNA methylation is inherited correctly to the next generation, several molecular mechanisms are involved [14], and many of these mechanisms have been studied in detail in Arabidopsis [41]. In Arabidopsis mutations in the fragment of methylation and demethylation pathways are not always lethal, yet it appears to be crucial during development and environmental stress responses in plants with complex genomes [40]. It was long established that in plants DNA methylation is not an active process rather disturbed stability of DNA methyltransferases may cause decreased level of DNA methylation [42]. In plants, domains rearranged methyltransferase 2 (DRM2) catalyses the de novo DNA methylation [40]. DRM2 is a homologue of DNA methyltransferase 3 [40], which is a family of de novo methyltransferases in mammals [41]. Unlike mammals, for de novo methylation in plants an RNA-directed DNA methylation (RdDM) pathway is of high importance [40]. In the canonical RdDM pathway as studied in Arabidopsis [43], RNA polymerase IV (POL IV) synthesizes short single stranded RNAs then RNA-dependent RNA polymerase 2 convert them to double stranded RNAs (dsRNAs) [40, 44]. Dicer-like protein 3 (DCL3) cleaves the dsRNAs into small interfering RNAs (siRNAs) and incorporated onto Argonaute (AGO) proteins, especially AGO4 and AGO6 [44]. The second stage of canonical RdDM pathway is established on RNA polymerase V dependent transcription of non-coding RNAs (ncRNAs) [44]. The ncRNAs are connected through the sequence complementarity of AGO4 and AGO6 proteins loaded with siRNAs [40]. Right after the AGO, siRNA, ncRNA and POL V ribonucleoprotein complex is assembled, DRM2 comes into play to target DNA methylation [40]. Canonical RdDM pathway is moderated by POL IV-dependent 24-nt siRNAs, small RNAs from various origins such as polymerase II (POL II) transcripts and viruses and moderate non-canonical RdDM pathways [45]. Initially, the species of RNA triggering RdDM was unknown, later studies found out involvement of siRNAs and long noncoding RNAs (lncRNAs) in this mechanism [14, 40]. In the centromeric region, and repetitive sequences of the Arabidopsis genome DNA methylation is heavily abundant [46], of which one-third of the methylated loci are highly associated with siRNAs which also points to the fact that siRNAs are involved in DNA methylation [47]. siRNAs are commonly associated with transcriptional silencing and targets CG or CHH cites for methylation and with each cycle of DNA replication, siRNAs preserves the pattern of CHH methylation in the daughter cells [43]. RdDM pathway directs DRM2, DNA methyltransferase 1 (MET1) and chromomethylase 3 (CMT3) to their target sites, as CHH methylation cannot be maintained by methyltransferases [43]. siRNAs play a key role in fine tuning any undesirable methylation pattern via RdDM pathway [14] and furthermore deep sequencing of siRNAs at various stages of plant development will reveal other key roles characterized by them. RdDM is involved in various biological processes such as, paramutation, repression of transposons activity, biotic and abiotic stress responses, and creation of methylation patterns during reproduction [40]. This pathway was first discovered in transgenic tobacco plants infected with viroid, non-protein-coding RNA molecules of few base pairs long which produced recombinant viroid sequences in the plant genome after replication mechanism [48]. Although there are some pathways involved in silencing untargeted sequences, DNA methylation maintenance pathways alter the action of epigenome [14]. To sustain the silenced state of transposons and to conserve the cell identity, stability of established global DNA methylation pattern is crucial [41]. Maintenance of DNA methylation in plants depends on the susceptibility of the methyltransferases towards different context of cytosine sequences such as CG, CHG and CHH [40]. CG methylation is maintained by DNA methyltransferase 1 (MET1 or DMT1), which is plant homologue of DNA methyltransferase 1 [41]. MET1 identifies the hemi-methylated CG context after DNA replication and converts the unmethylated cytosine to 5-mC in the daughter strand [49]. CHG methylation is maintained by CMT3 in a greater extent than by chromomethylase 2 (CMT2) [16, 41]. Methylated CHH context is maintained by DRM2 or CMT2 based on genomic region [40, 44], for instance DRM2 preserves the RdDM target sites while CMT2 catalyses heterochromatin containing histone H1 which is prohibited from RdDM [40]. It was also found that in case of DDM1 (decrease in DNA methylation 1) mediated RdDM pathways CMT2 catalyze CHH methylation [40]. Although the maintenance of methylation status by these pathways is well studied, the removal of DNA methylation by the same is not outlined accurately [41]. DNA methylation in angiosperms is well maintained by the plant genome itself, new information on establishment and maintenance of this has extended our knowledge on regulation of DNA methylation [14]. It is still very important to have a clear understanding of these molecular mechanisms to use them for improvising the epigenome for crop improvement programs.
DNA methylation and gene expression
The molecular mechanism behind transcriptional control during plant development is not yet completely understood. Studies have shown that in plants, DNA methylation suppresses DNA-transcription factor associations, which in turn regulates gene expression and various cellular processes [41]. Recent studies on whole genome methylation regarding gene expression have emphasised the effect of DNA methylation on gene regulation. Methylation commits to the inactivation of transposable elements or foreign DNA, thus maintains the stability of whole genome over non-homologous recombination and controls gene transcriptions [9]. DNA methylation also affects the developmental characters in plants. For example, linaria demonstrated variation in floral symmetry due to cytosine methylation [50]. The changes in floral symmetry resulted from epimutation and no changes were detected in the DNA sequence. However, these epigenetic changes were unstable and reverted to the original form after a certain number of generations. linaria epimutants produce radially symmetrical flowers instead of producing bilaterally symmetrical flowers. Due to the loss of function of linaria cycloidea-like gene (Lcyc), mutant flowers were produced in the same plant. It was found that in the mutant plants Lcyc gene persists in a highly methylated state, which causes the suppression of Lcyc gene function [50]. Although epigenetic variations are mostly non-heritable [4, 51], genes which are silenced by DNA methylation can be relocated to the alleles on sister chromatid or homologous genes on the other chromosomes. The process by which gene gets suppressed by DNA methylation and transfer of that to the sister chromatid is referred as paramutation [52]. Paramutation is unpredictable in nature and may lead to the production of variant phenotypes. In maize, paramutation has been well described in four genes, such as b1, r1, p11, and p1 which are responsible for encoding the transcription factors and biosynthesizing flavonoid pigments [53]. Among these four genes paramutation at b1 is extremely stable of all systems, because of which it is widely used as a model to study paramutation [54]. Paramutants are generated due to methylated B1 locus transferred to a non-methylated allele on the complementary gene, resulting in repression of gene expression [55]. DNA methylation can generate novel epiallelic state after transcription process which can provide a new avenue to give rise to phenotypic variation when it is lacking genetic mutation.
Transgenerational epigenetic memory and DNA methylation patterns
An organism’s epigenetic information may affect their phenotype, which can be feasibly stored and inherited following segregation as cytosine methylation [5]. DNA methylation may activate a gene which has been silenced by other mechanisms during embryo development. On the contrary, embryonic transcription may result in complete exclusion of DNA methylation machinery [11]. Heritability of methylation state is a spontaneous phenomena which supports the concept that DNA methylation could be included or excluded for specific cellular memory during development [11]. However, a growing number of evidences are available on heritable natural difference in the DNA methylation patterns of two individuals of the same species and it has a comprehensive capacity to confer to quantitative trait variation and crop improvement [5]. Evidences of epigenetic variations having an influence on natural variations can be seen in few classic examples, such as peloric mutant of linaria [50], a colorless non-ripening variant of tomato [56], and sex determination of melon [57]. These are the few cases where meiotically heritable epialleles give rise to morphological variations. Epigenetic alleles or epialleles are referred to the methylation level of certain region of genome which varies between individuals [40]. Within genetically diversified Arabidopsis population epiallelic variation has been discovered, however, it is still not clear if epialleles were emerged due to genetic variations [58]. It has been also found that RdDM pathways can affect protein-coding genes by silencing their activity and giving rise to epialleles which could be heritable by mitosis/meiosis [14]. Epialleles can possibly be generated during DNA methylation maintenance or mutagenesis, which in turn creates an avenue for crop improvement by using artificially generated epialleles.
DNA methylation as molecular basis of TCIV
Recent researches have been focused on detecting the frequency of epigenetic variations in the tissue culture system to assess the stability of clonally propagated plants [51]. Epigenetic variations were reported to be stable for more than a hundred years but occasionally they revert back to the original form [50]. During callus formation, cultures are successfully being established as experimental model systems [59] to unveil the dynamics of epigenetic changes during cell dedifferentiation [51], and eventually the regulation of developmental reprogramming [59]. Few well recognized examples of heritable and stable epigenetic modifications were already discussed, such as first natural plant variant linaria [50] and melon [57]. It has been speculated that the environment has an intense role in epigenetic variations [25]. For example, in Arabidopsis heat tolerance might get affected by CMT2-dependent CHH methylation, where it has been also showed that an allele at CMT2 locus exhibits a modified whole-genome CHH methylation pattern associated with temperature resilience [60]. It has been found that the Cmt2 mutants displayed higher level of heat tolerance, which in turn suggests genetic regulation of epigenetic mechanisms leading to natural adaptation to various levels of heat-stress [60]. According to the epigenetic theories, the interaction between an organism’s genes and environment affects an individual’s developmental process and eventually it’s heredity to the next generation [5]. In contrast to genetic alterations, epigenetic modifications may be heritable and can be influenced by the environment [5]. In the tissue culture system, the occurrence of epigenetic variations has been reported at several stages [10, 51, 61]. It was hypothesized by Phillips et al. [62] that DNA methylation could be the prime factor in TCIV. Several studies have reported TCIV in plants due to DNA methylation. For example, higher level of DNA methylation was noticed within maize callus and in vitro regenerated plants [10, 63]. Similarly, Brown et al. [64], Müller et al. [65], and Stroud et al. [66] found that high frequency DNA methylation and sequence variation were present in the progeny of the tissue culture-derived rice plants. In lowbush and hybrid blueberries, higher level of methylated CCGG sites were found in callus (215–258), while in the leaves methylated tetranucleotides sites were present in comparatively low number (75–100) [67]. However, analysis with methylation sensitive/insensitive restriction enzymes proved that DNA methylation does not play as the main factor behind all these changes.
It was reported that DNA methylation may affect gene expression by altering the chromatin structure via creating variation in methylation at specific sites which could result in alteration of gene expression sites in a positive or negative way [68]. Higher expression of OsSPL14 (Squamosa promoter binding protein like—14) promoter during the reproductive stage in rice due to hypermethylation increase panicle branching leading to higher grain yield [69]. In this study the authors have shown incorporation of the OsSPL14–WFP (Wealthy farmer’s panicle) allele in the commonly used rice variety ‘Nipponbare’ improved rice grain yield. This is an example of positive gene expression due to DNA methylation, on the other hand classic example of gene expression in a negative fashion due to DNA methylation is mantled in vitro-derived variants of oil palm [70]. Tissue culture-induced abnormality due to hypomethylation in oil palm fruit severely reduced the oil production and mainly affected the production of elite hybrids for oil production via micropropagation. However, there are various examples available on attaining superior agronomic quality due to DNA methylation, many other studies also suggested that crop species which avoid DNA methylation might be agronomically superior to the ones which are vulnerable to DNA methylation [9].
Stress induced in tissue culture system and DNA methylation
Even in the absence of an inducing stimulus, epigenetic mechanisms create an epigenetic memory during cell division to store the changes occurred in the surroundings [5]. It is well known that plant cells maintain their developmental plasticity during differentiation [42] and need to go through reprogramming to switch from differentiation to toti-/pluripotency [1]. Under tissue culture system, explants undergo either direct/indirect organogenesis or SE [1]. In case of indirect organogenesis process, the cells go through dedifferentiation, which incorporate chromatin level reprogramming to induce callus formation [1, 63]. Eventually the proliferating cells in the callus go through redifferentiation with the application of plant growth regulators (PGRs) in the culture medium leading to organogenesis or plantlet regeneration [51]. The dedifferentiation and redifferentiation process under an artificial condition during in vitro culture, imposes traumatic stress on the plant cells, which initiate mitotically and meiotically heritable genetic and epigenetic variations [8]. Stresses which arise during plantlet regeneration via tissue culture process affect the normal functioning of cell organelles, but plasma membrane and cell wall sense the stress first and produce reactive oxygen species (ROS) [71]. Overproduction of ROS by lipid autolytic peroxidation or antioxidative defences creates oxidative damages to the cells [72]. Cellular homeostasis is not conserved in tissue culture system under stress like cold and heavy metal and excess production of ROS may influence the decadence of DNA, proteins, lipids and pigments affecting cellular functioning [71] leading to TCIV [42]. Cassells, Curry [72] speculated that in plant tissue culture system various factors such as recalcitrance, hyperhydricity, poor physiological condition of explants are also involved behind inducing genetic and epigenetic variation, where oxidative-stress damage plays a major role [72]. Not only that but also TCIV can occur due to epigenetic regulation as it may results in permanent alteration of DNA methylation level [42]. Although there are many reports available on this area, transgenerational responses leading to TCIV due to environmental stress in maize is one of the classic instances [8]. The sources of variations can be categorized according to the time of occurrence in the explants, such as pre-existing or variation induced during in vitro process [35]. Although pre-existing variations such as chimeras in the explants could eventually induce stress differently and leads to variation [35], clear understanding of which including methylation pattern is very important to comprehend the changes arise during tissue culture process.
Permanent variations in the regenerants are caused by the presence of pre-existing variations in the source plants or may be due to the expression of novel variations as an effect of an unknown mechanism(s) in the genome [3]. The genotype, explant type, propagation method, use of PGRs, ploidy level of explant tissue, and the age of culture are main determining factors of precedent variations in tissue culture system [6]. TCIV have been studied extensively to date, yet this phenomenon is far from complete understanding. Pre-existing variations arise separately from the effect of mutations, epigenetic changes such as alteration in DNA methylation pattern or by the combined effect of genetic and epigenetic factors [8]. However, qualitatively and quantitatively inherited mutations, use of chimeric tissues as explants, sequence variations, chromosomal aberrations, regulation of cell cycle, and transposable element activation in explants are also reasons behind the presence of pre-existing variations in the explant tissues [35].
The frequency of appearance of somaclones and the nature of variation differ with the different explant sources used for clonal propagation [73]. It has been noticed that in profoundly differentiating tissues such as roots, leaves, and stems normally produce an increasing number of variations if used as explants rather than tissues with pre-existing meristem, for example shoot tips and axillary buds [74]. Regeneration from older and/or highly specialized structures generally recover higher amounts of variation in the regenerants [75]. Adventitious shoot regeneration directly from leaves, petioles, shoot internodes, segments of root, anthers, hypocotyls, cotyledons or indirectly through formation of callus from the same type of explants shows higher amounts of variations in the regenerants [76]. The presence of chimeric tissue in the explants [34], and the unstable behaviour of the explants in the tissue culture system due to in vitro-derived stresses are also involved in the induction of somaclonal variation [35]. Reportedly, type of explants as well as the age of the explants used in the tissue culture system are the major determinant factors of the plantlet regeneration rate [77]. In general lower rate of DNA methylation was observed in the cultures, when the explants were obtained from young plants of Pinus radiata D. Don [78]. In contradiction, higher proportion of methylated cytosines (22.4%) was noticed in the young microshoots tissues of Acacia mangium in comparison to the mature microshoots with lower methylation rate (20.7%) [79].
TCIV may also arise due to the presence of callus in the in vitro culture. Variants originating from the callus are sometimes denoted as “calliclones” and commercial laboratories try to avoid callus formation during micropropagation due to the occurrence of variation in in vitro-induced callus [80]. Initiation of culture and subsequent subculture cycles make explants vulnerable to oxidative stress which might induce mutations [72]. In addition to that, it is evident that unorganized growth in different levels of tissue culture process from highly organized meristem tip culture to callus formation in the ‘extreme’ process like protoplast culture also impose stress and causes somaclonal variation [34]. Generally, the extent of this stress depends on the technique of tissue culture [75]. Consequently, it was also found in eggplant that indirect regeneration via callus phase shows a higher rate of mutation than plantlet production through axillary branching [81]. The extent of DNA methylation is also reported to be dependent on the level of differentiation. It was observed that, DNA methylation level changes radically as calluses go through the dedifferentiation and redifferentiation process [82].
In various plant species, callus formation starts with the application of exogenous auxin and cytokinin [83]. Ideal concentration, and the ratio of auxin and cytokinin are most important for the efficient shoot, and root regeneration. Exogenously applied PGRs control primary events, which induce cell cycle disturbance leading to morphogenesis and the introduction of variations among the regenerants [83]. In genetically abnormal cells, PGRs increase the rate of cell division [84] and also the presence of PGRs in the media might induce somaclonal variation via cell-cycle disturbance [83]. Morao et al. [85] studied the comparison between DNA methylomes among various cell types where they found that mitotically active columella cells from root tips exhibits highest methylation level especially at CHH sites over transposons sequences implying strong RdDM activity. Comparative level of auxin and cytokinin influence the genetic composition of the cell population [86]. It has been observed that the application of 6-Benzylaminopurine in high concentration (15 mg/l), increases the chromosome number in the banana somaclone CIEN BTA-03 derived from a cultivar ‘Williams’ [87]. Similarly, a higher concentration of artificial auxin 2,4-Dichlorophenoxyacetic acid (2,4-D) also triggers somaclonal variations in soybean [88] and cotton [89]. This is possibly due to the frequent use of 2,4-D in callus and cell cultures inducing genetic anomalies such as polyploidy and endo-reduplication of DNA [90]. The effect of 2,4-D was also found in genome-wide cytosine methylation in carrots. The application of 2,4-D in the embryogenic culture of carrot promotes total cytosine methylation, therefore 2,4-D alters the pattern of methylation in the genome [91].
Induction of somaclonal variations during the in vitro condition also reportedly influenced by the number and length of the subculture cycles. With the increased number of subculture cycles, higher proliferation rate can be obtained in a relatively short period of time in tissue culture [92, 93]. In cell suspension and callus culture, the chances of somaclonal variation increases with a higher number of the subculture and duration in in vitro culture [92]. In micropropagated banana, somaclonal variation started to appear at 1.3% after the fifth subculture, and after 11th subculture increased to 3.8% [93]. In addition to subculture cycle, the length of the culture period also affects the rate of somaclonal variation occurrence [92]. Four months of in vitro storage was suggested as a solution to the unstable ploidy level observed in long-standing coffee cell culture [94]. The same phenomenon was observed in olive, where difference in the morphological characters was confirmed by randomly amplified length polymorphism (RAPD) analysis in long term cultures [95].
DNA methylation and organogenesis
During organogenesis, regulation of specific genes is very important and DNA methylation is described as one of the regulatory mechanism during this process [25]. Several reports are available on effects of DNA methylation in direct organogenesis. In bush lily for example, the rate of DNA methylation was higher in shoot tip regenerated plants than the plants from young leaves or petals [96]. However, along with the explants in vitro and ex vitro environmental factors also influence DNA methylation during organogenesis processes [61]. In apple shoot tips organogenesis, it was observed that DNA methylation was influenced by the prolonged exposure of the tissue to low (4 °C) temperature, though the DNA sequence or ploidy level did not change [97]. Effects of DNA methylation were also studied in indirect organogenesis along with direct organogenesis. In tissue culture-derived Arabidopsis callus, few chromatin modifying enzymes like MET1, KRYPTONITE, histone demethylase gene JUMONJI 14, and histone acetyltransferase WUSCHEL (WUS) were analyzed to identify their influences on degree of shoot regeneration [98]. Bisulfite sequencing (BS-seq) and ChIP results demonstrated that the methylation and histone modification at the promoter region of the gene resulted in altered expression of these genes and rate of shoot regeneration from the callus [98].
DNA methylation and SE
SE is one of the best studied plant tissue culture process [23] and it is affected by many factors. DNA methylation is regarded as a very crucial factor to control the induction and developmental process somatic embryos similarly to the zygotic embryos [99]. It is reported that DNA hyper and hypomethylation impact the SE process [30, 99, 100]. For example, using HPLC/MS/MS method association of DNA hypermethylation was detected with somatic embryo development in Acca sellowiana [101]. Likewise, in Coffea canephora increase in DNA methylation level found to be positively correlated with rate of SE [99]. Furthermore, DNA hypomethylation has been found to be related to somatic embryo development from asynchronous T87 cell culture of Arabidopsis [100]. Ji et al. [102] found that the level of genome-wide DNA methylation, most apparently for CHH context increases during globular-stage of somatic embryos collected from 6 weeks to 13 years of soybean continuous culture. It was also clearly shown that DNA hypomethylation was most abundant in the already silenced regions and it was coupled with the gene upregulation responsible for reinforcing RdDM pathway [102]. On the contradiction, in Siberian ginseng lower level of DNA methylation was detected using MSAP technique in embryogenic callus in comparison to the non-embryogenic callus [30]. The changes in DNA methylation pattern of plant cells and tissues arise during in vitro culture due to the exposure to different factors such as basal medium, PGR used, stages of culture, biotic and abiotic stresses due to wounding of the explant, nutrients used in the growth medium, physical factors, photoperiod, etc. [1, 23]. DNA methylation pattern found under these conditions regulate gene expression related to SE and plant regeneration processes [23]. For instance, in carrot cell suspension cultures, 16% methylated cytosine with stable DNA methylation pattern was seen during SE upon applying exogenous auxin. However, the application of hypomethylating drugs such as 5–azacytidine (5-azaC) and ethionine, blocked SE [91]. A similar trend was observed in alfalfa, application of 5-azaC in the embryogenic line decreased the formation of somatic embryos extensively [103]. Fraga et al [101] reported lower level of methylation in A. sellowiana cell culture when 5-azaC was added to the medium, but higher level of embryo induction was noticed when 5-azaC and 2,4-D were added in the culture in combination. It is also reported that at the preliminary stages of somatic embryo development, embryos contain comparatively lower levels of DNA methylation in comparison to the older stages [73]. Occasionally, SE is selected as a desirable pathway for regeneration [75] as direct embryogenesis provides genetically similar plants than shoot tip regeneration because at preliminary stage of SE DNA contains comparatively lower level of methylation [73].
Methods of detecting in vitro-induced DNA methylation
In large-scale commercial micropropagation, detection of variants is a matter of challenge due to the presence of a huge number of clones in a large area. Therefore, detection and exclusion of negative agronomic traits at early stages is very important to reduce economic loss to the growers [35]. At later stages of any crop improvement program, it is essential to evaluate the variants at different environmental conditions for the successful establishment of desirable traits over generations [74]. Although various methods are available to detect somaclonal variation, this review discusses the methods used for detection of DNA methylations in vitro. Modified AFLP techniques are mostly used to detect tissue culture-induced DNA methylation [51]. The principal of MSAP technique is constructed on the susceptibility of the pair of isoschizomers MspI and HpaII instead of MseI, as ‘frequent cutter’ enzymes, and EcoRI as a ‘rare cutter’ enzyme same as that used in the original AFLP protocol [15, 20, 104]. MspI and HpaII restrict the 5-CCGG-3 recognition site based on methylation of external or internal cytosine. MspI cleaves methylated internal cytosine residues (CmCGG) but not the external (mCCGG) whereas HpaII cleaves the hemimethylated external cytosine but remains inactive for fully methylated sequences [105]. Although this is an economically feasible, fast, and easy practicable process for non-model organisms, the selection of these restriction enzymes may results in inconsistent data interpretation, which may not agree with the previously acquired data [15]. This technique was first developed in dimorphic fungi [104] and later used to detect methylation in rice [106], banana [105], apple [107], Siberian ginseng [30], pepper [108], hop [7], Doritaneopsis orchid [12], freesia [109], and blueberry [67, 110]. Quantification of global DNA methylation can be done by high-performance separation techniques such as HPCE, HPLC [111]. These techniques involve genomic DNA digestion to nucleotide, nucleoside or nitrogenous bases through enzymatic hydrolysis, to isolate, and analyze 5-mC [51, 111]. Although both of this capillary hydrolysis based approaches are quite time consuming, they are highly specific and sensitive, which makes them useful for rapid quantification of global DNA methylation even from poorly isolated or low quality samples [111]. In some ornamental crops, such as Cedrus (Cedrus atlantica, and Cedrus libani) HPLC has been used to detect DNA methylation in axillary bud culture [22], whereas HPCE was used in in vitro shoot culture of pea to detect the global methylation pattern [21]. Unlike in other eukaryotic organisms, in case of plants, genome-wide application using ChIP methodology is usually scarce [18]. ChIP techniques such as, (ChIP)-chip and ChIP-seq detect DNA methylation by mapping the point of interaction between DNA and the protein of interest. The ChIP-chip technique identifies the sites of DNA-protein interaction in DNA while ChIP-seq detects cytosine methylation by combining immunoprecipitation with shotgun sequencing technique [18]. However, the huge amount of data generated through the high-throughput sequence creates a great difficulty to identify the protein binding sites in case of ChIP-based techniques [19]. ChIP-seq was used in Arabidopsis to detect DNA-transcription factor binding site during DNA methylation process [19]. Bisulfite modification is another efficient mechanism to identify methylated cytosine. During the process, genomic DNA treated with sodium bisulfite converts cytosine to uracil, while methylated cytosine does not change [16]. WGBS is believed to be the best procedure to detect DNA methylation in plant samples as this technique allows to detect single nucleotide resolution of 5-mC on a genome while other techniques fails to provide that [16, 112]. Currently this is the most updated and direct approach which allows to identify and detect the pattern of methylated cytosine within the whole genome [113]. However, this technique is still very costly for the plants with comparatively larger genome size than Arabidopsis or rice [113]. So far WGBS has been used to detect level of DNA cytosine methylation on various plant genomes, which further proved the fact that DNA methylation varies across plant species. This technique was used in the detection of DNA methylation in the whole genome of Arabidopsis. An unknown side of the Arabidopsis methylome was revealed in WGBS procedure following next generation sequencing [16]. Recently, this technique has been used to detect altered methylation level on long-term in vitro shoot culture and regenerants of apple [114] and plants regenerated from pineapple callus culture [112]. Methods detecting DNA methylation in some crops and plant species are listed in Table 1.
Tissue culture-induced DNA methylation and it’s implication on crop improvement
In any crop improvement program genetic variations are crucial factors. Conventional plant breeders develop new cultivars by combining genes of interest from well-established varieties or linked species by the process of sexual hybridization, thus developed new cultivars with better agronomic traits [4]. With an ever-increasing human population, the demand for sustainable food production came up as a challenge for conventional plant breeders. The aim of a crop improvement program is to select the improved varieties with heterosis and transgressive variations, including both genetic and epigenetic modifications [5]. Mutations caused by heritable epigenetic traits are known as epimutations and these epimutations are very difficult to detect without the intervention of whole genome structure analysis [41]. Examples of epimutations found in various plants are listed in Table 2. Several recent studies on Arabidopsis have shown that in a population of recombinant inbred lines (epiRILs) individual plants differ from each other on the basis of epigenetic information [5]. One of this studies demonstrated that the epiRILs were generated by exposing the genome to a mutation responsible for removal of DNA methylation and segregating away the mutation thus creating an altered methylation pattern in the segregated genomic segments [126]. Although there are only a few examples are available on using a genetic approach identical to epiRILs, it can be used as a technique to introduce variations in crop plants. Predominantly genetic mutations result in loss-of-function allele while epimutations often lead to gain-of-function alleles by the loss of epigenetic silencing [5]. Epialleles that originates from various genetic mutations such as transposon insertions are quite stable because of the continuous presence of reprogramming machinery of chromatin modification. On the other hand, naturally occurring epimutations are less stable due to the lack of assurance of reinstating epigenetic information [14]. Instability of the epimutants in comparison the genetic mutants can be a potential drawback of using epimutation in a crop improvement program. During a breeding program for crop improvement, if the epigenetic state only displays partial heritability, it can be difficult to stabilize the epigenetic state within the population and generate epimutants [5]. However, epimutations in clonally propagated plant species may allow scrutinizing epiallelic alterations in favour of novel allelic variations without depending upon DNA recombination [5, 135]. This would be specifically important in case of clonally propagated species [135], with either germplasm bottlenecks or limited recombination in some genomic region [5]. Another drawback of using epimutations for crop improvement program would be activation of transposable elements [136], which can lead to generation of production of deleterious alleles and higher rate of mutation resulting into diminished utility of the epimutants [5].
While some horticultural crop species are clonally propagated to maintain their trueness-to-type, in conventional crop improvement programs heritable genetic variations are important components, which often create stable inbred and hybrid varieties for further agricultural use [5]. Predominantly, plant breeders tend to select phenotypic variations over a specific molecular change, which occurs within a specific generation, e.g. non-heritable epigenetic variations [4, 5]. Stable genetic variations can arise due to the genetic changes, for example, gene duplication, and insertions of transposons in the genome [6]. However, as somaclones have widened the variability in crops, they can be used to improve many plant characteristics such as grain yield and quality, plant height, flowering, resistance to biotic and abiotic stresses such as insects-pests, diseases, cold, drought, salinity, and soil pH [6]. Several reports are available demonstrating desirable variations in somaclones that are already used in plant breeding programs regularly. For instance, Indian mustard which has high yielding capacity and shattering resistance [37], mint with increased oil and herb yield [137], neurotoxin devoid Lathyrus [138], early blight disease resistant potato [139], salinity and drought tolerant sugarcane [140], red rot disease resistant sugarcane [141], and aluminium toxicity tolerant rice [142].
DNA methylation is the only epigenetic factor for which conservation and stable inheritance pattern in the consecutive generations are well understood [41], e.g. that heritable TCIV in rice regenerants for subsequent generations were often due to DNA hypomethylation which also sometime effect the expression of nearby genes [66]. In another study on maize, it was found that differentially methylated regions were developed due to tissue culture process and among these regions hypomethylation was prevalent in comparison to hypomethylation [63]. TCIV due to DNA methylation was detected in Arabidopsis lineages, where variations were successfully inherited and segregated within R1 progenies, which provided the opportunity to study variations induced during tissue culture system in plants [143]. DNA methylation also plays an important role managing the developmental events and response features in the in vitro system for example, in Arabidopsis callus and suspension cultures regulation of undifferentiated state due to gene repression by DNA methylation in the promoter region of a particular single copy gene has been reported earlier [59]. In general, a typical crop improvement program takes 10–15 years to be fully completed and undergoes many stages from germplasm collection to crop production [75]. DNA methylation can be used in crop improvement programs in a controlled way to generate better agronomic traits without incorporating any foreign genes [25] (Fig. 2). Although in few cases, this method may not provide a extremely stable variant which will pass the long term conventional breeding procedure [5]. In Arabidopsis, diminished DNA methylation gives rise to plentiful morphological and phenotypic irregularities which includes reduced apical dominance, decreased plant size, leaf size and modification, lowered fertility, and irregular flowering time [13]. This may be due to the plants after going through the meiotic events, reportedly having decreased ability to reinstate the previous DNA methylation pattern [13]. Some clonally propagated strawberry cultivars were observed with hyper flowering and abnormal fruit setting. Hyper flowering was reported from ‘stipular buds’ at a particular position of leaf petiole. These buds showed high multiplication rate in vitro and an increasing number of flower production per inflorescence in ex vitro condition. This habit was speculated to be due to the DNA methylation and not the influence of any true mutation [144]. In lowbush blueberry, it has been reported that micropropagated plants are higher in polyphenols and flavonoids than the softwood cutting plants [145]. However, number of flower clusters, berries, fruit weight per plants diameter and height of individual fruits were significantly reduced in clonally propagated plants [145]. Although molecular assay of the tissue culture regenerants with simple sequence repeat (SSR) markers established their genetic fidelity with the softwood cutting counterparts, it was hypothesized that variations originating in the blueberry clones probably due to epigenetic variations induced during the in vitro culture process [145]. Later methylation analysis in micropropagated lowbush blueberry callus and leaves collected from the greenhouse grown plants confirmed that, in vitro derived calluses contained higher amounts (14–30%) of methylated cytosine in the genome, while the percentage of DNA methylation in leaves is comparatively lower (13–18%) [67]. Complete understanding of the molecular nature of TCIV is very important to exploit it further in crop breeding programs.

Potential implications of DNA methylation in the cycle of crop improvement via plant tissue culture
Future prospects
For many years, researchers have been discussing various possible ways to include epigenetic variations in crop breeding programs. Although there are not many reports available on using epimutations in crop improvements program, Thieme et al. [146] discussed inhibition of RNA POL II to decrease DNA methylation at heat responsive copia-like retrotransposon ONSEN to introduce novel genetic variation. However, there are some well-known cases on undesirable epimutations such as colourless non-ripening (CNP) variant of tomato [56], reduction in palm oil yields due to DNA hypomethylation in Karma retrotransposon [70]. Adaptation of tissue culture-induced (somaclonal) variation in crop improvement program have their advantages, for example, selection of somaclones in vitro may reduce the selection time and may fight biotic and abiotic stresses. TCIV has been successfully employed in many crop species with narrow genetic base and systems with limited genetic variations such as apomicts and vegetative reproducers [75]. Additionally, in the case of direct SE without any intervention of callus stage, embryos are formed from the individual somatic cell. Thus the chance of cellular mosaic formation is lower and originated plantlets are genetically similar with little variation [147]. For example, in clonally propagated ‘Grand Naine’ banana plants, SE has been used to reduce the chance of somaclonal variation [148]. As somatic embryos arise from single somatic cells, the chances of induction variation decrease. Later it has been confirmed that in banana variety, production of variants was lower (1.6–7.9%) when plantlets were produced via SE than from shoot tip culture (2.3–10.4%) [149]. In general, clones originating from axillary branching are reported to be true-to-type to the donor plants [2]. However, disadvantages of this process include chances of negative agronomic traits production, the occurrence of unpredictable changes, and the need for extensive field trials of the somaclones before the release as a variety [4]. Moreover, the desirability of the somaclones is not possible to predict and there is no way to tell that the character of interest will always be modified in an advantageous way [34]. Predominantly, somaclonal variations in the micropropagated plants are random and lack reproducibility. Therefore, the main concern about TCIV is to make it reproducible so that it can be used in regular crop breeding programs [4, 35]. It is not very easy to control TCIV as more than one factor are responsible for the induction of variations in the tissue culture regenerants [4]. These variations can be controlled by avoiding longer duration in in vitro condition and reducing the number of subculture cycles [94]. In micropropagated banana, increased rate of variation was reported with the increasing number of sub-culturing [35]. Not only the culture duration and several subculture cycles influence TCIV, mode of in vitro propagation method also affects the induction of variation. Reportedly, rice clones regenerated from protoplast culture gives rise to plant variants, which are different from conventionally propagated plants in terms of leaf morphology, flower characteristics, spikelet, and panicles [20]. Cellular organization of the explants is also a very important factor for somaclonal variation [34], generally the more breakdown of organizational structure (callus formation), the higher chances of occurrence of variation [35]. In clonally propagated ‘Honeycrisp’ apple, propagants shows some unexpected green and red colour patterns, which is found to be due DNA methylation at the promoter region of the MYB10 transcription factor involved in anthocyanin production [135]. Comprehensive knowledge of epialleles and epigenetic traits can help to crate a path of using DNA methylation profiling to identify the ‘off-types’ produced in tissue culture system [14]. Not only exclusion of deleterious alleles but also understanding epigenetic aspects of tissue culture regenerants will aid creating beneficial epialleles via epigenetic engineering.
Concluding remarks
Commercial micropropagation is based on trueness-to-type of the micropropagated plants to the donor plants. Various molecular approaches have been used to confirm the genetic fidelity in tissue culture plants. Although production of somaclones was thought to be originated from genetic variations, epigenetic factors are also found to be associated with phenotypic diversity. It is essential to understand the combining effects of genetic mutations and epigenetic variations on somaclonal variation to exploit it more efficiently in crop improvement programs. Even though somaclonal variation generates major problem regarding clonal fidelity in tissue cultured plants, it still provides a source for crop improvement in the plants with narrow genetic bases. Selection of the plants with desirable agronomic traits relies on induced variation during the tissue culture process. Recently, DNA methylation has been recognized as a major regulatory epigenetic mechanism which is associated with various regulatory gene functions during the tissue culture process. Although many studies are being performed on TCIV (including DNA methylation), the process is still far from being completely understood. Therefore, fully developed understanding of these processes will help to identify the hypervariable regions in the plant genome during tissue culture process, which could lead to the efficient control of somaclonal variations and their use in crop improvement programs on a regular basis.
References
Neelakandan AK, Wang K (2012) Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep 31:597–620
Debnath SC (2018) Thidiazuron in micropropagation of small fruits. In: Ahmad N, Faisal M (eds) Thidiazuron: from urea derivative to plant growth regulator. Springer Nature Singapore Pte Ltd, Singapore, pp 139–158. https://doi.org/10.1007/978-981-10-8004-3_6
Larkin PJ, Scowcroft W (1981) Somaclonal variation—a novel source of variability from cell cultures for plant improvement. Theor Appl Genet 60:197–214
Jain SM (2001) Tissue culture-derived variation in crop improvement. Euphytica 118:153–166. https://doi.org/10.1023/A:1004124519479
Springer NM (2013) Epigenetics and crop improvement. Trends Genet 29:241–247. https://doi.org/10.1016/j.tig.2012.10.009
Jain SM (1998) Plant biotechnology and mutagenesis for sustainable crop improvement. In: Behl RK, Singh DK, Lodhi GP (eds) Crop improvement for stress tolerance. CCSHAU, Hissar & MMB, New Delhi, India, pp 218–232
Peredo EL, Revilla MA, Arroyo-García R (2006) Assessment of genetic and epigenetic variation in hop plants regenerated from sequential subcultures of organogenic calli. J Plant Physiol 163:1071–1079
Kaeppler SM, Kaeppler HF, Rhee Y (2000) Epigenetic aspects of somaclonal variation in plants. Plant Mol Biol 43:179–188
Lukens LN, Zhan S (2007) The plant genome's methylation status and response to stress: implications for plant improvement. Curr Opin Plant Biol 10:317–322
Kaeppler SM, Phillips RL (1993) Tissue culture-induced DNA methylation variation in maize. Proc Natl Acad Sci 90:8773–8776
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Park SY, Murthy HN, Chakrabarthy D, Paek KY (2009) Detection of epigenetic variation in tissue-culture-derived plants of Doritaenopsis by methylation-sensitive amplification polymorphism (MSAP) analysis. In Vitro Cell Dev Biol Plant 45:104–108
Finnegan EJ, Peacock WJ, Dennis ES (1996) Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Acad Sci 93:8449–8454
Springer NM, Schmitz RJ (2017) Exploiting induced and natural epigenetic variation for crop improvement. Nat Rev Genet 18:563. https://doi.org/10.1038/nrg.2017.45
Fulneček J, Kovařík A (2014) How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet 15:2
Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452:215–219 http://www.nature.com/nature/journal/v452/n7184/suppinfo/nature06745_S1.html
Li R, Hu F, Li B, Zhang Y, Chen M, Fan T, Wang T (2020) Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications in response to drought stress. Sci Rep 10:8013. https://doi.org/10.1038/s41598-020-64975-5
Kaufmann K, Muino JM, Osteras M, Farinelli L, Krajewski P, Angenent GC (2010) Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat Protoc 5:457–472. https://doi.org/10.1038/nprot.2009.244
Wang C, Xu J, Zhang D, Wilson ZA, Zhang D (2010) An effective approach for identification of in vivo protein-DNA binding sites from paired-end ChIP-Seq data. BMC Bioinf 11:81–81. https://doi.org/10.1186/1471-2105-11-81
Vos P, Hogers R, Bleeker M, Reijans M, Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Smýkal P, Valledor L, Rodriguez R, Griga M (2007) Assessment of genetic and epigenetic stability in long-term in vitro shoot culture of pea (Pisum sativum L.). Plant Cell Rep 26:1985–1998
Renau-Morata B, Nebauer SG, Arrillaga I, Segura J (2005) Assessments of somaclonal variation in micropropagated shoots of Cedrus: consequences of axillary bud breaking. Tree Genet Genomes 1:3–10
Karim R, Nuruzzaman M, Khalid N, Harikrishna JA (2016) Importance of DNA and histone methylation in in vitro plant propagation for crop improvement: a review. Ann Appl Biol 169:1–16. https://doi.org/10.1111/aab.12280
Steward FC, Ammirato PV, Mapes MO (1970) Growth and development of totipotent cells some problems, procedures, and perspectives. Ann Bot 34:761–787
Us-Camas R, Rivera-Solís G, Duarte-Aké F, De-la-Peña C (2014) In vitro culture: an epigenetic challenge for plants. Plant Cell Tissue Organ Cult 118:187–201
Vidal J, Rama J, Taboada L, Martin C, Ibanez M, Segura A, González-Benito M (2009) Improved somatic embryogenesis of grapevine (Vitis vinifera) with focus on induction parameters and efficient plant regeneration. Plant Cell Tissue Organ Cult 96:85
Sripaoraya S, Marchant R, Power JB, Davey MR (2003) Plant regeneration by somatic embryogenesis and organogenesis in commercial pineapple (Ananas comosus L.). In Vitro Cell Dev Biol Plant 39:450–454
Santana-Buzzy N, Canto-Flick A, Barahona-Pérez F, del Carmen Montalvo-Peniche M, Zapata-Castillo PY, Solís-Ruiz A, Zaldívar-Collí A, Gutiérrez-Alonso O, de Lourdes Miranda-Ham M (2005) Regeneration of habanero pepper (Capsicum chinense Jacq.) via organogenesis. HortScience 40:1829–1831
Biswas M, Dutt M, Roy U, Islam R, Hossain M (2009) Development and evaluation of in vitro somaclonal variation in strawberry for improved horticultural traits. Sci Hortic 122:409–416
Chakrabarty D, Yu K-W, Paek KY (2003) Detection of DNA methylation changes during somatic embryogenesis of Siberian ginseng (Eleuterococcus senticosus). Plant Sci 165:61–68
Ghosh A, Hossain MM, Sharma M (2014) Mass propagation of Cymbidium giganteum Wall. ex Lindl. using in vitro seedlings. Indian J Exp Biol 52:905–911
Debnath SC (2014) Bioreactor-induced adventitious shoot regeneration affects genotype-dependent morphology but maintains clonal fidelity in red raspberry. In Vitro Cell Dev Biol Plant 50:777–788. https://doi.org/10.1007/s11627-014-9632-2
Ghosh A, Igamberdiev AU, Debnath SC (2018) Thidiazuron-induced somatic embryogenesis and changes of antioxidant properties in tissue cultures of half-high blueberry plants. Sci Rep 8:16978. https://doi.org/10.1038/s41598-018-35233-6
Karp A (1994) Origins, causes and uses of variation in plant tissue cultures. In: Plant cell and tissue culture. Kluwer, Dordrecht, pp 139–151
Bairu MW, Aremu AO, Staden J (2010) Somaclonal variation in plants: causes and detection methods. Plant Growth Regul 63:147–173. https://doi.org/10.1007/s10725-010-9554-x
Braun AC (1959) A demonstration of the recovery of the crown-gall tumor cell with the use of complex tumors of single-cell origin. Proc Natl Acad Sci 45:932–938
Katiyar RK, Chopra VL (1995) A somaclone of Brassica juncea is processed into a variety and is released for commercial cultivation in India. Cruciferae Newsl 17:92–93
Ahloowalia BS (1986) Limitations to the use of somaclonal variation in crop improvement. In: Somaclonal variations and crop improvement. Springer, pp 14–27
Breiman A, Rotem-Abarbanell D, Karp A, Shaskin H (1987) Heritable somaclonal variation in wild barley (Hordeum spontaneum). Theor Appl Genet 74:104–112
Zhang H, Lang Z, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev 19:489–506
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204
Bednarek PT, Orłowska R (2020) Plant tissue culture environment as a switch-key of (epi) genetic changes. Plant Cell Tissue Organ Cult 140:245–257
Zhang H, Zhu J-K (2011) RNA-directed DNA methylation. Curr Opin Plant Biol 14:142–147. https://doi.org/10.1016/j.pbi.2011.02.003
Gallego-Bartolomé J (2020) DNA methylation in plants: mechanisms and tools for targeted manipulation. New Phytol 227:38–44
Cuerda-Gil D, Slotkin RK (2016) Non-canonical RNA-directed DNA methylation. Nat Plants 2:1–8
Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39:61
Lister R, Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133:523–536. https://doi.org/10.1016/j.cell.2008.03.029
Wassenegger M, Heimes S, Riedel L, Sänger HL (1994) RNA-directed de novo methylation of genomic sequences in plants. Cell 76:567–576. https://doi.org/10.1016/0092-8674(94)90119-8
He XJ, Chen T, Zhu JK (2011) Regulation and function of DNA methylation in plants and animals. Cell Res 21:442–465
Cubas P, Vincent C, Coen E (1999) An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401:157–161
Miguel C, Marum L (2011) An epigenetic view of plant cells cultured: somaclonal variation and beyond. J Exp Bot 62:3713–3725. https://doi.org/10.1093/jxb/err155
Chandler VL (2010) Paramutation’s properties and puzzles. Science 330:628–629
Sidorenko LV, Peterson T (2001) Transgene-induced silencing identifies sequences involved in the establishment of paramutation of the maize p1 gene. Plant Cell 13:319–335
Stam M, Belele C, Dorweiler JE, Chandler VL (2002) Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev 16:1906–1918
Soppe WJJ, Jacobsen SE, Alonso-Blanco C, Jackson JP, Kakutani T, Koornneef M, Peeters AJM (2000) The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell 6:791–802
Manning K, Tor M, Poole M, Hong Y, Thompson AJ, King GJ, Giovannoni JJ, Seymour GB (2006) A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet 38:948–952 http://www.nature.com/ng/journal/v38/n8/suppinfo/ng1841_S1.html
Martin A, Troadec C, Boualem A, Rajab M, Fernandez R, Morin H, Pitrat M, Dogimont C, Bendahmane A (2009) A transposon-induced epigenetic change leads to sex determination in melon. Nature 461:1135–1138 http://www.nature.com/nature/journal/v461/n7267/suppinfo/nature08498_S1.html
Vaughn MW, Tanurdžić M, Lippman Z, Jiang H, Carrasquillo R, Rabinowicz PD, Dedhia N, McCombie WR, Agier N, Bulski A, Colot V, Doerge RW, Martienssen RA (2007) Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol 5:e174. https://doi.org/10.1371/journal.pbio.0050174
Berdasco M, Alcázar R, García-Ortiz MV, Ballestar E, Fernández AF, Roldán-Arjona T, Tiburcio AF, Altabella T, Buisine N, Quesneville H (2008) Promoter DNA hypermethylation and gene repression in undifferentiated Arabidopsis cells. PLoS One 3:e3306
Shen X, De Jonge J, Forsberg SK, Pettersson ME, Sheng Z, Hennig L, Carlborg Ö (2014) Natural CMT2 variation is associated with genome-wide methylation changes and temperature seasonality. PLoS Genet 10:e1004842
De-la-Pena C, Nic-Can G, Ojeda G, Herrera-Herrera JL, Lopez-Torres A, Wrobel K, Robert-Diaz ML (2012) KNOX1 is expressed and epigenetically regulated during in vitro conditions in Agave spp. BMC Plant Biol 12:203. https://doi.org/10.1186/1471-2229-12-203
Phillips RL, Kaeppler SM, Peschke VM (1990) Do we understand somaclonal variation? In: Progress in plant cellular and molecular biology. Kluwer, Dordrecnt, pp 131–141
Stelpflug SC, Eichten SR, Hermanson PJ, Springer NM, Kaeppler SM (2014) Consistent and heritable alterations of dna methylation are induced by tissue culture in maize. Genetics 198:209. https://doi.org/10.1534/genetics.114.165480
Brown PTH, Kyozuka J, Sukekiyo Y, Kimura Y, Shimamoto K, Lörz H (1990) Molecular changes in protoplast-derived rice plants. Mol Gen Genet 223:324–328
Müller E, Brown PTH, Hartke S, Lörz H (1990) DNA variation in tissue-culture-derived rice plants. Theor Appl Genet 80:673–679
Stroud H, Ding B, Simon SA, Feng S, Bellizzi M, Pellegrini M, Wang G-L, Meyers BC, Jacobsen SE (2013) Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2:e00354
Ghosh A, Igamberdiev A, Debnath S (2017) Detection of DNA methylation pattern in thidiazuron-induced blueberry callus using methylation-sensitive amplification polymorphism. Biol Plant 61:511–519
Jain SM, Brar DS, Ahloowalia B (2013) Somaclonal variation and induced mutations in crop improvement, vol 32. Springer Science & Business Media
Miura K, Ikeda M, Matsubara A, Song X-J, Ito M, Asano K, Matsuoka M, Kitano H, Ashikari M (2010) OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat Genet 42:545–549 http://www.nature.com/ng/journal/v42/n6/suppinfo/ng.592_S1.html
Ong-Abdullah M, Ordway JM, Jiang N, Ooi S-E, Kok S-Y, Sarpan N, Azimi N, Hashim AT, Ishak Z, Rosli SK (2015) Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 525(7570):533
Das K, Roychoudhury A (2014) Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front Environ Sci 2:53. https://doi.org/10.3389/fenvs.2014.00053
Cassells A, Curry R (2001) Oxidative stress and physiological, epigenetic and genetic variability in plant tissue culture: implications for micropropagators and genetic engineers. Plant Cell Tissue Organ Cult 64:145–157. https://doi.org/10.1023/A:1010692104861
Sahijram L, Soneji JR, Bollamma KT (2003) Analyzing somaclonal variation in micropropagated bananas (Musa spp.). In Vitro Cell Dev Biol Plant 39:551–556
Duncan RR (1996) Tissue culture-induced variation and crop improvement. Adv Agron 58:201–240
Krishna H, Alizadeh M, Singh D, Singh U, Chauhan N, Eftekhari M, Sadh RK (2016) Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 6:1–18
Pijut PM, Beasley RR, Lawson SS, Palla KJ, Stevens ME, Wang Y (2012) In vitro propagation of tropical hardwood tree species—A review (2001–2011). Propag Ornamental Plants 12:25–51
George EF, Hall MA, De Klerk G-J (2007) Plant propagation by tissue culture: the background, vol 1. Springer Science & Business Media
Fraga MF, Cañal M, Rodríguez R (2002) Phase-change related epigenetic and physiological changes in Pinus radiata D. Don. Planta 215:672–678
Baurens FC, Nicolleau J, Legavre T, Verdeil J-L, Monteuuis O (2004) Genomic DNA methylation of juvenile and mature Acacia mangium micropropagated in vitro with reference to leaf morphology as a phase change marker. Tree Physiol 24:401–407
Skirvin RM, McPheeters KD, Norton M (1994) Sources and frequency of somaclonal variation. HortScience 29:1232–1237
Zayova E, Vassilevska-Ivanova R, Kraptchev B, Stoeva D (2010) Somaclonal variations through indirect organogenesis in eggplant (Solanum melongena L.). Biodivers Conserv 3:1–5
Huang H, Han S, Wang Y, Zhang X, Han Z (2012) Variations in leaf morphology and DNA methylation following in vitro culture of Malus xiaojinensis. Plant Cell Tissue Organ Cult 111:153–161. https://doi.org/10.1007/s11240-012-0179-9
Peschke VM, Philips RL (1992) Genetic implications of somaclonal variation in plants. Adv Genet 30:41–75
Bayliss MW (1980) Chromosomal variation in plant tissues in culture. Int Rev Cytol Suppl 11:113–144
Morao AK, Bouyer D, Roudier F (2016) Emerging concepts in chromatin-level regulation of plant cell differentiation: timing, counting, sensing and maintaining. Curr Opin Plant Biol 34:27–34
d'Amato F (1975) The problem of genetic stability in plant tissue and cell cultures. In: Frankel OH, Hawkes JG (eds) Crop genetic resources for today and tomorrow. Cambridge University Press, Cambridge, pp 338–348
Giménez C, García E, Enrech NX, Blanca I (2001) Somaclonal variation in banana: cytogenetic and molecular characterization of the somaclonal variant cien BTA-03. In Vitro Cell Dev Biol Plant 37:217–222. https://doi.org/10.1007/s11627-001-0038-6
Gesteira AS, Otoni WC, Barros EG, Moreira MA (2002) RAPD-based detection of genomic instability in soybean plants derived from somatic embryogenesis. Plant Breed 121:269–271
Jin S, Mushke R, Zhu H, Tu L, Lin Z, Zhang Y, Zhang X (2008) Detection of somaclonal variation of cotton (Gossypium hirsutum) using cytogenetics, flow cytometry and molecular markers. Plant Cell Rep 27:1303–1316
Bouman H, De Klerk GJ (2001) Measurement of the extent of somaclonal variation in begonia plants regenerated under various conditions. comparison of three assays. Theor Appl Genet 102:111–117. https://doi.org/10.1007/s001220051625
LoSchiavo F, Pitto L, Giuliano G, Torti G, Nuti-Ronchi V, Marazziti D, Vergara R, Orselli S, Terzi M (1989) DNA methylation of embryogenic carrot cell cultures and its variations as caused by mutation, differentiation, hormones and hypomethylating drugs. Theor Appl Genet 77:325–331. https://doi.org/10.1007/BF00305823
Bairu MW, Fennell CW, van Staden J (2006) The effect of plant growth regulators on somaclonal variation in Cavendish banana (Musa AAA cv. ‘Z elig’). Sci Hortic 108:347–351. https://doi.org/10.1016/j.scienta.2006.01.039
Rodrigues PHV, Tulmann Neto A, Cassieri Neto P, Mendes BMJ (1997) Influence of the number of subcultures on somaclonal variation in micropropagated nanicao (Musa spp., AAA Group). Acta Hortic 490:469–474
Clarindo WR, Carvalho CR, Mendonça MAC (2012) Ploidy instability in long-term in vitro cultures of Coffea arabica L. monitored by flow cytometry. Plant Growth Regul 68:533–538. https://doi.org/10.1007/s10725-012-9740-0
Farahani F, Yari R, Masoud S (2011) Somaclonal variation in Dezful cultivar of olive (Oleaeuropaea subsp. europaea). Genet Conserv 40:216–233
Wang QM, Wang YZ, Sun LL, Gao FZ, Sun W, He J, Gao X, Wang L (2012) Direct and indirect organogenesis of Clivia miniata and assessment of DNA methylation changes in various regenerated plantlets. Plant Cell Rep 31:1283–1296. https://doi.org/10.1007/s00299-012-1248-6
Hao YJ, Deng XX (2003) Genetically stable regeneration of apple plants from slow growth. Plant Cell Tissue Organ Cult 72:253–260
Li W, Liu H, Cheng ZJ, Su YH, Han HN, Zhang Y, Zhang XS (2011) DNA Methylation and histone modifications regulate de novo shoot regeneration in Arabidopsis by modulating WUSCHEL expression and auxin signaling. PLoS Genet 7:e1002243. https://doi.org/10.1371/journal.pgen.1002243
Nic-Can GI, Lopez-Torres A, Barredo-Pool F, Wrobel K, Loyola-Vargas VM, Rojas-Herrera R, De-la-Pena C (2013) New insights into somatic embryogenesis: leafy cotyledon1, baby boom1 and WUSCHEL-related homeobox4 are epigenetically regulated in Coffea canephora. PLoS One 8:e72160. https://doi.org/10.1371/journal.pone.0072160
Kwiatkowska A, Zebrowski J, Oklejewicz B, Czarnik J, Halibart-Puzio J, Wnuk M (2014) The age-dependent epigenetic and physiological changes in an Arabidopsis T87 cell suspension culture during long-term cultivation. Biochem Biophys Res Commun 447:285–291. https://doi.org/10.1016/j.bbrc.2014.03.141
Fraga HPF, Vieira LN, Caprestano CA, Steinmacher DA, Micke GA, Spudeit DA, Pescador R, Guerra MP (2012) 5-Azacytidine combined with 2,4-D improves somatic embryogenesis of Acca sellowiana (O. Berg) Burret by means of changes in global DNA methylation levels. Plant Cell Rep 31:2165–2176. https://doi.org/10.1007/s00299-012-1327-8
Ji L, Mathioni SM, Johnson S, Tucker D, Bewick AJ, Do Kim K, Daron J, Slotkin RK, Jackson SA, Parrott WA, Meyers BC, Schmitz RJ (2019) Genome-wide reinforcement of dna methylation occurs during somatic embryogenesis in soybean. Plant Cell 31:2315. https://doi.org/10.1105/tpc.19.00255
Santos D, Fevereiro P (2002) Loss of DNA methylation affects somatic embryogenesis in Medicago truncatula. Plant Cell Tissue Organ Cult 70:155–161
Reyna-Lopez GE, Simpson J, Ruiz-Herrera J (1997) Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol Gen Genet 253:703–710
Peraza-Echeverria S, Herrera-Valencia VA, Kay A-J (2001) Detection of DNA methylation changes in micropropagated banana plants using methylation-sensitive amplification polymorphism (MSAP). Plant Sci 161:359–367
Wang X, Wu R, Lin X, Bai Y, Song C, Yu X, Xu C, Zhao N, Dong Y, Liu B (2013) Tissue culture-induced genetic and epigenetic alterations in rice pure-lines, F1 hybrids and polyploids. BMC Plant Biol 13:77. https://doi.org/10.1186/1471-2229-13-77
Li X, Xu M, Korban SS (2002) DNA methylation profiles differ between field- and in vitro-grown leaves of apple. J Plant Physiol 159:1229–1234
Portis E, Acquadro A, Comino C, Lanteri S (2004) Analysis of DNA methylation during germination of pepper (Capsicum annuum L.) seeds using methylation-sensitive amplification polymorphism (MSAP). Plant Sci 166:169–178
Gao X, Yang D, Cao D, Ao M, Sui X, Wang Q, Kimatu J, Wang L (2010) In vitro micropropagation of Freesia hybrida and the assessment of genetic and epigenetic stability in regenerated plantlets. J Plant Growth Regul 29:257–267. https://doi.org/10.1007/s00344-009-9133-4
Goyali JC, Igamberdiev AU, Debnath SC (2018) DNA methylation in lowbush blueberry (Vaccinium angustifolium Ait.) propagated by softwood cutting and tissue culture. Can J Plant Sci 98:1035–1044
Berdasco M, Fraga MF, Esteller M (2009) Quantification of global DNA methylation by capillary electrophoresis and mass spectrometry. In: DNA methylation. Springer, pp 23–34
Lin W, Xiao X, Zhang H, Li Y, Liu S, Sun W, Zhang X, Wu Q (2019) Whole-genome bisulfite sequencing reveals a role for dna methylation in variants from callus culture of pineapple (Ananas comosus L.). Genes 10:877. https://doi.org/10.3390/genes10110877
Chwialkowska K, Korotko U, Kosinska J, Szarejko I, Kwasniewski M (2017) Methylation sensitive amplification polymorphism sequencing (MSAP-Seq)—A method for high-throughput analysis of differentially methylated CCGG sites in plants with large genomes. Front Plant Sci 8:2056
Gulyás A, Dobránszki J, Kiss E, Da Silva JAT, Posta K, Hidvégi N (2019) Changes in DNA methylation pattern of apple long-term in vitro shoot culture and acclimatized plants. J Plant Physiol 239:18–27. https://doi.org/10.1016/j.jplph.2019.05.007
Rakocevic A, Mondy S, Tirichine L, Cosson V, Brocard L, Iantcheva A, Cayrel A, Devier B, Abu El-Heba GA, Ratet P (2009) MERE1 a low-copy-number copia-type retroelement in Medicago truncatula active during tissue culture. Plant Physiol 151:1250. https://doi.org/10.1104/pp.109.138024
Rosati P, Mezzetti B, Ancherani M, Foscolo S, Predieri S, Fasolo F (1990) In vitro selection of apple rootstock somaclones with Phytophthora cactorum culture filtrate. International Society for Horticultural Science (ISHS), Leuven, Belgium, pp 409–416. https://doi.org/10.17660/ActaHortic.1990.280.66
Gillis K, Gielis J, Peeters H, Dhooghe E, Oprins J (2007) Somatic embryogenesis from mature Bambusa balcooa Roxburgh as basis for mass production of elite forestry bamboos. Plant Cell Tissue Organ Cult 91:115–123
Bednarek PT, Orłowska R, Koebner RMD, Zimny J (2007) Quantification of the tissue-culture induced variation in barley (Hordeum vulgare L.). BMC Plant Biol 7:10
Guo W, Wu R, Zhang Y, Liu X, Wang H, Gong L, Zhang Z, Liu B (2007) Tissue culture-induced locus-specific alteration in DNA methylation and its correlation with genetic variation in Codonopsis lanceolata Benth. et Hook. f. Plant Cell Rep 26:1297–1307. https://doi.org/10.1007/s00299-007-0320-0
Schellenbaum P, Mohler V, Wenzel G, Walter B (2008) Variation in DNA methylation patterns of grapevine somaclones (Vitis vinifera L.). BMC Plant Biol 8:78–78. https://doi.org/10.1186/1471-2229-8-78
Baránek M, Křižan B, Ondrušíková E, Pidra M (2010) DNA-methylation changes in grapevine somaclones following in vitro culture and thermotherapy. Plant Cell Tissue Organ Cult 101:11–22
Sharma SK, Bryan GJ, Winfield MO, Millam S (2007) Stability of potato (Solanum tuberosum L.) plants regenerated via somatic embryos, axillary bud proliferated shoots, microtubers and true potato seeds: a comparative phenotypic, cytogenetic and molecular assessment. Planta 226:1449–1458
Momose M, Abe Y, Ozeki Y (2010) Miniature inverted-repeat transposable elements of stowaway are active in potato. Genetics 186:59. https://doi.org/10.1534/genetics.110.117606
Nassar AMK, Kubow S, Leclerc YN, Donnelly DJ (2014) Somatic mining for phytonutrient improvement of ‘russet burbank’ potato. Am J Potato Res 91:89–100. https://doi.org/10.1007/s12230-013-9334-z
Sato M, Hosokawa M, Doi M (2011) Somaclonal variation is induced de novo via the tissue culture process: a study quantifying mutated cells in Saintpaulia. PLoS One 6:e23541. https://doi.org/10.1371/journal.pone.0023541
Bender J, Fink GR (1995) Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 83:725–734. https://doi.org/10.1016/0092-8674(95)90185-X
Jacobsen SE, Meyerowitz EM (1997) Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277:1100–1103
Saze H, Kakutani T (2007) Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J 26:3641–3652
Schmitz RJ, Schultz MD, Lewsey MG, O’Malley RC, Urich MA, Libiger O, Schork NJ, Ecker JR (2011) Transgenerational epigenetic instability is a source of novel methylation variants. Science 334:369–373
Durand S, Bouché N, Perez Strand E, Loudet O, Camilleri C (2012) Rapid establishment of genetic incompatibility through natural epigenetic variation. Curr Biol 22:326–331. https://doi.org/10.1016/j.cub.2011.12.054
Peschke VM, Phillips RL, Gengenbach BG (1987) Discovery of transposable element activity among progeny of tissue culture—derived maize plants. Science 238:804–807. https://doi.org/10.1126/science.238.4828.804
Peschke VM, Phillips RL (1991) Activation of the maize transposable element Suppressor-mutator (Spm) in tissue culture. Theor Appl Genet 81:90–97. https://doi.org/10.1007/BF00226117
Miura K, Agetsuma M, Kitano H, Yoshimura A, Matsuoka M, Jacobsen SE, Ashikari M (2009) A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc Natl Acad Sci 106:11218–11223
Zhang L, Cheng Z, Qin R, Qiu Y, Wang J-L, Cui X, Gu L, Zhang X, Guo X, Wang D (2012) Identification and characterization of an epi-allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 24:4407–4421
Telias A, Lin-Wang K, Stevenson DE, Cooney JM, Hellens RP, Allan AC, Hoover EE, Bradeen JM (2011) Apple skin patterning is associated with differential expression of MYB10. BMC Plant Biol 11:93. https://doi.org/10.1186/1471-2229-11-93
Mirouze M, Reinders J, Bucher E, Nishimura T, Schneeberger K, Ossowski S, Cao J, Weigel D, Paszkowski J, Mathieu O (2009) Selective epigenetic control of retrotransposition in Arabidopsis. Nature 461:427. https://doi.org/10.1038/nature08328
Kukreja AK, Dhawan OP, Mathur AK, Ahuja PS, Mandal S (1991) Screening and evaluation of agronomically useful somaclonal variations in Japanese mint (Mentha arvensis L.). Euphytica 53:183–191
Yadav VK, McHta SL (1995) Lathyrus sativus: a future pulse crop free of neurotoxin. Curr Sci 68:288–292
Rodriguez NV, Kowalski B, Rodriguez LG, Caraballoso IB, Suarez MA, Perez PO, Quintana CR, Gonzalez N, Ramos RQ (2007) In vitro and ex vitro selection of potato plantlets for resistance to early blight. J Phytopathol 155:582–586
Yadav PV, Suprasanna P, Gopalrao KU, Anant BV (2006) Molecular profiling using RAPD technique of salt and drought tolerant regenerants of sugarcane. Sugar Tech 8:63–68. https://doi.org/10.1007/BF02943744
Singh G, Sandhu SK, Meeta M, Singh K, Gill R, Gosal SS (2008) In vitro induction and characterization of somaclonal variation for red rot and other agronomic traits in sugarcane. Euphytica 160:35–47
Roy B, Mandal AB (2005) Towards development of Al-toxicity tolerant lines in indica rice by exploiting somaclonal variation. Euphytica 145:221–227
Jiang C, Mithani A, Gan X, Belfield EJ, Klingler JP, Zhu J-K, Ragoussis J, Mott R, Harberd NP (2011) Regenerant Arabidopsis lineages display a distinct genome-wide spectrum of mutations conferring variant phenotypes. Curr Biol 21:1385–1390
Boxus P, Jemmali A, Terzi JM, Arezki O (2000) Drift in genetic stability in micropropagation: the case of strawberry. International Society for Horticultural Science (ISHS), Leuven, Belgium, pp 155–162. https://doi.org/10.17660/ActaHortic.2000.530.16
Goyali JC, Igamberdiev AU, Debnath SC (2015) Propagation methods affect fruit morphology and antioxidant properties but maintain clonal fidelity in lowbush blueberry. HortScience 50:888–896
Thieme M, Lanciano S, Balzergue S, Daccord N, Mirouze M, Bucher E (2017) Inhibition of RNA polymerase II allows controlled mobilisation of retrotransposons for plant breeding. Genome Biol 18:134. https://doi.org/10.1186/s13059-017-1265-4
Deverno LL (1995) Evaluation of somaclonal variation during somatic embryognesis. Somatic embryogenesis in woody plants. Plant Cell Rep 10:425–430
Shchukin A, Ben-Bassat D, Israeli Y (1996) Plant regeneration via somatic embryogenesis in Grand Nain banana and its effect on somaclonal variation. In: III International Symposium on In vitro and Horticultural Breeding, pp 317–318
Shchukin A, Ben-Bassat D, Israeli Y (1998) Somaclonal variation and horticultural performance of ‘Grand Naine’ bananas multiplied via somatic embryogenesis or shoot-tip culture. In: Plant biotechnology and in vitro biology in the 21st century. International Association for Plant Tissue Culture Abstracts, Jerusalem, pp 14–19
Acknowledgements
Authors’ research in this area is supported by the St. John’s Research and Development Centre, Agriculture and Agri-Food Canada, St. John’s, Newfoundland and Labrador, Canada.
Funding
Agriculture and Agri-Food Canada St. John’s Research and Development Centre contribution No. 240.
Author information
Authors and Affiliations
Contributions
A.G. outlined, wrote the manuscript, prepared the tables and the figures. A.U.I. revised and reviewed the manuscript. S.C.D. conceptualized, helped in writing, revised, and reviewed the manuscript. All authors have approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest/competing interests.
Consent to participate
This research did not involve any human participants and/or animals.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ghosh, A., Igamberdiev, A.U. & Debnath, S.C. Tissue culture-induced DNA methylation in crop plants: a review. Mol Biol Rep 48, 823–841 (2021). https://doi.org/10.1007/s11033-020-06062-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-06062-6