
Abstract
Genetic diversity and population structure in Indian featherback fish, Chitala chitala (Hamilton, 1822) was investigated by combined analyses of two full mitochondrial genes, ATPase 6/8 and Cytochrome b. A total of 403 individuals, collected from 14 rivers yielded 61 haplotypes. Hierarchical partitioning analysis identified 19.01% variance ‘among’ and 80.99% variance ‘within groups and populations’. The mean coefficient of genetic differentiation (FST) was observed to be significant 0.26 (p < 0.05). Mantel tests rejected the hypothesis that genetic and geographic distances were correlated. The patterns of genetic differentiation, AMOVA and principal coordinate analyses indicated that natural populations were sub-structured and comprised of four genetic stocks of C. chitala in Indian rivers. The results also supported the higher resolution potential of concatenated gene sequences. The knowledge of genetic variation and divergence, from this study, can be utilized for its scientific conservation and management in the wild.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Order Osteoglossiformes represents an ancestral teleost lineage, whose fossil records have been retrieved from the deposits, belonging to late Jurassic or early Cretaceous period [1]. The family Notopteridae comprises of ten species' belonging to four genera, distributed in the freshwaters of Africa and South Asia. The Indian featherback fish, Chitala chitala (Hamilton, 1822) [2], is widely distributed in Asian countries including India, Bangladesh, Sri Lanka, Pakistan, Nepal [3]. According to Taverne [4] the species exhibited wider distribution during prehistoric period, than today.
In India, C. chitala constitutes a substantial component of the inland fisheries. It has high demand as ornamental trade, food, sport and is considered a potential candidate species for aquaculture [5]. The increased sedimentation, environmental degradation, changes in river courses and overexploitation [6] have caused decline in its capture fisheries, due to which the species has been categorized as 'Near Threatened' by the International Union for Conservation of Nature [7]. Several studies have been conducted in C. chitala, which include development of captive breeding protocols, larval rearing and age-growth relationship [8]. In an investigation, morphometric and meristics tools were employed to identify stocks of C. chitala in Indian rivers [9].
However, the efficient management requires the knowledge of natural genetic diversity and population structure [10]. Molecular data can provide critical inputs for determining genetic divergence and connectivity [11]. Mitochondrial DNA markers are proven tools to effectively characterize populations and capture demographic signatures due to their unique characteristics, such as high copy number, faster mutation rate and low effective population size [12]. Mitochondrial genes, particularly ATPase 6/8 and Cytochrome b (Cytb) have been widely used to reveal intraspecific genetic variability and differentiation in fishes belonging to several taxonomic orders, such as Osteoglossiformes, Perciformes and Cypriniformes, etc. In C. chitala, previous studies had indicated the genetic variability, inferred from two different mitochondrial regions, partial Cytb and D-loop [5]. Molecular studies, based on ATPase 6/8 genes, revealed genetic divergence in other notopterid Notopterus notopterus [13]. Till now, most genetic studies have used single and partial mitochondrial genes for population level analyses. However, recently, there has been a growing inclination towards using longer gene sequences that can provide sufficient resolution and robust computational support.
In the present study, we analyzed a concatenated data set of two full mitochondrial genes (ATPase 6/8 and Cytb) of C. chitala to investigate patterns of genetic variability, differentiation and population structure. The demographic history and genetic constraints were also assessed. The baseline data, generated from this study, will be a useful resource for the conservation and management of Chitala fisheries.
Materials and methods
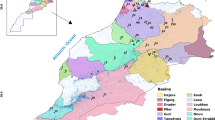
The tissue samples of C. chitala individuals (n = 403) were collected from 14 rivers belonging to five river basins (Indus, Ganges, Brahmaputra, Mahanadi, and Narmada) (Fig. 1). Sampling strategy covered a wide natural range of distribution. The details of collection, GPS coordinates and year of sampling are provided in Table 1.

Sampling locations of C. chitala from Indian rivers covering wide geographical distribution range. (1.Satluj River (Harike Pattan, Punjab); 2.Gandak River (Valmiki Nagar, Bihar); 3.Ghaghra River (Mehmoodabad, UP); 4.Chauka River (UP); 5.Sharda River (Sharda Barrage, Lakhimpur Kheri, UP) 6.Gomti River (Lucknow, UP); 7.Tons River (Rewa, MP); 8.Tons River (Chakghat, UP); 9.Son River (Bansagar, Beohari, Madhya Pradesh); 10.Ken River (Rangua, MP); 11.Hooghly River (Nabadeep, WB); 12.Hooghly River (Beldanga, WB); 13.Hooghly River (Satui, WB); 14.HooghlyRiver (Behrampur, WB); 15.Padma River (Farakka, WB); 16.Padma River (Manikchak, WB); 17.Brahmaputra River (Kolongpar, Assam); 18.Brahmaputra River (Uzan Bazaar, Assam); 19.Brahmaputra River (Guwahati, Assam); 20.Mahanadi River (Hirakud Dam, Odisha); 21.Mahanadi River (Sonepur, Odisha); 22.Mahanadi River (Jobra Barrage, Odisha); 23.Narmada River (Hoshangabad, MP)
Genomic DNA extraction, amplification, and sequencing
The total genomic DNA was extracted from tissue/fin-clip/blood (ethanol preserved) using the modified phenol–chloroform method [14]. DNA quality was checked on 0.8% agarose gel stained with Nucleic Acid Safe Dye (G Biosciences) and visualized in gel documentation system (UVP Imaging System, Cambridge, UK). The purity and concentration was examined on DeNovix® DS-11 spectrophotometer. The DNA concentration was adjusted to 50 ng/µl and stored at 40C.
Two complete mitochondrial genes, Cytb (1139 bp) and ATPase 6/8 (842 bp) were amplified using primer pairs L14724: 5′-CGA GAT CTG AAA AAC CAT CGT TG-3′; H15915: 5′-AAC TGC AGT CAT CTC CGG TTT ACA AGA A-3′ [15] and ATP8.2L8331: 5′-AAA GCR TYR GCC TTT TAA GC-3′; CO3.2H9236: 5′- GTT AGT GGT CAK GGG CTT GGR TC-3′ [16], respectively. Gene amplification was performed in a final reaction volume of 25 µl, which contained 1X reaction buffer (10 mM Tris, 50 mM KCl, 0.01% gelatin, pH 9.0), 200 µM of each dNTP, 1.5 mM MgCl2, 5 pmol of each primer, 3U Taq polymerase (Genei, India) and approximately 50 ng of template DNA. PCR conditions included: denaturation at 940C for 5 min, 30 cycles of 940C for 30 s, 550C for 1 min and 720C for 1 min 30 s; final extension at 720C for 10 min. The amplicons were purified and sequenced bidirectionally on ABI automated sequencer (Applied Biosystems).
Genetic analysis
The sequences were edited and aligned using BioEdit sequence alignment editor version 7.2 [17]. We conducted a partition homogeneity test in PAUP* 4.0 [18] with heuristic search and 100 replicates (optimality criterion = parsimony) on all 1981 nucleotide characters. This was done to examine whether these two genes (ATPase 6/8 and Cytb) could be combined in a unique data matrix. The results showed no incongruence between the two gene data sets, and therefore could be analyzed jointly. MEGA X [19] was used to estimate gene characteristics such as, nucleotide composition, conserved/variable sites and transition/transversion ratio. Number of haplotypes, haplotype diversity (Hd, probability that two randomly chosen haplotypes are different in a sample) and nucleotide diversity (π, the mean no. of nucleotide differences between two randomly chosen DNA sequences) were estimated using DnaSP v 5.0 [20]. Median-joining network [21] was drawn using Network v5.0 software (Fluxus Technology, Ltd.) to determine the linkages among haplotypes.
Historical patterns of population dynamics were estimated through mismatch distributions and neutrality tests (Tajima's D and Fu's Fs) implemented in Arlequin 3.5. This analysis demonstrates whether the population had undergone expansion in the past. The parameters of demographic expansion, θ0 and θ1, and τ (mode of mismatch distribution) were calculated using the τ = 2µt equation. The raggedness index (r) and the sum of squared deviations (SSD) for each population were calculated according to Schneider and Excoffier [22]. These parameters quantify mismatch distribution pattern, where non-significant result indicates population expansion. Gene flow (Nm), was estimated, for each population, using Arlequin 3.5 [23] with 1000 replicates. It is a product of the average size (N) and proportion of immigrants (m) in each population. The levels of gene flow indicate the extent to which each local population may act as an independent evolutionary unit.
The hierarchical partitioning of genetic variation was assessed using analysis of molecular variance (AMOVA). The samples were grouped according to their geographic relevance as well as river basins (10,000 permutations) for testing levels of significance. Mantel test [24] was performed in XL-STAT ver. 2010 (Addinsoft, France), with 10,000 permutations, to determine the extent of isolation by distance (IBD). This analysis was done using two matrices, (pair-wise FST) and geographical distances (km) between populations.
Population structure was estimated using two clustering approaches, Principal Coordinate Analysis (PCoA), implemented in GenAlex 6 [25] and Bayesian assignment method through BAPS version 6.0 [26]. PCoA plot was prepared based on the covariance of the genetic distance matrix. In BAPS, samples were grouped according to river basins to estimate the pattern of clustering in concatenated mitochondrial genes. We employed a mixture model and tested K from 1 to 14 (maximum no. of clusters) with 1000 replicates per K. The number of reference individuals was set to 200.
Temporal stability of genetic variation
Demonstrating temporal stability is a powerful way of confirming the reliability of observed spatial genetic patterns. In this investigation, we used historical samples of C. chitala from three rivers, Son, Hooghly and Padma. Because of substantial intervening time between tissue sampling from these rivers, temporal stability of C. chitala populations was studied, so that these samples could be analyzed together with contemporary data. The samples from Son were collected during 2011–2012 (n = 31) and 2015–2017 (n = 35), while Padma (n = 29) samples during 2001–2003, 2006 and 2016 (n = 11), with almost a gap of two to five generations. The collections of Hooghly samples had two sampling points, 2001–2002 (21) and 2016 (12). The pair-wise FST values were calculated for each population, collected at different time intervals. The results showed low and insignificant genetic differentiation between different sampling points (for each river), therefore the samples were merged river-wise, for further data analysis. Hereafter, the samples from Son1 & 2, Hooghly1 & 2 and Padma1 & 2 were referred to as Son, Hooghly and Padma, respectively (Suppl Table 1).
Results
Genetic variability
Sequence analyses of ATPase 6/8 (842 bp) and Cytb (1139 bp) yielded 23 and 30 haplotypes, respectively. ATPase 8 spanned from 1 to 168 bp, whereas ATPase 6 from159—842 bp, with overlapping region of 10 bp. ATPase 6/8 genes were characterized by 22 variable sites, 13 parsimony informative and 9 singleton sites, whereas Cytb had 26 variable sites, 17 parsimony informative and 9 singletons. The partition homogeneity test indicated no incongruence between the two genes and therefore, we concatenated the sequences (5′ATPase 8-ATPase 6-Cytb-3′) as a data set for further analysis. A total of 61 haplotypes were identified for concatenated mitochondrial DNA sequences. The results of individual genes, as well as the concatenated sequence (ATPase 6/8 + Cytb), were concordant. The average frequency of nucleotides were A: 30.7%, T: 27.1%, G: 12.6%, C: 29.6%, with a transition to transversion ratio (Ts:Tv) of 6.479. The nucleotide composition of both individual genes as well as the concatenated displayed bias towards A + T content. The haplotype diversity Hd ranged from 0.000 (Mahanadi) to 0.933 (Chauka). Mean nucleotide diversity (π) of 0.002 was observed, with a range of 0.000 (Mahanadi) to 0.002 (Chauka) (Table 2).
Phylogenetic relationships among haplotypes
The phylogeographic inferences were performed using the median-joining network of concatenated data set (Fig. 2). Haplotypes H3, H10 and H12 were the most common, out of total 61 haplotypes. Haplotype H3 was shared between all populations except Gomti and Tons, Haplotype H10 (in populations, except Chauka, Ken, Mahanadi, and Narmada) while H12 was represented by 11 populations except for Satluj, Sharda and Mahanadi. Population-specific haplotypes were found in Satluj (5), Ghaghra (6), Sharda (1), Son (11), Hooghly (3), Padma (3), Brahmaputra (9) and Narmada (4). Samples from Son exhibited the maximum number of haplotypes (19), followed by Brahmaputra (18) and Ghaghra (15). River Satluj was represented by 10 haplotypes, Narmada (6), whereas Mahanadi was represented by only one haplotype. A total of 42 haplotypes were population-specific, while 19 were shared.

Median joining network of 61 concatenated haplotypes of C. chitala (Network V 5.0), with star-like topologies, indicating population expansion. (The size of nodes are proportional to the number of individuals. Perpendicular tick marks on the lines joining haplotypes represent the number of nucleotide substitutions)
Demographic history and neutrality tests
Cytb gene did not show any significant negative values for Tajima and Fu's Fs, whereas ATPase 6/8 showed significant negative values for Fu's Fs and Tajima's D in Padma and Brahmaputra. Significant negative values of Fu's Fs were observed in two rivers, Sharda and Son. Pairwise mismatch distribution, Tajima’s D and Fu's Fs tests were performed for combined mitochondrial genes for each population. The parameters used to estimate the pattern of population dynamics are shown in Table 3. The mean value of Tajima's D and Fu's Fs were 0.103 and -2.068, respectively. Based on combined (1.98 kb) gene analysis, the genetic constraints for C. chitala were evident in rivers Satluj, Narmada and five rivers (Ghaghra, Son, Hooghly, Padma, and Brahmaputra) of Ganga–Brahmaputra basin. The mismatch distribution plot, for all populations together, displayed a multimodal pattern, revealing demographic equilibrium or a stable population [27] (Fig. 3). Based on the sum of squared deviations, all populations were non-significant except Gomti and Brahmaputra. The raggedness indices were also calculated under the demographic expansion model for each population, wherein all populations were non-significant (Table 3). The rivers belonging to Ganga basin, when analyzed separately, presented negative significant Fu's Fs for all rivers, except Sharda.

Mismatch graph (DnaSP v5.0) illustrating multimodal pattern for overall locations based on pairwise sequence differences. The overall pattern displays demographic equilibrium or stable population (The solid line and dotted line represent the curves for expected and observed values)
Genetic differentiation among populations
The overall coefficient of genetic differentiation (FST) was 0.263 for combined data set. The pair-wise FST ranged from 0.000 (Sharda-Son) to 0.866 (Satluj-Mahanadi) (Table 4). The mean FST of Ganga basin reduced remarkably (0.074) when analyzed separately. The pair-wise population genetic differentiation in concatenated genes was similar to both the individual genes, when analyzed separately (Suppl Table 2).
The Principal Coordinate Analysis (PCoA), based on the pair-wise genetic distances between populations, resulted in existence of three distinct clusters, and a mixed cluster of rivers belonging to Ganga basin (Fig. 4). The eigenvalue for PC1 was found to be significant (1.116) and contributed 74.62%, while PC2 was found to be non-significant, contributing 10.31%. The cumulative total variation by PC1 and PC2 was 84.94%. Basin-wise, principal component scores, (PC1, PC2) for Satluj (0.691, 0.134), Mahanadi (−0.484, 0.301) and Narmada (− 0.403, − 0.150) suggested that they are genetically distinct, while these scores were comparable for rivers of Ganga basin (Gandak, Ghaghra, Chauka, Sharda, Gomti, Tons, Son, Ken, Hooghly and Padma) and Brahmaputra. These rivers formed a mixed cluster, distant from three other basins (Suppl Table 3).

Principal coordinate analysis (PCoA) (GenAlex 6) using pairwise distances between populations of C. chitala. The pattern showed existence of three distinct clusters of Satluj, Mahanadi and Narmada and a mixed cluster of rivers belonging to Ganga basin and Brahmaputra
Bayesian clustering partitions the individuals into genetically distinct units without prior group assignment. In this study, BAPS 6.0 was used to ascertain the number as well as the putative mixing within the basins. Based on log likelihood estimates, K = 4 was found to be the best partition (Fig. 5). Cluster I, comprised haplotypes from the Ganges; Cluster II, haplotypes from Ganges and Narmada; Cluster III, haplotypes from the Ganges, Mahanadi, and Satluj; Cluster IV, haplotypes from Ganges and Satluj. The representation of Ganges samples in all the clusters was observed. The lack of river basin wise clustering was obvious from the results. However, the Mahanadi population was genetically differentiated and therefore the most distinct (Fig. 6).

Plot of K (x axis) against log likelihood value (y axis) for BAPS in C. chitala

Histogram of the assignment test using BAPS6.0
A three way AMOVA was performed, considering four groups namely, Satluj, Ganga, Mahanadi and Narmada. The hierarchical analysis of molecular variance demonstrated that 19.01% of the total genetic variance was due to differences among groups (FCT = 0.190, p < 0.05), 73.71% was due to differences within populations (FST = 0.263, p < 0.05) and 7.27% was attributed to variation among populations within groups (FSC = 0.090, p < 0.05) (Table 5).
Results of Isolation by Distance (IBD) were not statistically significant when all populations were considered together, showing thereby that there existed no correlation between genetic and geographical distance matrices (R2 = 0.008, r = 0.179; p = 0.121).
Population genetic connectivity
Gene flow estimates (number of migrants, Nm) showed moderate to high values (Table 4). According to Nei (1987), Nm values (> 1) contributed positively against genetic differentiation among populations. In present study, higher gene flow was observed among the rivers of Ganga basin (Gandak, Ghaghra, Chauka, Sharda, Gomti, Tons, Son, Ken, Hooghly, Padma). The Brahmaputra showed moderate to higher gene flow for Gangetic rivers. It was observed that rivers Satluj, Mahanadi and Narmada presented low to moderate levels of gene flow, however with pronounced genetic differentiation.
Discussion
Information of genetic variability and population structure can be utilized in identifying distinct management units which is needed for effective conservation of fisheries resources. This study provides useful inputs and delineates stock boundaries of C. chitala. A combined dataset (ATPase 6/8 + Cytb) revealed high haplotype diversity and low nucleotide diversity across all riverine populations, except for river Mahanadi. The haplotype and nucleotide (%) diversity levels are considered high if the value is greater than 0.5 [28]. This pattern, particularly (high Hd and low π) might be attributed to demographic expansion that occurred after a reduction in effective population size, retaining new mutations [29]. Similar trend in molecular diversities are concordant to other notopterids, Chitala lopis [30], Chitala chitala [5] and N. notopterus [13, 31]. Overall, the pattern of haplotype sharing among the river basins indicated genetic structuring in C. chitala. In Mahanadi population, a single haplotype was fixed in all individuals. This might indicate a possibility of small effective population size and inbreeding. Similar results were observed in another notopterid, N. notopterus, where the lack of genetic connectivity in Mahanadi and Indo-Gangetic Rivers was noticed [13]. It is likely that the fragmentation of Mahanadi populations during the early Pleistocene [32] might be responsible for present loss of haplotype sharing. In our earlier study, the partial sequence of Cytb (307 bp) revealed only one haplotype in Satluj samples [5]. However, in the present investigation, the concatenated gene set yielded five population-specific haplotypes, of which two haplotypes were found to be ancestral, giving rise to other related haplotypes. This supports the potential of longer gene sequences for genetic analysis [33]. However, higher genetic variability in river Satluj might also be due to the comparatively larger sample size, used in this study. Small sample size might affect the frequency and distribution of variation as well as amount of uncertainty in the estimates and resolution. In present study, though the sample sizes from four rivers Gandak, Chauka, Sharda and Tons were small, the patterns of haplotype sharing, frequency and clustering were consistent with other rivers of the basin.
At population level, genealogical relationships among haplotypes are preferred to phylogenetic methods, because the network accommodates both, existent ancestral sequence as well as alternative connections [21]. In present study, haplotype network presented three haplogroups displaying star-like topologies, indicating genetic bottleneck and population expansion [34]. These results are important in view of adaptive potential and probability of population persistence [35]. The pattern of molecular diversity levels show signatures of population expansion in Satluj, Ghaghra, Son, Hooghly, Padma, Brahmaputra and Narmada.
Neutrality tests are considered sensitive measures of demographic events, where significant negative values indicate signatures of population expansion [36]. The results of Fu’s Fs showed concordance with diversity patterns and explained the events of population expansion in these rivers. The population expansion was also confirmed by non significant raggedness index. Tajima’s D values were negative all the seven populations, indicating the excess of rare nucleotide site variants. Moreover, on analyzing Gangetic rivers separately, significantly negative Fu's Fs were observed in all populations except Sharda, which may be due to small sample size, analyzed in present study. It is likely that founder effect may be responsible for genetic constraints in these populations.
The FST indicates the genetic differentiation between populations. In general, a higher value (> 0.25) is correlated with a high genetic differentiation between populations [37], which is consistent with our study (FST = 0.26) and with other freshwater species like N. notopterus [13]. In C. chitala individuals from river Satluj, Mahanadi, and Narmada showed significant differentiation from other rivers. These three basins presented significant pair-wise FST, suggesting limited gene flow. The degree of gene flow presented high level of genetic connectivity among rivers of Ganga basin; and these populations maintained genetic homogeneity, probably due to ancestral mixing. However, in another congener Chitala lopis, the low level genetic differentiation and corresponding higher gene flow were observed [30]. Sodsuk and Sodsuk (2000) assayed 28 allozyme loci for C. lopis, but no locus exhibited polymorphism [38].
Barby et al. [39] illustrated the biogeography of notopteridae, wherein the arrival of Chitala species in Asian continent had been estimated to be about 55 Mya. This corresponded Eocene epoch, the estimated entry period of several other spacies including carps and catfishes [40]. The contemporary geological events were considered responsible for formation of Indus and Ganga, with its Himalayan tributaries. Satluj samples showed presence of distinct haplotypes, however limited connectivity might have allowed sharing of haplotypes with Gangetic rivers. Our analysis supported this hypothesis and Satluj populations were distinct from others. Chauhan et al. [41] have described the connectivity between Satluj and rivers of Ganga system via erstwhile river Ghaggar. Not enough variance and differentiation were observed for component rivers of Ganga system, in current study. All the rivers (Gandak, Ghaghra, Chauka, Sharda, Gomti, Tons, Son, Ken, Hooghly and Padma) clustered together, which is indicative of common gene pool in Gangetic system. The Ganges and the Brahmaputra river system flow through very different regions for most of their lengths and join upstream at the Bay of Bengal. This may be the reason for the genetic homogeneity between tributaries of the Ganges and Brahmaputra. Similar results of genetic proximity between Ganga and Brahmaputra have been reported in C. chitala [5] and another freshwater species Sperata seenghala [42].
River Narmada, is among the longest rivers in India, which originates from Amarkantak (M.P., India). The occurrence of physical barriers seems to contribute towards genetic fragmentation and dispersal capabilities of C. chitala. Earlier, Khedkar et al. [43] described the genetic impoverishment of two species (Catla catla and Mastacembelus armatus) owing to asymmetrical dispersal in river Narmada. Moreover, a study conducted on alluvial plain sedimentation for river Narmada envisaged that it had been a much bigger channel during late Pleistocene with higher discharge than present [44]. This might also contribute to genetic fragmentation of resources in river Narmada.
The Mahanadi river flows eastwards and drains into Bay of Bengal. Historically, the tributaries of Narmada river, i.e. Mahanadi and Godavari used to flow westward, but due to rise in plateau and tilt of peninsula during the middle Pleistocene, they changed their course [45] which resulted in rivers, as seen today, flowing as independent river systems, southward through Eastern Ghats [32]. Such phylogeographic events affect long term histories in populations like Mahanadi. This became explicit, when Mahanadi samples were excluded from analysis. Out of a total of 26.28% genetic differentiation, over 6% was caused due to Mahanadi samples. It is likely that the population might be experiencing selection pressure. Moreover, this warrants for larger sample sizes, analyzed with multiple marker systems.
In a study, Martinez et al. [46] correlated the impact of habitat type with the diversity levels in fishes. Lower diversity levels in River Gomti might be attributed to multiple factors related to habitat. This river has been reported to have poor vegetation cover in the banks, siltation and low water velocity. Present findings are also supported by a recent investigation, wherein the negative allometric growth with poor condition factor of C. chitala in river Gomti, was reported [47]. Moreover, Verma and Dalela [48] pointed out the decline in featherbacks and carps due to the rise in pollution in river Gomti caused by domestic and industrial wastes. Similar decline in genetic diversity due to habitat modification has been observed in C. chitala in Mekong river [49]. River Brahmaputra is characterised by large and variable flow, enhanced rate of sediment discharge and distinct river morphology. Additionally, reduction in efficiency of energy utilization from primary energy to fish, was reported [50]. The primary consumers (both herbivore and detritivore) contributed only 17.1% of total energy. Moreover, anthropogenic activities such as dam construction might have caused alterations in fish breeding grounds resulting in the loss of genetic diversity.
The analysis of molecular variance also confirmed the sub-structuring in C. chitala poupulations. Similar patterns of genetic differentiation and variance partitioning had been obtained using Cytb and control region [5]. Bayesian clustering produced the highest log likelihood values for four clusters, however failed to resolve river basin wise. The Mantel tests in C. chitala populations rejected the hypothesis that genetic differentiation and geographic distance were mutually correlated. This lack of interdependence confirmed that the genetic differentiation among populations was not determined by the effect of distance between populations. The resulting genetic divergence in C. chitala from different river basins appeared to be an outcome of restricted gene flow between populations as well as phylogeographic events. These findings highlight that both geographic and environmental factors contributed towards mitochondrial variability patterns. Overall, the patterns of pairwise genetic differentiation, gene flow, PCoA and AMOVA indicated the existence of four management units namely, Ganges–Brahmaputra, Satluj, Mahanadi and Narmada. This baseline information will be useful in conservation and management of C. chitala, which has significant conservation and aquaculture value.
Conclusion
Understanding genetic structure of a species is among the key components, which is essential for planning management strategies. The resource managers should be aware with management units of important fish species. C. chitala is important for conservation and aquaculture viewpoint. This study unraveled high degree of genetic differentiation with evidences for presence of four natural genetic stocks. Demographic parameters indicate that there is need to assess a fine scale structuring through use of multilocus markers, particularly in Gangetic rivers. Two other rivers, Gomti and Brahmaputra, displayed lower diversities, while Mahanadi presented absence of alternate haplotype. These populations are important for conservation, and the resource managers need to adopt suitable measures, in this context. This may be achieved through cohesive approaches of policy makers with multiple stakeholders and researchers.
References
Hilton EJ, Lavoué S (2018) A review of the systematic biology of fossil and living bony-tongue fishes, Osteoglossomorpha (Actinopterygii: Teleostei). Neotropical Ichthyology 16(3):180031
Hamilton F (1822) An account of the fishes found in the river Ganges and its branches. Printed for A, Constable and company
Froese R, Pauly D (2003) Fish Base WWW Electronic Publication. World Fish. https://www.fishbase.org
Taverne L (1998) Les ostéoglossomorphes marins de l’Éocène du Monte Bolca (Italie): Monopteros Volta, 1796, Thrissopterus Heckel, 1856 et Foreyichthys Taverne, 1979. Considérations sur la phylogénie des téléostéens ostéoglossomorphes. Studi e ricerche sui giacimenti terziari di Bolca 7:67–158
Mandal A, Mohindra V, Singh RK, Punia P, Singh AK, Lal KK (2012) Mitochondrial DNA variation in natural populations of endangered Indian Feather-Back Fish. Chitala chitala. Molecular biology reports. 39(2):1765–1775
Sarkar UK, Deepak PK, Negi RS, Singh S, Kapoor D (2006) Captive breeding of endangered fish Chitala chitala (Hamilton-Buchanan) for species conservation and sustainable utilization. In Marine, Freshwater, and Wetlands Biodiversity Conservation (pp. 211–221). Springer, Dordrecht
Chaudhry S (2010) Chitala chitala. The IUCN Red List of Threatened Species 2010: e.T166510A6225101
Sarkar UK, Negi RS, Deepak PK, Lakra WS, Paul SK (2008) Biological parameters of the endangered fish Chitala chitala (Osteoglossiformes: Notopteridae) from some Indian rivers. Fish Res 90(1–3):170–177
Sarkar UK, Deepak PK, Negi RS (2009) Length–weight relationship of clown knifefish Chitala chitala (Hamilton 1822) from the River Ganga basin. India J App Ichthyol 25(2):232–233
Ward RD, Grewe PM (1994) Appraisal of molecular genetic techniques in fisheries. Rev Fish Biol Fisheries 4(3):300–325
Crandall KA, Posada D, Vasco D (1999, November) Effective population sizes: missing measures and missing concepts. In Animal Conservation forum (Vol. 2, No. 4, pp. 317–319). Cambridge University Press
Goios A, Alvarez L (2013) Mitochondrial Genetics/Evolution. In: Stanley M, Kelly H (eds) Brenner’s Encyclopedia of Genetics, 2nd edn. Elsevier, London p, pp 439–440
Gupta A, Lal KK, Mohindra V, Singh RK, Punia P, Dwivedi AK, Gupta BK, Luhariya RK, Masih P, Mishra RM, Jena JK (2013) Genetic divergence in natural populations of bronze featherback, Notopterus notopterus (Osteoglossiformes: Notopteridae) from five Indian rivers, analyzed through mtDNA ATPase6/8 regions. Meta gene 1:50–57
Singh RK, Lal KK, Mohindra V, Punia P, Sah RS, Kumar R, Gupta A, Das R, Lakra WS, Ayyappan S (2012) Genetic diversity of Indian Major Carp, Labeo calbasu (Hamilton, 1822) populations inferred from microsatellite loci. Biochem Syst Ecol 44:307–316
Xiao W, Zhang Y, Liu H (2001) Molecular Systematics of Xenocyprinae (Teleostei: Cyprinidae): Taxonomy, Biogeography, and Coevolution of a Special Group Restricted in East Asia. Mol Phylogenet Evol 18(2):163–173
Sivasundar A, Bermingham E, Ortí G (2001) Population structure and biogeography of migratory freshwater fishes (Prochilodus: Characiformes) in major South American rivers. Mol Ecol 10(2):407–417
Hall TA (1999, January) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic acids symposium series (Vol. 41, No. 41, pp. 95–98). [London]: Information Retrieval Ltd., c1979-c2000
Swofford D L (2001) Paup*: Phylogenetic analysis using parsimony (and other methods) 4.0. B5
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37–48
Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152(3):1079–1089
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mole Ecol Res 10(3):564–567
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Can Res 27(2):209–220
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in Excel Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295
Corander J, Marttinen P, Sirén J, Tang J (2013) BAPS: Bayesian analysis of population structure. Department of Mathematics and statistics, University of Helsinki, Finland, Manual, p 6
Ray N, Currat M, Excoffier L (2003) Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 20(1):76–86
Hobbs JPA, Van Herwerden L, Jerry DR, Jones GP, Munday PL (2013) High genetic diversity in geographically remote populations of endemic and widespread coral reef angelfishes (genus: Centropyge). Diversity 5(1):39–50
Avise JC, Neigel JE, Arnold J (1984) Demographic influences on mitochondrial DNA lineage survivorship in animal populations. J Mol Evol 20(2):99–105
Wibowo A, Affandi R, Soewardi K, Sudarto S (2017) Genetic Differentiation of the Kampar river’s Giant Featherback (Chitala lopis Bleeker 1851) Base on Mitochondrial DNA Analysis. Indonesian Fisheries Research Journal 16(2):49–58
Takagi AP, Ishikawa S, Nao T, Song SL, Hort S, Thammavong K, Saphakdy B, Phomsouvanhm A, Nishida M, Kurokura H (2010) Genetic differentiation and distribution routes of the bronze featherback Notopterus notopterus (Osteoglossiformes: Notopteridae) in Indochina. Biol J Lin Soc 101(3):575–582
Menon AGK (1951) Further studies regarding Hora’s Satpura hypothesis. The Role of Eastern Ghats in distribution of the Malayan flora and fauna to Peninsular India. Proceedings of the national Academy of Sciences. India. 17:475–497
Kim M, Morrison M, Yu Z (2011) Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J Microbiol Methods 84(1):81–87
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129(2):555–562
Frankham R, Ballou JD, Briscoe DA (2004) A primer of conservation genetics. Cambridge University Press, United Kingdom, p 220
Delrieu-Trottin E, Maynard J, Planes S (2014) Endemic and widespread coral reef fishes have similar mitochondrial genetic diversity. Proceedings of the Royal Society B: Biological Sciences 281(1797):20141068
Hartl DL, Clark AG, Clark AG (1997) Principles of population genetics, vol 116. Sinauer associates, Sunderland, MA
Sodsuk PK, Sodsuk, (2000) Gentic diversity of featherback fish in Thailand. Agriculture and Natural resources 34(2):227–239
Barby FF, Ráb P, Lavoué S, Ezaz T, Bertollo LAC, Kilian A, Maruyama SR, Aguiar de Oliveira E, Artoni RF, Santos MH, Ilesanmi Jegede O (2018) From chromosomes to genome: insights into the evolutionary relationships and biogeography of Old World knifefishes (Notopteridae; Osteoglossiformes). Genes 9(6):306
Daniels RR (2001) Endemic fishes of the Western Ghats and the Satpura hypothesis. Current Science-Bangalore 81(3):240–244
Chauhan T, Lal KK, Mohindra V, Singh RK, Punia P, Gopalakrishnan A, Sharma PC, Lakra WS (2007) Evaluating genetic differentiation in wild populations of the Indian major carp, Cirrhinus mrigala (Hamilton–Buchanan, 1882): evidence from allozyme and microsatellite markers. Aquaculture 269(1–4):135–149
Acharya AP, Pavan-Kumar A, Gireesh-Babu P, Joshi CG, Chaudhari A, Krishna G (2019) Population genetics of Indian giant river-catfish, Sperata seenghala (Sykes, 1839) using microsatellite markers. Aquat Living Resour 32:4
Khedkar GD, Jamdade R, Kalyankar A, Tiknaik A, Ron TB, Haymer D (2014) Genetic fragmentation in India’s third longest river system, the Narmada. Springer Plus 3(1):385
Bhandari S, Maurya DM, Chamyal LS (2005) Late Pleistocene alluvial plain sedimentation in lower Narmada Valley, Western India: palaeoenvironmental implications. J Asian Earth Sci 24(4):433–444
Briggs JC (2003) The biogeographic and tectonic history of India. J Biogeogr 30(3):381–388
Martinez AS, Willoughby JR, Christie MR (2018) Genetic diversity in fishes is influenced by habitat type and life-history variation. Ecology and Evolution 8(23):12022–12031
Chandan R, Singh RK, Singh A, Paul P, Sah RS, Kumar R, Mohindra V, Lal KK, Jena JK (2020) Spatio-temporal variations in length-weight relationship and condition factor of two notopterids, Chitala chitala (Hamilton, 1822) and Notopterus notopterus (Pallas, 1769). Indian J Fish 67(2):120–124
Verma SR, Dalela RC (1975) Studies on the Pollution of the Kalinadi by Industrial Wastes near Mansurpur Part 2: Biological Index of Pollution and Biological Characteristics of the River. Actahydrochimica et hydrobiologica 3(3):259–274
Tran NN, Thanh VC, Hirata Y (2005) Genetic diversity and conservation of the threatened freshwater Clown Knifefish Notopterus chitala through artificial propagation in the Mekong Delta. Biosphere conservation: for nature, wildlife, and humans 6(2):61–66
Vass K, Das M, Tyagi R, Katiha P, Samanta S, Srivastava N, Bhattacharjya B, Suresh V, Pathak V, Chandra G, Debnath D (2011) Strategies for sustainable fisheries in the Indian part of the Ganga-Brahmaputra river basins. International Journal of Ecology and Environmental Sciences 37(4):157–218
Acknowledgments
This work has been carried out under the project scheme of the Indian Council for Agricultural Research, India entitled, “Outreach Activity on Fish Genetic Stocks Phase-II”. The authors are thankful to Director, ICAR-National Bureau of Fish Genetic Resources, Lucknow, India for providing the facilities needed for executing this work. The authors are also grateful to Sh Pradipta Paul, Dr. L.K Tyagi, Dr. Santosh Kumar, Sh Kantharajan G, Dr. Achal Singh, and Sh Rajkumar for their help in sampling and geo-referenced riverine map preparations. Sh R.S. Sah and Sh Rajesh Kumar are thankfully acknowledged for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. The methodologies were approved by the Institutional Animal Ethical Committee (IAEC), ICAR-NBFGR, Lucknow, India vide No. G/CPCSEA/IAEC/2015/2 dated 27 October, 2015.
Informed consent
Informed consent was obtained from all the authors who were involved in this piece of work. The manuscript is submitted after forwarding through the prescribed procedures of the institute with the approval of the competent authority.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dutta, N., Singh, R.K., Pathak, A. et al. Mitochondrial DNA markers reveal genetic connectivity among populations of Osteoglossiform fish Chitala chitala. Mol Biol Rep 47, 8579–8592 (2020). https://doi.org/10.1007/s11033-020-05901-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05901-w