
Abstract
The Deccan mahseer, Tor khudree (Sykes 1839), belonging to family Cyprinidae is an important food and a game fish distributed in peninsular India. Due to overfishing and habitat destruction, the species is declared endangered and placed on the IUCN red list. Therefore, a well-designed conservation program may be essential to get this species protected in its natural habitat. We used a total of 152 samples from four rivers of peninsular India to assess the genetic diversity and structure of the mahseer using concatenated sequences of two mitochondrial genes, ATPase 6/8 (790 bp) and Cyt b (1000 bp). High haplotypic diversity was seen with 44 haplotypes. Individual gene wise haplotypes included 10 and 21 haplotypes for ATPase6/8 and Cyt b, respectively. AMOVA revealed most of the genetic variations (71.02%) to be within the populations. Significant genetic differentiation was observed between all population pairs, with FST values ranging from 0.121 to 0.372, with minimum between Tunga and Tungabhadra population and maximum between Tunga and Periyar population. Haplotype network showed one ancestral haplotype (TKACH04). Significant negative Fu’s F and unimodal mismatch distribution suggested recent demographic expansion. The results of the present study would serve as a useful resource for further research on population genetics and conservation programs of the species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Mahseer, an iconic group of fish in the Cyprinidae family, is characterized by large scales and a large body was once considered a single species (Day 1876), is now distributed under three genera Tor, Neolissochilus, and Naziritor (Ogale 2002; Sati et al. 2013; Eschmeyer et al. 2017; Froese and Pauly 2018). However, members only under the genus Tor are considered true mahseers (Desai 2003; Nguyen et al. 2008) and there are 16 valid species, all of which exhibit highly potamodromous behaviors, with upstream spawning migration (Nautiyal et al. 2001, 2008; Shrestha 1997). Eight out of 16 species are reported in India (Pinder et al. 2019). The “Deccan Mahseer,” Tor khudree (Sykes 1839) (Order: Cypriniformes, Family: Cyprinidae) is one of the charismatic mahseer species distributed in Peninsular India (Menon 1992; Jayaram 1994, 2005; Basavaraja 2011; Raghavan et al. 2011). This mahseer is an esthetic food and game fish endemic to the Southwestern part of India (Day 1889). They are commonly called “King of Indian Freshwater systems” (Sati et al. 2013; Sarkar et al. 2015). The slow-growing fish with a long life span and high nutritional value (Day 1876; Froese and Pauly 2010; Sarkar et al. 2015) is overexploited for food by the forest-dwelling communities in central and western peninsular regions of India (Raghavan et al. 2008; Basavaraja 2011). Due to its close resemblance with Tor malabaricus from Peninsular India, they were considered synonymous (Menon 1992, 1999; Pinder et al. 2019). Now malabaricus is considered a valid species with the present name (Silas et al. 2005; Raghavan and Ali 2013). The similarity in the morphology of both species has also led the breeders to misidentify the species. Moreover, overfishing due to food and sports are responsible for the decrease in khudree population. Habitat destruction due to the building of dams has also been the cause of concern for the declining population of the species. Some of the minor and major dams and reservoirs of the important rivers of Deccan India include Tunga Anicut Dam, Tunga, Tungabhadra reservoir, Tungabhadra dam, Harangi dam/reservoir, Cauvery and Bhoothathankettu dam, and Periyar. The highest number (~ 100) of dams present is known for River Cauvery. Due to its declining status, it has been recorded as an endangered species on the Red List of the International Union for Conservation of Nature (Pinder and Raghavan 2013). Their presence indicates the health of the rivers as a little change in the environment would lead to a decline in their number (Basavaraja 2011) and decline in number of natural populations might lead to eradication of unique genotypes that are not found anywhere else (Parmaksiz 2019). Knowledge of the genetics of the species is lacking and research is limited to taxonomy and phylogeny of the species (Nguyen et al. 2008). Information on the stock structure of the species is also very essential before designing an effective management program, conservation strategy, standardized breeding, or selective breeding program. Economic fish species provide both a source of food and an opportunity of employment for people (Parmaksiz 2023). Therefore, the purpose of this study was to determine the genetic variation and population structure of the endangered mahseer across the Deccan riverine systems of India. The effective stock characterization is possible now due to the usage of Molecular markers as they have been utilized in various studies to determine effective population size, population demography, and gene flow (Habib et al. 2012) in fish populations. Maternal inheritance and absence of recombination are a few of the important properties of mitochondrial DNA that make it useful in tracing evolution history, founder events, introductions, and population bottlenecks (Harrison 1989).
The mitochondrial gene has demonstrated its importance as a maker in species identification through DNA barcoding (Mohanty et al. 2015; Bhattacharya et al. 2016), thus helping in overcoming the uncertainty in the morphological identification of species. The mitochondrial gene COI has been declared a basic tool for molecular taxonomy and identification (Ratnasingham and Hebert 2007) and we have used it to resolve the morphological ambiguities that we faced during the sample collection. We have also used concatenated mitochondrial genes: ATPase 6/8 and Cyt b as the marker of choice for the population studies in khudree (Sahoo et al. 2019), since it is effective in showing considerable among and within-population variation, which provides information on the structuring of populations (Avise et al. 1987). Hence, the present study is done to comprehend the genetic diversity and population structure among four different populations of khudree as a step toward understanding the genetics of this fish that might help further in the management and conservation of the species.
Material and Methods
Sample Collection and DNA Extraction
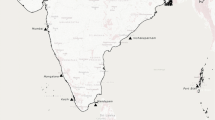
Fin clipping of the species was performed between April 2015 and March 2018 from River Tungabhadra, River Tunga, River Periyar, and River Cauvery (Table 1, Fig. 1). Live fish were caught with prior permission from the concerned Department of Forests and wild life at the protected area randomly and after careful collection of fin tissue, they were released back into the water. Fin clippings from 174 collected specimens were preserved in 95% ethanol at −20 °C until further use. The congeneric species Neolissochilus hexagonalepis from GenBank was chosen as the outgroup (KU380329) in Phylogenetic analysis. High molecular weight genomic DNA was extracted using the standard phenol–chloroform method (Sambrook et al. 1989), resuspended in Tris–EDTA buffer, and stored at −20 °C until use. The integrity of the DNA was checked using agarose gel electrophoresis and Nanodrop Biospectrometer (Eppendorf India Pvt. Ltd.). The DNA was then diluted to 25 ng/µl working solutions.

Schematic map of Indian Rivers showing the place of collection of samples
PCR Amplification and Sequencing
To avoid any ambiguities in the identification of the species during collection, all 174 samples were verified using DNA barcoding which involved PCR amplification of a 600 bp fragment of the COI gene using universal primers, FishF1-5′ TCA ACC AAC CAC AAA GAC ATT GGC AC 3′ and FishR2-5′ ACT TCA GGG TGA CCG AAG AAT CAG AA- 3′ (Ward et al. 2005). A 25 µl PCR mixture contained 1.0–2.0 µl of DNA template (25 ng/ml), 2.5 µl 10X PCR buffer, 1 µl of each primer (10 pmol/µl), 1.5 µl MgCl2 (20 mM), 2 µl dNTP mix (2.5 mM each), and 0.25 U of Taq DNA polymerase. The reactions were performed on a GeneAmp PCR System (Applied Biosystems Inc., Foster City, CA) for 35 cycles under the conditions: one cycle of initial denaturation at 94 °C for 5 min followed by 30 s at 94 °C, 45 s at 58 °C, and 30 s at 72 °C for COI, with a final extension at 72 °C for 7 min. The PCR products were purified using Qiagen PCR purification kit followed by bidirectional cycle sequencing on ABI 3100 PE automated capillary sequencer.
Partial ATPase 6/8 and Cyt b genes from the verified samples were amplified using universal primers, ATPase 6/8—L8331 5′AAA GCR TTR GCC TTT TAA GC 3′ and H9236 5′ GTT AGT GGT CAK GGG CTT GGR TC 3′ (Thai et al. 2005) and a combination of Universal forward primer (Kocher et al. 1989) and carp-specific reverse primer (Das et al. 2014) in case of Cyt b—L14841 5′ AAA AAG CTT CCA TCC AAC ATC TCA GCA TGA TGA AA 3′ and CIFA-Cyt b-R2 5′ CTC ATT CTA GTG CTT TAT TTT C 3′. A 25 µl PCR mixture contained 1.0–2.0 µl of DNA template (25 ng/ml), 2.5 µl PCR buffer, 1 µl of each primer (10 pmol/µl), 1.5 µl MgCl2 (20 mM), 2 µl dNTP mix (2.5 mM each), and 0.25 U of Taq DNA polymerase. The reactions were performed on a GeneAmp PCR System (Applied Biosystems Inc., Foster City, CA) for 35 cycles under the conditions: one cycle of initial denaturation at 94 °C for 5 min followed by 30 s at 94 °C, 1 m at 55 °C, and 2 m at 72 °C for ATPase 6/8 and 30 s at 94 °C, 40 s at 50 °C, and 45 s at 72 °C for Cyt b, with final extension at 72 °C for 7 min. The PCR products were purified using Qiagen PCR purification kit followed by bidirectional cycle sequencing on ABI 3100 PE automated capillary sequencer.
Species Identification, Generation of Consensus Sequences
The COI sequences obtained were edited in BioEdit version 7.0.5.3 (Hall 1999). The edited sequences were fed in FASTA format into BOLD Systems v3 (https://v3.boldsystems.org/) for species identification. The sequences obtained after the sequencing of ATPase 6/8 and Cyt b genes of the confirmed individuals were edited and aligned using CLUSTALW in BioEdit version 7.0.5.3 (Hall 1999) to get the final set of consensus sequences.
Genetic Diversity and Population Differentiation Study
The consensus sequences of mitochondrial genes were concatenated manually using BioEdit version 7.0.5.3 (Hall 1999) based on the concept of supergene topology (Havird and Santos 2014). The sequential genetic information was estimated, which included the number of polymorphic sites, parsimony informative sites, Haplotype diversity, nucleotide diversity, private haplotypes using the DnaSP version 6 (Rozas et al. 2017). Genetic differentiation among the Tor khudree populations was calculated using the F-statistics (FST) which was estimated by using conventional method using haplotypic frequencies with 1023 nonparametric permutations. The significance of FST was determined by the p value generated in Arlequin v3.1.1 (Excoffier and Lischer 2006) and hierarchical partitioning of genetic diversity within and between populations was determined by Analysis of Molecular Variance as implemented in Arlequin v3.1.1.A median-joining network was also constructed using PopART v. 1.7 (Bandelt et al. 1999) to better illustrate the association between the haplotypes.
Phylogenetic Analysis, Population Demography
The phylogenetic analysis was conducted using MEGA 10 (Kumar et al. 2018). A maximum-likelihood tree was constructed with HKY + GAMMA + I model as the best-predicted model from jModelTest 2.1.10. The congeneric species Neolissochilus hexagonalepis was taken as the outgroup (KU380329). The robustness of the test was found out using the bootstrap method of 1000 replications. Only the haplotypes of the concatenated sequences were included for the generation of a phylogenetic tree.
Demographic history was calculated for Tor khudree by distribution of the number of differences in site between the pair of sequences referred as mismatch distribution. For a model of constant population size, calculated expected values were plotted against the observed values using DnaSP6 program. To check whether the observed distribution deviated significantly from the expected values under the population expansion model, Harpending’s raggedness index (Hri) Harpending (1994) and the sum of squared deviation (SSD) were calculated using Arlequin. The Tajima’s D test (Tajima 1989) and Fu’s Fs test (Fu 1997) were used to test the hypothesis of neutrality of the concatenated mitochondrial genes in DnaSP v.6.
Results
Sequence Analysis, Genetic Diversity, and Population Differentiation Study in T. khudree
Out of 174 samples collected, 152 samples, i.e., 39 out of 39 in Tungabhadra, 47 out of 47 in Tunga, 27 out of 43 in Periyar, and 39 out of 45 in Cauvery got verified as Tor khudree. Consensus sequences of 790 bp of ATPase 6/8 gene (MZ057291–MZ057442) and 1000 bp of Cyt b gene (MZ057443–MZ057594) were obtained from the sequencing of the PCR products. Analysis of the ATPase 6/8 and Cyt b genes revealed 10 and 21 different haplotypes, respectively. The genetic diversity analysis of both the genes is described in the supplementary data 1 (S1). Analysis of Molecular Variance (AMOVA) using ATPase 6/8 gene revealed 50.83% among population variation and 49.17% within population variation with pairwise FST ranging from −0.01624 to 0.70977 (p < 0.05). Significant genetic differentiation was observed among all population pairs except Periyar-Cauvery (−0.01624). Similarly, AMOVA using the Cyt b gene showed 14.87% among population variation and 85.13% within population variation with pairwise FST ranging from 0.05 to 0.28 (p < 0.05) indicating moderate to high genetic differentiation among all populations.
Analysis of concatenated sequences of both genes in four populations revealed 44 haplotypes. Tungabhadra has the highest with 24 haplotypes followed by Cauvery with 14, Tunga with 11, and Periyar with 07 haplotypes. Out of 1790 sites in the concatenated genes, 27 sites were polymorphic, 14 were parsimony informative sites, and 13 were singleton variable sites. The average nucleotide composition was 27.2% T, 29.2% C 30.0% A, and 13.6% G and it was biased toward the A + C content. The haplotype diversity (Hd) and nucleotide diversity (π) ranged from 0.792 to 0.968 and 0.00068 to 0.00157, respectively. Periyar population showed the lowest Hd and π and Tungabhadra showed the highest Hd and π (Table 1). The frequency of each haplotype in a population is provided in Supplementary data 2 (S2).
AMOVA illustrated the population structure using the concatenated sequences of both genes. The results revealed 28.98% among population variation and 71.02% within population variation (Table 2). Pairwise FST ranged from 0.121 to 0.372 (p < 0.05) indicating significant genetic differentiation among all population pairs. The highest genetic differentiation was observed between Tunga and Periyar populations and it was lowest between Tunga and Tungabhadra populations (Table 3).
Median-joining tree revealed Haplotype 4 (TKACH04) to be found in most of the individuals of the three populations: Tungabhadra, Tunga, and Periyar as it was present in the center of the network but absent in the Cauvery population (Fig. 2A). Haplotype 9 was also shared by the above three populations. Haplotype10 was shared by three populations (Tungabhadra, Tunga, and Cauvery). Haplotype 17 was shared by three populations (Tungabhadra, Periyar, and Cauvery). Haplotypes 27 and 29 were shared by two populations (Tunga and Cauvery). Haplotypes 34 and 35 were shared by two populations (Periyar and Cauvery). Many unique haplotypes were found not shared by any populations; the highest number of unique haplotypes was seen in the Tungabhadra population (20) and the lowest was seen in the Periyar population (1). Haplotypic distribution has been shown in the sampling locations (Fig. 2B).

A Median-Joining Network of 44 haplotypes observed in T. Khudree populations B Mitochondrial haplotype distribution at sampling locations
Phylogenetic Analysis, Population Demography Analysis
Phylogenetic analysis revealed the monophyly of the Tor khudree haplotypes that were separated a hundred percent from the outgroup species Neolissochilus hexagonolepis (Fig. 3). The results of the mismatch analysis and neutrality tests along with the associated p-values are represented in Table 4. The Tajima’s D was negative for all populations, which indicated the presence of an excess of rare nucleotide variants compared to what was to be expected under the neutral model of evolution. However, the results (−0.27, p > 0.05) showed no significant deviations from the null hypothesis of selective neutrality for all the populations. The individual population also showed similar results. In contrast, Fu’s Fs tests for the overall population and individual stocks, which are based on the distribution of haplotypes, though showed negative values had a significant deviation from neutrality (−8.718, p < 0.05) suggesting populations are showing population size expansion and/or purifying selection.

Phylogenetic relationship among Tor khudree populations using concatenated sequences of ATPase 6/8 and Cyt b genes. A maximum Likelihood tree with 1000 replicates, and HKY + G + I model with Neolissochilus hexagonolepis as outgroup
Mismatch distribution Supplementary data 3 (S3) for the overall population as well as individual population showed a unimodal plot which indicated that the population had undergone a recent population expansion after a bottleneck. The estimated values of Harpending’s raggedness index and SSD were non significant which suggested that the data is a good fit between the observed and expected values of the pairwise distributions. The data supported the recent population expansion.
Discussion
Our study aimed at revealing the population genetic structure of Deccan mahseer across the Deccan region of peninsular India. The utility of maternally inherited Mt DNA has been used in the present study for species identification as well as to assess the genetic diversity and population structure of the endangered species, endemic to India. The highly conserved mitochondrial gene, COI, was used in species identification through barcoding (Ward et al. 2005; Lakra et al. 2011). Out of collected 174 samples, 152 got confirmed as Tor khudree, and the rest were found to be mostly Tor malabaricus, followed by Tor putitora and Tor tor. The least number of samples that got confirmed was from the Kerala population also indicated the coexistence of two types of mahseer in Periyar waters, Tor khudree and Tor malabaricus (Raghavan et al. 2011). Since mitochondrial genes have a high rate of evolution, it allows for distinguishing between haplotypes in small sample sizes and threatened species (Habib et al. 2012; Sharma et al. 2019). They have been used in the study to examine the genetic variability and structure of the species. Analysis using the two mitochondrial genes revealed both the genes are AT rich as reported in other studies (Johns and Avise 1998). Genetic variability (haplotypes diversity and nucleotide diversity) of ATPase 6/8 gene was lower than the Cyt b gene due to high conservness of ATPase 6/8 gene during evolution (Sati et al. 2013). We have also concatenated both the genes based on the super gene concept to get a better resolution on the population genetics of Tor khudree since mtDNA markers show different levels of rate of evolution (Reeb and Avise 1990; Havird and Santos 2014; Kim et al. 2014; Sahoo et al. 2019). The combined mitochondrial sequences have proved to be very useful in delineating genetic diversity and differentiation in many aquatic species including mahseers (Kim et al. 2014; Sahoo et al. 2019; Sharma et al. 2019; Sah et al. 2020). The nucleotide composition in the concatenated sequences was biased toward A + T as seen in other fishes (Johns and Avise 1998). The order of base composition was A > C > T > G where nucleotide G was least represented similar to other carps (Bej et al. 2012). The presence of low Guanine is a special feature of the mitochondrial genome (Cantatore et al. 1994; Zhu et al. 2017; Balasubramaniam et al. 2022). Genetic diversity study using the concatenated mitochondrial genes revealed the presence of 44 haplotypes with the Tungabhadra population having the highest number of haplotypes (24). The number of haplotypes ranged from 7 to 24. Due to a large number of haplotypes and polymorphic sites, the Tungabhadra population could be considered a highly divergent population. Sharing of haplotypes was seen to be limited to three populations as evident from the median-joining network (Fig. 2A). This indicated the common evolutionary history among the individuals of those populations as well as gene flow between populations (Das et al. 2014). A close genetic relationship is suggestive among the populations, though the oldest haplotype is absent in the Cauvery population. Many haplotypes were also having limited distribution to a single population; out of 44 haplotypes, 36 were private haplotypes (81.8%) which suggested notable population differentiation, as well as the absence of population admixture (Sharma et al. 2019), or these haplotypes, may have arisen due to independent mutation events (Das et al. 2018). These unique haplotypes may have arisen due to environmental changes at their respective locations (Balasubramaniam et al. 2022). These private haplotypes could be used as a marker for stock identification (Sahoo et al. 2019). The distribution of mitochondrial haplotypes is reported to be proportional to their age (Neigel and Avise 1993; Kim et al. 2020). Haplotype 4 (TKACH04) is present in most of the individuals and perhaps the ancestral haplotype which is evident from the median-joining network as it is present in the center of the network. Haplotype diversity ranged from 0.792 to 0.968 and π ranged between 0.00068 and 0.00157 which was comparable with the other mahseers (Sati et al. 2015; Sharma et al. 2019) in terms of high haplotypic diversity and low nucleotide diversity. This may be due to sudden population growth and accumulation of mutation after diverging from a population facing bottleneck events (Avise et al. 1984; Grant and Bowen 1998). The range of Hd and π seen in khudree is similar to the range observed in other freshwater fishes (Habib et al. 2012).
Population Differentiation and Stock Structure
FST is one of the most widely used estimates for detecting genetic differentiation between and among populations. This measure provides information on the evolutionary events that may cause genetic differentiation among populations (Holsinger and Weir 2009). In general, FST values of 0–0.05 represent little differentiation; values of 0.05–0.25 indicate moderate differentiation and values higher than 0.25 indicate great differentiation among populations (Wright 1978). In this study, the FST between the four populations ranged from 0.121 to 0.372, indicating a moderate to high level of genetic differentiation (Sati et al. 2013; Sahoo et al. 2019; Sharma et al. 2019). The overall FST was found to be 0.289, p < 0.05 suggesting high and significant genetic differentiation among the khudree populations and freshwater fishes tends to exhibit higher levels of genetic differentiation as compared to marine fishes (Ward et al. 1994). AMOVA using ATPase 6/8 gene revealed high among population variation as compared to high within population variation using Cyt b. The results based on the concatenated sequences also showed high intrapopulation variation, concurrent with the information on the Cyt b from our study and the other study on Tor mahseer (Sah et al. 2020), suggested possible gene flow among the populations and this result is comparable to other mahseers where high among population variation is observed (Esa and Abdul Rahim 2013; Sati et al. 2013; Sharma et al. 2019). This observation is also in congruence with Vrijenhoek 1998, who described that low among population variation and high within population variation is expected in migratory fishes like mahseer.
Phylogenetic Study and Population Demography
Phylogenetic relationships using the HYK + I + G model did not show the populations to separate into clusters instead mixed clusters were seen indicating the sharing of haplotypes among the population. The neutrality test of Tajima’s D revealed nonsignificant negative values for all populations of T. khudree which supported the changes in concatenated Cyt b and ATPase 6/8 for the species in the collection sites are selectively neutral. However, the neutrality test of Fu’s F revealed significant negative values and the mismatch distribution of all the population was seen to be unimodal thus suggesting possibilities of recent population expansion or a purifying selection (Hudson et al. 1992; Rogers and Harpending 1992). The unimodal pattern could also be due to range expansion with high levels of migration in between the demes (Ray et al. 2003; Excoffier 2004).
Conclusion
Our work on genetic diversity of Tor khudree using the mitochondrial markers with samples from four different rivers of Deccan India has been done for the first time. The result suggested the importance of mitochondrial markers for studying the variations within and among population of Tor khudree. High genetic differentiation was seen in all khudree populations. The highly divergent Tungabhadra population can serve as conservation unit. The within population variation in khudree would provide wide genetic base for any genetic improvement program. The results from this study can be compared with the results obtained on using highly advanced markers such as SSR and SNP for better understanding the genetic differentiation and genetic structure of the esthetic Deccan mahseer.
Data Availability
The data that support the findings of this study are openly available in “NCBI” at https://www.ncbi.nlm.nih.gov/nucleotide
References
Avise JC, Neigel JE, Arnold J (1984) Demographic influences on mitochondrial DNA lineage survivorship in animal populations. J Mol Evol 20(2):99–105
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigel JE, Reeb CA, Saunders NC (1987) Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annu Rev Ecol Evol Syst. https://doi.org/10.1146/ANNUREV.ES.18.110187.002421
Balasubramaniam S, Soman M, Katneni VK, Tomy S, Gopalapillay G, Vijayan KK (2022) Mitochondrial DNA based diversity studies reveal distinct and sub-structured populations of pearlspot, Etroplus suratensis (Bloch, 1790) in Indian waters. J Genet 101(1):1–15. https://doi.org/10.1007/s12041-021-01341-y
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Basavaraja N (2011) The Deccan Mahseer–biology, breeding and conservation. In Seminar on Aquaculture and Mahseer Biodiversity Conservation at CIFE, Mumbai. PMCid: PMC3391906
Bej D, Sahoo L, Das SP, Swain S, Jayasankar P, Das PC, Routray P, Swain SK, Jena JK, Das P (2012) Complete mitochondrial genome sequence of Catla catla and its phylogenetic consideration. Mol Biol Rep 39(12):10347–10354. https://doi.org/10.1007/s11033-012-1912-5
Bhattacharya M, Sharma AR, Patra BC, Sharma G, Seo EM, Nam JS, Chakraborty C, Lee SS (2016) DNA barcoding to fishes: current status and future directions. DNA Mapp Seq Anal 27(4):2744–2752. https://doi.org/10.3109/19401736.2015.1046175
Cantatore P, Roberti M, Pesole G, Ludovico A, Milella F, Gadaletal MN, Saccone C (1994) Evolutionary analysis of cytochrome b sequences in some Perciformes: evidence for a slower rate of evolution than in mammals. J Mol Evol 39(6):589–597. https://doi.org/10.1007/BF00160404
Das SP, Bej D, Swain S, Mishra CK, Sahoo L, Jena J, Jayasankar P, Das P (2014) Population divergence and structure of Cirrhinus mrigala from peninsular rivers of India, revealed by mitochondrial cytochrome b gene and truss morphometric analysis. DNA Mapp Seq Anal 25(2):157–164. https://doi.org/10.3109/19401736.2013.792055
Das SP, Swain S, Jena JK, Das P (2018) Genetic diversity and population structure of Cirrhinus mrigala revealed by mitochondrial ATPase 6 gene. DNA Mapp Seq Anal 29(4):495–500. https://doi.org/10.1080/24701394.2017.1310852
Day F (1876) On some of the fishes of the Deccan. Zool J Linn Soc 12(64):565–578. https://doi.org/10.1111/j.1096-3642.1876.tb00232.x
Day F (1889) Fauna of British India, including Ceylon and Burma. Fishes 1:1–548. https://doi.org/10.1080/00222939009460788
Desai VR (2003) Synopsis of biological data on the tor mahseer Tor tor (Hamilton, 1822) (No. 158). Food & Agriculture Org
Esa Y, Abdul Rahim KA (2013) Genetic structure and preliminary findings of cryptic diversity of the Malaysian Mahseer (Tor tambroides Valenciennes: Cyprinidae) inferred from mitochondrial DNA and microsatellite analyses. BioMed Res Int. https://doi.org/10.1155/2013/170980
Eschmeyer WN, Fricke R, Van der Laan R (2017) Catalog of fishes: genera, species, references
Excoffier L (2004) Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol 13(4):853–864. https://doi.org/10.1046/j.1365-294X.2003.02004
Excoffier L, Lischer H (2006) An integrated software package for population genetics data analysis. Computational and Molecular Population Genetics Lab (CMPG), Institute of Zoology, University of Berne, Switzerland
Froese R, Pauly D (2010) FishBase Fishbase: World Wide Web Electronic Publication. http://www.fishbase.org/search.php
Froese R, Pauly D (2018) FishBase. Version 06/2018. World Wide Web Electronic Publication. https://www.fishbase.org/. Assessed on 14 Nov 2019
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147(2):915–925. https://doi.org/10.1093/genetics/147.2.915
Grant WAS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89(5):415–426. https://doi.org/10.1093/jhered/89.5.415
Habib M, Lakra WS, Mohindra V, Lal KK, Punia P, Singh RK, Khan AA (2012) Assessment of ATPase 8 and ATPase 6 mtDNA sequences in genetic diversity studies of Channa marulius (Channidae: Perciformes). Proc Natl Acad Sci India B 82(4):497–501. https://doi.org/10.1007/s40011-012-0061-x
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic acids symposium series, vol. 41, no. 41. Information Retrieval Ltd.: London, pp. 95–98, c1979–c2000. https://doi.org/10.14601/Phytopathol_Mediterr-14998u1.29
Harpending RC (1994) Signature of ancient population growth in a low resolution mitochondrial DNA mismatch distribution. Hum Biol 66: 591–600
Harrison RG (1989) Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol Evol 1:6–11. https://doi.org/10.1016/0169-5347(89)90006-2
Havird JC, Santos SR (2014) Performance of single and concatenated sets of mitochondrial genes at inferring metazoan relationships relative to full mitogenome data. PLoS ONE 9(1):84080. https://doi.org/10.1371/journal.pone.0084080
Holsinger KE, Weir BS (2009) Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet 10(9):639–650. https://doi.org/10.1038/nrg2611
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA sequence data. Genet 132(2):583–589. https://doi.org/10.1093/genetics/132.2.583
Jayaram KC (1994) The Krishna River system: a bioresources study, vol 160. The Survey
Jayaram KC (2005) Deccan Mahseer fishes: their ecostatus and threat percepts. Zoological Survey of India
Johns GC, Avise JC (1998) A comparative summary of genetic distances in the vertebrates from the mitochondrial cytochrome b gene. Mol Biol Evol 15(11):1481–1490. https://doi.org/10.1093/oxfordjournals.molbev.a025875
Kim WJ, Dammannagoda ST, Jung H, Baek IS, Yoon HS, Choi SD (2014) Mitochondrial DNA sequence analysis from multiple gene fragments reveals genetic heterogeneity of Crassostrea ariakensis in East Asia. Genes Genom 36:611–624. https://doi.org/10.1007/s13258-014-0198-5
Kim MJ, Cho Y, Wang AR, Kim SS, Choi SW, Kim I (2020) Population genetic characterization of the black-veined white, Aporia crataegi (Lepidoptera: Pieridae), using novel microsatellite markers and mitochondrial DNA gene sequences. Conserv Genet 21(2):359–371. https://doi.org/10.1007/s10592-020-01257-7
Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, Villablanca FX, Wilson AC (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA 86(16):6196–6200. https://doi.org/10.1073/pnas.86.16.6196
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547. https://doi.org/10.1093/molbev/msy096
Lakra WS, Verma MS, Goswami M, Lal KK, Mohindra V, Punia P, Gopalakrishnan A, Singh KV, Ward RD, Hebert P (2011) DNA barcoding Indian marine fishes. Mol Ecol Res 11(1):60–71. https://doi.org/10.1111/j.1755-0998.2010.02894.x
Menon AK (1992) Taxonomy of Mahseer fishes of the genus Tor Gray with description of a new species from the Deccan. J Bombay Nat Hist Soc 89(2):210–228
Menon AGK (1999) Check list – fresh water fishes of India. Records of the Zoological Survey of India. Occasional Paper No. 175:i–xxix– 1–366
Mohanty M, Jayasankar P, Sahoo L, Das P (2015) A comparative study of COI and 16 S rRNA genes for DNA barcoding of cultivable carps in India. DNA Mapp Seq Anal 26(1):79–87. https://doi.org/10.3109/19401736.2013.823172
Nautiyal P, Bahuguna SN, Thapliyal RP (2001) The role of ecological factors in governing the direction, time and purpose of migration in Himalayan Mahseer Tor putitora (Ham.). Appl Fisher Aquac 1:133–138
Nautiyal P, Rizvi AF, Dhasmanaa P (2008) Life-history traits and decadal trends in the growth parameters of golden Mahseer Tor putitora (Hamilton 1822) from the Himalayan Stretch of the Ganga River System. Turkish J Fish Aquat Sci 8(1):125–132
Neigel JE, Avise JC (1993) Application of a random walk model to geographic distributions of animal mitochondrial DNA variation. Genet 135:1209–1220. https://doi.org/10.1093/genetics/135.4.1209
Nguyen TT, Na-Nakorn U, Sukmanomon S, ZiMing C (2008) A study on phylogeny and biogeography of mahseer species (Pisces: Cyprinidae) using sequences of three mitochondrial DNA gene regions. Mol Phylogenet Evol 48(3):1223–1231. https://doi.org/10.1016/j.ympev.2008.01.006
Ogale SN (2002) Mahseer breeding and conservation and possibilities for commercial culture. The Indian Experience, Cold Water Fisheries in the Trans-Himalayan Countries, Food and Agriculture Organization of the United Nations (FAO), Rome, pp 186–205
Parmaksiz A (2019) Population genetic diversity of yellow barbell (Carasobarbus luteus) from Kueik, Euphrates and Tigris Rivers based on mitochondrial DNA D-loop sequences. Turk J Fish Aquat Sc 20(1):79–86. https://doi.org/10.4194/1303-2712-v20_1_08
Parmaksiz A (2023) Genetic diversity and population structure analysis of Capoeta trutta (Heckel, 1843) in Turkey and Iraq based on mitochondrial d loop gene. Gene Rep 31:101761
Pinder AC, Raghavan R (2013) Conserving the endangered mahseers (Tor spp) of India: the positive role of recreational fisheries. Curr Sci 104(11):1472–1475
Pinder AC, Britton JR, Harrison AJ, Nautiyal P, Bower SD, Cooke SJ, Lockett S, Everard M, Katwate U, Ranjeet K, Raghavan R (2019) Mahseer (Tor spp) fishes of the world: status, challenges and opportunities for conservation. Rev Fish Biol Fish 29(2):417–452. https://doi.org/10.1007/s11160-019-09566-y
Raghavan R, Prasad G, Ali PH, Pereira B (2008) Fish fauna of Chalakudy River, part of Western Ghats biodiversity hotspot, Kerala, India: patterns of distribution, threats and conservation needs. Biodivers Conserv 17(13):3119–3131. https://doi.org/10.1007/s10531-007-9293-0
Raghavan R, Ali A, Dahanukar N, Rosser A (2011) Is the Deccan Mahseer, Tor khudree (Sykes, 1839) (Pisces: Cyprinidae) fishery in the Western Ghats Hotspot sustainable? A participatory approach to stock assessment. Fish Res 110(1):29–38. https://doi.org/10.1016/j.fishres.2011.03.008
Raghavan R, Ali A (2013) Tor malabaricus, The IUCN Red List of Threatened Species 2013, T172457A6895822
Ratnasingham S, Hebert PD (2007) BOLD: the barcode of life data system. Mol Ecol Notes 7(3):355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
Ray N, Currat M, Excoffier L (2003) Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 20(1):76–86. https://doi.org/10.1093/molbev/msg009
Reeb CA, Avise JC (1990) A genetic discontinuity in a continuously distributed species: mitochondrial DNA in the American oyster, Crassostrea virginica. Genetics 124:397–406
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9(3):552–569. https://doi.org/10.1093/oxfordjournals.molbev.a040727
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302. https://doi.org/10.1093/molbev/msx248
Sah P, Mandal S, Singh RK, Kumar R, Pathak A, Dutta N, Srivastava JK, Saini VP, Lal KK, Mohindra V (2020) Genetic structure of natural populations of endangered Tor mahseer, Tor tor (Hamilton, 1822) inferred from two mitochondrial DNA markers. Meta Gene 23:100635. https://doi.org/10.1016/j.mgene.2019.100635
Sahoo L, Mohanty M, Meher PK, Murmu K, Sundaray JK, Das P (2019) Population structure and genetic diversity of hatchery stocks as revealed by combined mtDNA fragment sequences in Indian major carp. Catla Catla DNA Mapp Seq Anal 30(2):289–295. https://doi.org/10.1080/24701394.2018.1484120
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press
Sarkar UK, Mahapatra BK, Saxena SR, Singh AK (2015) Mahseer in India: An overview on research status and future priorities. J Ecophysiol Occup Health 15(1 & 2):45–52. https://doi.org/10.18311/jeoh/2015/1650
Sati J, Sah S, Pandey H, Ali S, Sahoo PK, Pande V, Barat A (2013) Phylogenetic relationship and molecular identification of five Indian Mahseer species using COI sequence. J Environ Biol 34(5):933
Sati J, Kumar R, Sahoo PK, Patiyal RS, Ali S, Barat A (2015) Genetic characterization of Golden mahseer (Tor putitora) populations using mitochondrial DNA markers. DNA Mapp Seq Anal 26(1):68–74. https://doi.org/10.3109/19401736.2013.823170
Sharma L, Ali S, Barat A, Kumar R, Pande V, Laskar MA, Sahoo PK, Sumer S (2019) Molecular identification and genetic diversity analysis of Chocolate mahseer (Neolissochilus hexagonolepis) populations of Northeast India, using mitochondrial DNA markers. DNA Mapp Seq Anal 30(3):397–406. https://doi.org/10.1080/24701394.2018.1526929
Shrestha TK (1997) The Mahseer in the rivers of Nepal disrupted by dams and ranching strategies
Silas EG, Gopalakrishnan A, John L, Shaji CP (2005) Genetic identity of Tor malabaricus (Jerdon) (Teleostei: Cyprinidae) as revealed by RAPD markers
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genet 123(3):585–595. https://doi.org/10.1093/genetics/123.3.585
Thai BT, Burridge CP, Pham TA, Austin CM (2005) Using mitochondrial nucleotide sequences to investigate diversity and genealogical relationships within common carp (Cyprinus carpio L.). Anim Genet 36(1):23–28. https://doi.org/10.1111/j.1365-2052.2004.01215.x
Vrijenhoek RC (1998) Conservation genetics of freshwater fish. J Fish Biol 53:394–412
Ward RD, Woodwark M, Skibinski DOF (1994) A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol 44(2):213–232. https://doi.org/10.1111/j.1095-8649.1994.tb01200.x
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PD (2005) DNA barcoding Australia’s fish species. Philos Trans R Soc Lond B 360(1462):1847–1857. https://doi.org/10.1098/rstb.2005.1716
Wright S (1978) Variability within and among natural populations. Evolution and the genetics of populations
Zhu S, Li X, Yang J, Li Y, Chen F, Li J, Wu Z (2017) Complete mitochondrial genome of the freshwater fish, Carassioides acuminatus (Cypriniformes, cyprinidae). DNA Mapp Seq Anal 2(1):207–209. https://doi.org/10.1080/23802359.2017.1310602
Acknowledgements
The authors are thankful to the Director, ICAR-CIFA, Bhubaneswar, for providing the laboratory facility to carry out the present work and Indian Council of Agricultural Research, Government of India, New Delhi, for funding the work. Heartfelt thanks to Dr. V. S. Basheer, Principal Scientist. ICAR National Bureau of Fish Genetic Resources, Kochi center. Dr. Annam Pavan Kumar, Senior Scientist, ICAR-Central Institute of Fisheries Education, Mumbai. Mrs. K.T. Darshan, Senior Assistant Director of Fisheries, Kodagu, and Mr. C. P. Aiyappa, Coorg Wildlife Society, Kodagu and Mr. Vivek Viswasrao, Chief Horticulturists, TATA power hatchery, Lonavala, for their constant support during sample collection.
Funding
The work has been funded by Indian Council of Agricultural Research, Government of India, New Delhi under the Outreach Activity Network Project.
Author information
Authors and Affiliations
Contributions
GD: Lab work and manuscript writing. SPD, LS, SS, RC: Sample collection and processing. SKS, PJ, JKS: Intellectual research inputs. PD: Original research idea and manuscript writing All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Ethical Approval
All applicable international, national, and/or institutional guidelines for sampling, care and experimental use of fishes for the study have been followed and that all necessary approvals have been obtained. Prior permission was received from respective wildlife and fisheries department of the state from where the samples were collected. Noninvasive method was used during sample collection which included collection of the caudal fin tissues.
Consent for Publication
All the authors have gone through the work done and have given consent for publication of the work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Das, G., Das, S.P., Sahoo, L. et al. High Genetic Differentiation and Genetic Diversity in Endangered Mahseer Tor khudree (Sykes, 1839) as Revealed from Concatenated ATPase 6/8 and Cyt b Mitochondrial Genes. Biochem Genet (2024). https://doi.org/10.1007/s10528-023-10623-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10528-023-10623-2