
Abstract
MicroRNA (miRNA)-regulated gene expression plays an important role in various plant metabolic processes. Although adventitious roots are critical to plant growth in lotus, the role of miRNA in AR formation remains unclear. Expression profiling of miRNAs was carried out during three different developmental stages of ARs in lotus: no induction of AR stage, initial stage of ARs, and maximum number of ARs. These data are referenced with the whole lotus genome as already identified through high-throughput tag-sequencing. 1.3 × 107 tags were achieved, of which 11,035,798, 11,436,062, and 12,542,392 clean tags were obtained from each stage, respectively. miRNA analysis revealed that miRNAs were less than 10% among all small RNAs. In total, 310 miRNAs (240 up-regulated and 70 down-regulated miRNAs) exhibited expression changes from the no induction stage to the initial stage. Moreover, expression of 140 miRNAs was increased and that of 123 miRNAs was decreased between the initial and maximum AR stages, mostly by ~ − 4–4-fold. miRNAs involved in metabolic pathways differed between the initial stage/no induction stage and the maximum number stage/initial stage. Several miRNAs in the initial stage/no induction stage were related to plant hormone metabolism and pyruvate and MAPK pathways, while major miRNAs in the maximum number stage/initial stage were involved in carbohydrate metabolism. All differentially expressed miRNAs associated with AR formation from the initial stage to maximum stage were also analyzed. The expression of 16 miRNAs was determined using qRT-PCR. This work provides a general insight into miRNA regulation during AR formation in lotus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lotus belongs to the dicotyledons and originates from China and India. Lotus is a member of Nelumbonaceae, which has one genus, Nelumbo, and two species: N. nucifera and N. lutea [1, 2]. Lotus, which is rich in nutrients, is very popular in some Asian countries as a vegetable, and thus its cultivated area is larger than that of any other aquatic plant in China. Lotus is also used in various processed food products, including salted lotus, boiled lotus, glutinous rice lotus root, lotus drink, and lotus leaf tea, and is an ingredient of traditional medicine used for clearing heat, stanching bleeding, and elimination of phlegm.
Adventitious roots (ARs) play an important role in water and nutrition uptake during plant growth and development, because the principal root of lotus is under developed, possibly due to degeneration during evolution. ARs usually emerge on the hypocotyls of seedlings or at the internodes of the rhizome underground. It has been shown that ARs located on the internodes of the rhizome outnumber those on the hypocotyl, suggesting that more water and nutrition support are required during the reproductive stages of plant. Therefore, regulation of the number and emergence of ARs, which affect early plant growth of lotus (when the principal root is not well developed), is a task that urgently needs solving in lotus breeding. Asexual propagation is commonly used in lotus production, with new traits often formed through hybridization, and useful phenotypes fixed through asexual propagation. The developmental process of ARs includes three periods: the induction of the primordial, initiation of ARs, and the emergence of ARs from the hypocotyl [3, 4]. During the first period, meristematic tissue conducive to AR formation is differentiated from normal cells [4], and the primordial root is established. In the subsequent periods, the emergence of roots from stems or the hypocotyl occur [5]. The formation and development of ARs is controlled by intrinsic factors (genetic material) and environmental factors (light, plant hormones, damage). Gene expression can affect the formation of ARs in plants. MicroRNA (miRNA), commonly ~ 18–24 nucleotides in length, is believed to be a major source regulating gene expression by degrading mature mRNA [6, 7]. In recent years, miRNA has been found to participate in various biological process by regulating gene expression in plants, such as floral development, organogenesis, and adaptation to stress [7,8,9,10,11]. There is evidence that auxin and miRNA abundance are required during AR formation in Arabidopsis. Several miRNAs (miR160, miR164, miR167, miR1670, miR172, and miR393) have been shown to be involved in root development (root initiation or elongation) by regulating auxin responsive factors [12]. Further research shows that significant change in the expression of miR172 leads to AR development [13], and miR160, miR164, and miR393 control AR formation by affecting the expression of MbARF16, MbNAC1, and MbTIR1. This suggests that most miRNAs have high correlation with genes involved in the IAA signal transduction pathway.
ARs are a kind of secondary root and are responsible for water and nutrient uptake during the entire period of lotus development, but especially during the seedling stage. Any factor which affects AR formation will directly result in a growth delay. In recent years, a lot of experiments have been carried out to analyze AR formation in lotus. These studies have found that AR formation in lotus was controlled by various genes, exogenous applied IAA, and mechanical damage [14,15,16]. In this study, miRNAs were isolated from different developmental stages of lotus seedlings, and their functions were analyzed with the goal of uncovering a comprehensive regulatory pathways related to AR formation.
Materials and methods
Plant materials
The material selected in this experiment was the rhizome of Taikong lotus 36. The plant was cultivated on a protected agricultural field of Yangzhou University, Southeast China, with conventional field management in April. Plants were planted in a pool in the soil, and the water depth was maintained at about 30 cm for the whole growth season. The average temperature was kept at 26–28 °C during the day and 18–20 °C during the night. Seeds were harvested in October, and stored in a repertory.
For water absorption, the seed coats used in the experiment were broken and then placed in a container with water for germination at 28 °C. The material was selected after the cotyledon had emerged from the seed. The detailed time points for samples used in RNA sequencing included 0 day: no induction of ARs, 1 day: inducted ARs, 10 days: maximum number of ARs.
Preparation of paraffin sections
The treatment of seeds of Taikong lotus were performed as above, and then the seeds were put into water for germination. Hypocotyls from eight time points (12 h for each sample) within 5 days were collected. Hypocotyls were fixed with 10% formalin for 24–48 h at room temperature, and fixed tissues were trimmed into an appropriate size and shaped in embedding cassettes (this included putting these tissues into 50%, 60%, 70%, 80%, 90% and 100% ethanol for 1 h with two changes, respectively, and then placing them into xylene for about 1.5 h with three changes). The treated tissues were placed into paraffin wax overnight, and paraffin blocks were prepared with embedded tissues. The paraffin blocks were cut into sections of 8 µm, and transferred to a flat plate for drying at 37 °C overnight. Safranin and fast green were used to stain tissue for about 2–4 h respectively, and then 95% ethanol was used to remove redundant safranin and fast green. The dyed film was sealed with neutral gum after placing a cover glass. The sealed film was stored in a box at room temperature.
Library construction and sequencing of small RNAs
Three different stages were chosen for miRNA analysis: a C0 library (germinating seeds), C1 library (AR induction stage: about 1 day seedlings), and C2 library (maximum stage of ARs: about 10 days). The conventional cetyltrimethylammonium Ammonium Bromide CTAB method was used to extract total RNA of hypocotyls at 4 °C. DNA contamination was entirely removed after DNaseI treatment. To separate RNA segments of different sizes by PAGE gel, 200 ng−1 µg of RNA sample was used, and a 18–30 nt (14–30 ssRNA Ladder Marker, TAKARA) stripe and recycle was selected. A 3′ and 5′ adaptor system connection was prepared (Tru Seq Small RNA Sample Pre Kit, Illumina), and then the cDNAs from these RNAs were obtained after amplification. Some of the PCR procedures and processes used were referred to those reported by Fang et al. [17] and Luan et al. [18]. The PCR products were purified, and then analyzed using a HiSeq 2000 Sequencing System (Illumina, San Diego, CA, USA).
Obtaining known and new miRNAs
This project was performed at the Beijing Institute of Genomics (BIG) using special constructs. After getting data by PCR, these raw tags, including 5′ primer contaminants, no insert tags, oversized insertions, low quality tags, poly A tags, and small tags were filtered into clean reads, and then the length distribution of these clean tags was summarized, which was helpful to see the composition of the small RNA sample (the normal length of small RNA segments was between 18 and 30 nt). For reads mapping, clean reads were mapped to the reference genome and another sRNA database using the Bowtie 2 tool. RNA tags were mapped to more than one category with the priority rule: miRNA > piRNA > snoRNA > Rfam > other sRNAs. For miRNA prediction, the RIPmiR tool was applied to analysis of the secondary structure, the dicer cleavage site, and the minimum free energy of the sRNA tags that could be mapped to the genome. The small RNA expression level was calculated by using TPM (transcripts per kilobase million), and this method was also directly used to compare the difference of gene expression among samples. Computational prediction of miRNA targets was a critical initial step in identifying miRNA; mRNA target interactions for experimental validation and special software (target finder) were used during this process.
Screening of differentially expressed small miRNAs
The NOISeq method was used to screen small RNAs arising from the above three libraries with the following steps. For the first step, the level of gene expression of every library was counted into log2 (fold-change) M and absolute different value D to establish a noise distribution model. For the second step, the average expression level including control average and treated average in three libraries was counted. For the third step, the fold change (MA = log2((treat_avg)/(control_avg))) and absolute different value D (DA = |control_avg − treat_avg|) were obtained. Differentially expressed miRNAs were defined when MA and DA divergence were significantly different. Fold change ≥ 2 and diverge probability ≥ 0.8 were used as a threshold to judge whether the miRNAs were differentially expressed.
Annotation of differentially expressed small miRNAs
The miRNAs obtained in this experiment were annotated by GO tool with three ontologies: molecular function, cellular component, and biological process. All the differentially expressed miRNAs were enriched and classified into various biological functions after compared against the lotus genome released in the NCBI database. First, all the differentially expressed miRNAs were compared to the database (http://www.geneontology.org/), and the number of miRNAs were calculated for the three ontologies detailed above. These differentially expressed miRNAs were then input into a list of significantly enriched GO terms by hypergeometric test. For pathway (biological function) analysis, the KEGG tool was applied to organize the enrichment analysis of differentially expressed miRNAs. Therefore, all the differentially expressed small RNAs were grouped into different metabolic pathways after comparison with the lotus genome.
qRT-PCR validation of differentially expressed miRNAs
In order to identify the results of tag-sequencing, models of miRNA expression at three periods were carried out using the qRT-PCR method. The method of RNA extraction and cDNA synthesis was the same as mentioned above. Mx 3000P (STRATAGENE, http://www.stratagene.com) was used to perform the qRT-PCR reaction, and mRNA levels were identified by SYBR Green Master Mix according to Mx instructions. Primers of miRNA were designed by primer 5.0 software, and are listed in Supplementary Additional Table S1. In this experiment, we used β-Actin as the reference gene, which had the following primers, up-stream: 5′-AACCTCCTCCTCATCGTACT-3′, and down-stream: 5′-GACAGCATCAGCCATGTTCA-3′. The mixture used for PCR amplification was 25 µl including 12.5 µl of 2 × Tli RNaseH Plus, 10 µM of primers (upstream and downstream), 2 µl of cDNA, and 8.5 µl of ddH2O. The PCR reaction program included 30 s at 94 °C, 5 s at 95 °C (40 cycles), and 60 s at 60 °C. The relative expression level of miRNAs was counted using the 2−△△Ct method. For the first step, △Ct value was counted in each sample when the Ct (target) and Ct (actin) value in treated plants (△Ct (target)) and control (△Ct (normal)) was obtained. For the second step, △△Ct value was calculated according to the result of △Ct (target) and △Ct (normal). At the last step, 2−△△Ct (relative expression level) was determined.
Results
Observations on the development of ARs in lotus
The morphological changes during the entire AR developmental process were identified under normal growth conditions (26 °C/day and 20 °C/night). ARs began to be induced from the cortex for about 5 days, and the number reached a maximum (about 20 ARs) within 10 days after seed germination (Fig. 1a). In addition, we also found that the principal root was not well developed since the principal root was not induced during the seedling stage. In order to further monitor AR formation, microstructure analysis was carried out using paraffin sections before ARs broke through the seed coat (within 5 days). It was shown that root primordium already existed even though the material was collected at 0 h after germination, suggesting that the ARs of lotus belong to latent primordial forms. The root primordium began to develop after 24 h, and several root primordia could be formed after 60 h of cultivation. ARs started to break through the seed coat at 84 h, which could be observed at the hypocotyl surface (Fig. 1b). Therefore, the three key time points of 0 day (C0 stage), 1 day (C1 stage), and 10 days (C2 stage) were used for further analysis of expression profiling of miRNAs during AR formation.

Developmental processes of lotus ARs. a ARs formation in hypocotyls (0 day, 2 days, 5 days and 10 days) of lotus under normal growth condition. b The observation of microstructure during ARs development by paraffin section technique
Identification of small RNAs during AR formation
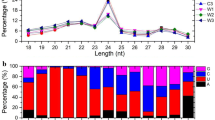
Plant growth and development was controlled by small RNAs by regulated mRNA expression. Therefore, three stages of ARs formation were selected to construct expressed libraries (C0 library, C1 library, and C2 library) with the aim to monitor post-transcriptional regulation of miRNAs involved in AR formation in lotus seedlings. In this study, a total of 12,338,713, 12,569,713, and 13,498,313 reads were achieved in the C0, C1, and C2 libraries respectively with 11,035,798 (90.75%), 11.436,062 (92.33%), and 12,542,392 (94.61%) clean reads in each library after screening out low quality reads (3′ adaptor null, insert null, 5′ adaptor null, small RNAs, smaller than 18 nt, and PolyA) (Table 1). It was elucidated that 4.26% and 13.43% of these clean reads were expressed specifically in the C0 and C1 stages in the C1/C0 libraries, and 16.77% and 9.57% of clean reads were specific to the C1 and C2 stages in the C2/C1 libraries, respectively. At the same time, we found that most of clean read lengths were distributed between 21 and 24 nt (Fig. 2a).

Detail information of miRNAs obtained by HiSeq deep sequencing technique. a Length distribution of tags in C0, C1 and C2 libraries. b Distribution of small RNA among different categories in C0, C1 and C2 libraries. c Nucleotide bias at each position of novel miRNA candidates in C0, C1 and C2 libraries
All the clean reads obtained were compared against the Rfam database, and we observed that most of clean reads (more than 45%) belonged to un-annotated small RNAs, and only ~ 5–10% of clean reads belonged to miRNAs in three libraries (Fig. 2b). Analysis of the nucleotide components for these miRNAs showed that the number of nucleotides in different cleavage sites of miRNA sequences was different in C0, C1, and C2 libraries. The number of nucleotide U at the 5′ end in C0 library (about 80%) was greater than that of the C1 library (about 30%) and C2 library (about 30%), and G at the 3′ end in the C1 library (about 80%) was greater than the C0 library (about 25%) and C2 library (about 30%). In addition, we also found that A was the main nucleotide from the ~ 8–11 cleavage site in the C0 library (Fig. 2c).
Differentially expressed miRNA in three libraries
Three libraries relevant to AR development were constructed and differentially expressed miRNAs were screened from these libraries to monitor metabolism changes regulated by miRNAs. It was shown that 18,416,132 (82.31%) reads had similar expression profiling from C0 stage to C1 stage, and 956,555 (4.26%) and 3,018,993 (13.43%) reads were only expressed in the C0 library and C1 library. From C1 stage to C2 stage, 17,661,660 had the same expression characteristic in both libraries, and 4,020,932 (16.77%) and 2,295,862 (9.57%) reads were specially expressed in the C2 stage and C1 stage (Additional Fig. 1).
At the same time, we observed that 310 miRNAs altered their transcriptional level in C1/C0 libraries, among which 240 were up-regulated (including 116 known miRNAs and 124 novel miRNAs) and 70 were down-regulated (including 20 known miRNAs and 50 novel miRNAs). From C1 stage to C2 stage, 291 miRNAs, including 140 up-regulated miRNAs (69 known miRNAs and 71 novel miRNAs) and 133 down-regulated miRNAs (48 known miRNAs and 75 novel miRNAs) altered the expression. From the C0 stage to the C2 stage, 353 miRNAs including 264 up-regulated and 89 down-regulated genes changed expression profiling (Fig. 3a; Table 2). We also found that some miRNAs exhibited different expression patterns, although most of miRNAs showed similar expression tendencies in C1/C0 libraries, C2/C1 libraries, and C2/C0 libraries. Twelve novel miRNAs (miRNA1876, miRNA1172.2, miRNA1205C, miRNA396F, miRNA3444a-5p, miRNA6284, miRNA845d, miRNA7739-5p, miRNA3946-3p, miRNA5595a, miRNA9492, and miRNA1876820a) and 39 known miRNAs were found to be specifically expressed in C1/C0 libraries and C2/C1 libraries, suggesting that metabolism was different in these two stages of AR development (Fig. 3b).

Expression profiling of miRNAs in C0 versus C1 libraries, C1 versus C2 libraries and C0 versus C2 libraries. a The number of differentially expression miRNAs in C1/C0 libraries, C2/C1 libraries and C2/C0 libraries. b The change of expression level of these differentially expression miRNAs in C1/C0 libraries, C2/C1 libraries and C2/C0 libraries
GO and KEGG analysis of the differentially expressed miRNAs
Differentially expressed known miRNAs and novel miRNAs involved in AR formation in lotus were analyzed by GO and KEGG. There were significant differences between miRNAs and novel miRNAs in C1/C0 libraries.
The largest number of differentially expressed known miRNAs was associated with plant hormone signal transduction, and was different for the novel miRNAs in the C1/C0 libraries (Fig. 4a). The expression of novel miRNAs participating in pyruvate metabolism, followed by MAPK signal pathway, were obviously changed (Fig. 4b).

KEGG analysis of differentially expressed miRNAs in C1/C0 libraries. a The number of differentially expressed known miRNAs involved in biochemical pathways and signal transduction pathways in C1/C0 libraries. b The number of differentially expressed novel miRNAs involved in biochemical pathways and signal transduction pathways in C1/C0 libraries
For KEGG analysis, all differentially expressed known and novel miRNAs were classified into three groups: biological process (nine and seven pathways of known and novel miRNAs), cellular component (eight and seven pathways of known and novel miRNAs) and molecular function (five and three pathways of known and novel miRNAs). It was elucidated that these differentially expressed known and novel miRNAs showed similar number tendencies in the three groups. The number of known and novel miRNAs involved in cellular process for biological process, cell part for cellular component, and binding or catalytic activity for molecular function were more than that of other pathways (Additional Figs. 2, 3).
Function analysis of the most folds changed miRNAs
We found that three groups contained the largest number of differentially expressed known and novel miRNAs, so these miRNAs were selected to monitor the alteration of metabolism involved in AR formation in lotus. In total, 25 miRNAs were found to change expression at the C1/C0 stages. Among them were 12 known miRNAs involved in plant hormone metabolism (auxin and brassinosteroid (BR) metabolism or response), and six (two known miRNAs and four novel miRNAs) and seven novel miRNAs were involved in MAPK signal pathway and pyruvate metabolism, respectively.
It was shown that seven (miR160a, miR393b-5p, miR160f-5p, miR171d-5p, miR162-3p, miR1026a, and miR171b-3p) and five (miR157a-5p, miR9748, miR2105, miR390a-5p, and miR9748) miRNAs were related to auxin metabolism and BR metabolism, respectively. Expression of some miRNAs participating in energy metabolism (novel_mir_257, novel_mir_1582, novel_mir_310, novel_mir_506, and novel_mir_24) was also found to be changed (Table 3).
The expression of miRNAs related to AR formation
The data obtained in this study were compared with these well-defined miRNAs with the aim to identify miRNA involved in AR formation in C1/C0 libraries (Fig. 5). We observed that 13 miRNAs were changed in expression, and ten miRNAs (including miR171d-5p, miR395a, miR396b-5p, novel_miR_407, novel_miR_141, novel_miR_24, novel_miR_340, miR160a, miR162-3p, novel_miR_133) enhanced expression, and three miRNAs (novel_miR_340, novel_miR_611, and novel_miR_499) decreased expression in C1/C0 libraries. Among these changed known miRNAs, miR396b-5p changed expression level the most (12 times), followed by miR160a. In addition, the most changed novel miRNA expression level was novel_miR_133, which showed about an eightfold enhancement in expression (Fig. 5a). The secondary structure of the above six novel miRNAs related with AR formation in lotus are shown in Fig. 5b.

miRNAs relevant to ARs formation. a The change of expression of known or novel miRNAs related with ARs formation. b The secondary structure of six novel miRNAs relevant to ARs formation
In this study, 16 miRNAs (miR397a, iR408-5p, miR398b, miR482d, miR8558b, miR168a-5p, miR1863, miR952b, miR157a-5p, miR171b-3p, miR162-3p, miR1026a, miR394b-3p, miR160f-5p, miR160a, and miR5077) were used to further analyze expression using the qRT-PCR method (Fig. 6). miRNA expression in the C0, C1, and C2 stages had a similar trend with those found in tag sequencing. Therefore, we believed that the data resulting from tag sequencing were a real reflection of miRNA regulation during AR development.

Expression profiling analysis of 16 different expressed miRNAs in germinated stage, induced stage and maximum stage during adventitious root development
miRNAs involved in IAA and BR metabolism
We found that the most changed of the miRNAs were involved in plant hormone metabolism, and most of these miRNAs were related with IAA and BR metabolism. A total of five miRNAs, which regulated TIR1 (miR393b-5p), ARF (miR160a and miR9748), GH3 (miR171d-5p), and SAUR (miR162-3p) biological processes were involved in the IAA pathway. In addition, five miRNAs, such as miR157a-5p, miR9748, miR2105, miR390a-5p, and miR393b-5p participated in the BR pathway (Fig. 7). Therefore, we believe that miRNAs regulate AR formation in lotus by affecting IAA and BR metabolism processes, which regulate downstream gene expression. However, how miRNAs affect these two biological processes is not certain, and further study should be carried out to testify it.

miRNAs involved in IAA and brassinosteroid metabolism. Red box represents the genes in the biological pathway which was regulated by miRNAs. (Color figure online)
Discussion
Data analysis for miRNA sequencing
For plants with undeveloped principal roots, ARs can function to increase water uptake and absorb nutrients in support of plant growth. In total, three developmental processes were included: AR induction stage, formation of ARs, and emergence of ARs from stem or hypocotyls [3, 5]. It was shown that this biological process is strictly regulated by many exogenous or intrinsic factors. Therefore, in this study, lotus seedlings from three stages (0 day: seed germinated, 1 day: ARs induced, 10 days: maximum number of ARs) were selected to monitor the changes in miRNAs expression during AR formation (Fig. 1a, b).
Based on Solexa technology, the RNA-seq technique has already been extensively used as an efficient approach to explore novel miRNAs and the regulation of plant growth and development at a transcriptional level by miRNA. In recent years, we have identified a change of gene mRNA levels and protein content during AR formation [15, 16]. In this study, three libraries (a C0 library, C1 library, and C2 library) were constructed using RNA-seq technology, and the RNA-seq technique was applied to analyze miRNA expression for different developmental stages with the aim of exploring miRNA regulation of AR formation. In a total, about 1.2 × 107 total reads were obtained in each library. These raw tags were then filtered with reference sequences to get clean tags (about 1.1 × 107, with more than 90% clean tag rate) (Table 1). In addition, some miRNAs which changed expression in the C1/C0 libraries and C2/C1 libraries were selected (Fig. 3; Table 2, Additional File 1). From the data obtained in this experiment, we found that there was a significant difference in expression of miRNA from seedling stage to induced AR stage (C1/C0 libraries), and from initial AR formation stage to maximum AR stage (C2/C1 libraries), suggesting that the mechanism of miRNA regulation in these different developmental stages might be different.
Plant hormone metabolism and response during AR formation
The formation of ARs is controlled by plant hormones, and IAA was believed to be a key factor affecting AR formation. The process of AR development is promoted when IAA content is improved or the activity of IAA synthesis-related enzymes decline. It is evidence that the effect of IAA on AR development mainly originates from the acceleration of cell division relevant to primordia formation [19, 20], which is very similar to another reports [21]. Aside from promotion of AR formation, another type of growth regulating substance (NAA) can also enhance the number of roots [22]. Further study shows that the effect of auxin on AR development occurs at the induction stage, because the response of the plant to auxin is more sensitive at this stage than other stages [23]. Many genes which are responsible for IAA metabolism are explored using the high throughput sequencing technique [24]. IAA transport is a critical component of the IAA regulated network, and is performed by two kinds of carrier (influx carriers and efflux carriers) [25]. PIN is believed to be an efflux carrier, and auxin, an influx carrier. Both kinds of carriers have been shown to exert great influence on lateral root development [26, 27]. There is evidence that Arabidopsis miR10515 can regulate SUR1, which encodes an indole glucosinolate biosynthetic enzyme that improves IAA content (KONG) [28]. Similarly, the miR393 family in rice can target the auxin signaling F BOX (OsTIR1 and OsAFB2), and directly affect crown root growth (BIAN) [29]. We found that miR160a, miR393b-5p, miR160f-5p, miR162-3p, and miR1026a related with auxin response factor and IAA metabolism altered expression from seedling stage to AR induction stage (Fig. 4; Table 3). Based on these data, IAA appears to be a key plant hormone in regulating AR formation in lotus.
It is reported that AR formation is possibly regulated by brassinosteroid. Nakamura et al. report that BR-insensitive mutants had more ARs and shorter root length than those of a normal plant. Other experiments demonstrate that BR may participate in cell differentiation, which leads to AR formation [30]. At the same time, transgenic OsCKI1, which is induced by BR, causes plants to have fewer adventitious roots [31], also suggesting that BR is involved in AR development. New evidence shows that a crosstalk exits between auxin and BR to control plant growth. Auxin metabolism is changed after accumulation of BR, and different amount of auxin in BR mutants has an influence on plant phenotypes [32]. In our experiment, we found that several miRNAs (miR157a-5p, miR9748, and miR2105) were involved in regulating protein brassinosteroid insensitive 1, and an miRNA (miR9748) involved in regulating increased brassinosteroid resistant 1 expression. Therefore, based on previous studies, we believe BR might negatively affect AR formation in lotus. Three members (MAPKKK, MAPKK, and MAPK) are included in the mitogen-activated protein kinase family, and these proteins are involved in many biological processes in plants. It is reported that mitogen-activated protein kinase is involved in various biological processes by affecting ethylene biosynthesis [33], which is relevant to AR formation [23]. New evidence shows that MPK6 is required for the inhibition of root growth through its effect on H2S toxicity [34]. In addition, MAPK, which is possibly activated by NO, is thought to be involved in the auxin-signaling pathway that regulates AR formation [35]. In our experiment, the expression of several miRNAs, such as miR5077, novel_mir_1646, novel_mir_133, miR394b-3p, novel_mir_310, and novel_mir_340, was changed in C1/C0 libraries. However, among these miRNAs, miR394b-3p, novel_mir_310, and novel_mir_340 showed decreased expression, while miR5077, novel_mir_1646, and novel_mir_133 showed increased expression (Table 3, Additional File 1), suggesting that different miRNAs have different functions during AR formation in lotus.
miRNA related to anaerobic adaptation and energy metabolism
Some plants have special adaptations (e.g., micro-structural, physiological or biochemical, or molecular) to accommodate anaerobic condition during growth, otherwise emergence conditions could lead to disaster for their productivity or food quality [36]. Aquatic plants show more resistance to flooding conditions than land vegetables partially because of different expression of some genes [37]. Alcohol dehydrogenase, often considered as key enzyme in adaptation to flooding conditions, shows different activity in different types of plants [38]. In this study, we found that several miRNAs related with alcohol dehydrogenase metabolism showed increased abundance during AR formation (Table 3), suggesting that alcohol dehydrogenase was also necessary for lotus growth.
We found that miRNAs relevant to H+-transporting ATPase (miR9552b-3p), Ca2 + transporting ATPase (miR9748), malate dehydrogenase (novel_mir_24), glutamate synthase (novel_mir_333), and S-glutathione dehydrogenase (NADPH/NADH) (novel_mir_506) had improved expression in C0/C1 libraries (Table 3; Fig. 5), suggesting that energy metabolism is needed for AR formation. Energy metabolism is an undoubtedly an important biological process which can provide energy for various activities of the plant [39,40,41]. NADPH/NADH and ATP are generated by biochemical reaction in plants, and their delivery is necessary for energy metabolism. NADPH/NADH and ATP are often transported to vigorous growth organs by malate/oxaloacetate shuttles. Alteration of energy metabolism is often achieved through malate valves, which can be derived from a change of mRNA levels in plants [42,43,44,45]. Malate dehydrogenase plays an essential role in malate valves, and is also involved in energy metabolism in response to stress conditions. The expression of MDH (L8940) was enhanced in C0/C1 libraries. New evidence shows that AR formation is related to energy metabolism. CsARN6.1, an AAA ATPase, is involved in AR formation in cucumber [46]. Therefore, these data show that energy metabolism may be a required process for lotus to form ARs and adapt to submergence stress.
In addithon, 16 genes for qRT-PCR analysis in this study were selected to identify the data accuracy obtained by RNA-seq technique (Fig. 6). Some of these selected miRNAs for qRT-PCR were relevant to plant hormone signal transduction, and some were randomly selected from these different expressed miRNAs in C1/C0 libraries. We found that the result of qRT-PCR for these selected miRNAs were completely identical in expressing tendency with that of high throughput sequencing technology. Therefore, the next step of following experiment was to analysis the role of some important miRNAs including miR396b-5p, miR160a and novel_mir_133 during formation of ARs in lotus.
Conclusion
In this study, analysis of miRNA expression involved in ARs formation was carried out to study miRNA regulation. About 1.3 × 107 tags were obtained including 11035798, 11436062 and 12542392 clean tags in C0, C1 and C2 libraries respectively. We found that 310 miRNAs were changed expression in C1/C0 libraries including 240 up-regulated miRNAs and 70 miRNAs down-regulated miRNAs. At the same time, 140 miRNAs were enhanced expression and 123 miRNAs were decreased expression in C2/C1 libraries. It was shown that metabolism processes, of which the differentially expressed miRNAs participated, were different between C1/C0 libraries and C2/C1 libraries. A total of 13 miRNAs relevant to ARs formation were changed in expression in C1/C0 libraries including 10 up-regulated miRNAs and three down-regulated miRNAs.
References
Shen-Miller J (2002) Sacred lotus, the long-living fruits of China Antique. Seed Sci Res 12:131–143
Ming R, Van Buren R, Liu YL, Yang M, Han YP, Li LT et al (2013) Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol 14:R41
Kevers C, Hausman JF, Faivre-Rampant O, Evers D, Gaspar T (1997) Hormonal control of adventitious rooting: progress and questions. J Appl Bot Angew Bot 71:71–79
Da Costa CT, de Almeida MR, Ruedell CM, Schwambach J, Maraschin FS, Fett-Neto AG (2012) When stress and development go hand in hand: main hormonal controls of adventitious rooting in cuttings. Front Plant Sci 4:133
Li SW, Xue L, Xu S, Feng H, An L (2009) Mediators, genes and signaling in dventitious rooting. Bot Rev 75:230–247
Brodersen P, Voinnet (2009) Revisiting the principles of microRNA target recognition and mode of action. Nature 10:141–148
Mathieu J, Yant LJ, Murdter F, Kuttner F, Schmid M (2009) Repression of flowering by the miR172 target SMZ. PLoS Biol 7:e1000148
Sun P, Zhang ZL, Zhu QF, Zhang GY, Xiang P, Lin YL, Lai ZX, Lin JK (2018) Identification of miRNAs and target genes regulating catechin biosynthesis in tea (Camellia sinensis). J Integ Agri 17:1154–1164
Hussain K, Mungikar K, Kulkarni A, Kamble A (2018) Identification, characterization and expression analysis of pigeonpea miRNAs in response. Gene 653:57–64
Zhang XQ, Li KC, Xing RG, Liu Song, Chen XL, Yang HY, Li PC (2018) miRNA and mRNA expression profiles reveal insight into chitosan mediated regulation of plant growth. J Agric Food Chem 66:3810–3822
Chen JL, Zheng Y, Qin L, Wang Y, Chen LF, He YJ, Fei ZJ, Lu G (2016) Identification of miRNAs and their targets through high throughput sequencing and degradome analysis in male and female Asparagus officinalis. BMC Plant Biol 16:80
Gleeson M, Constantin M, Carroll BJ, Mitter N (2014) MicroRNAs as regulators of adventitious root development. J Plant Biochem Biotechnol 23:339–347
Levy A, Szwerdszarf D, Abu-Abied M, Mordehaev I, Yaniv Y, Riov J, Arazi T, Sadot E (2014) Profiling microRNAs in Eucalyptus grandis reveals no mutual relationship between alterations in miR156 and miR172 expression and adventitious root induction during development. BMC Genom 15:524
Cheng LB, Jiang RZ, Yang ML, Li LJ, Li SY (2017) A comparative proteomic analysis for adventitious root formation in lotus root (Nelumbo nucifera Gaertn). ZNC 72:181–196
Cheng LB, Jiang RZ, Yang JJ, Xu XY, Zeng HT, Li SY (2018) Transcriptome rofiling reveals an IAA-regulated response to adventitious root formation in lotus seedling. ZNC 73:229–240
Cheng LB, Liu HY, Jiang RZ, Li SY (2018) A proteomics analysis of adventitious root formation after leaf removal in lotus (Nelumbo nucifer Gaertn.). ZNC. https://doi.org/10.1515/znc-2018-0011
Fang YN, Zheng BB, Yang W, Wu XM, Xu Q, Guo WW (2016) High-throughput sequencing and degradome analysis reveal altered expression of miRNAs and their targets in a male-sterile cybrid pummelo Citrus grandis. BMC Genom 17:591
Luan YS, Cui J, Zhai JM, Li J, Han L, Meng J (2015) High-throughput sequencing reveals differential expression of miRNAs in tomato inoculated with Phytophthora infestans. Planta 241:1405–1416
Strader LC, Chen GL, Bartel B (2010) Ethylene directs auxin to control root cell expansion. Plant J 64:874–884
Kohli A, Sreenivasulu N, Lakshmanan P, Kumar PP (2013) The phytohormone crosstalk paradigm takes center stage in understanding how plants respond to abiotic stresses. Plant Cell Rep 32:945–957
Leyser O (2001) Auxin signalling: the beginning, the middle and the end. Curr Opin Plant Biol 4:382–386
Yan YH, Li JL, Zhang XQ, Yang WY, Wan Y, Ma YM, Zhu YQ, Peng Y, Huang LK (2014) Effect of naphthalene acetic acid on adventitious root development And associated physiological changes in stem cutting of hemarthria compressa. PLoS ONE 9:e90700
Druege U, Franken P, Lischewski S, Ahkami AH, Zerche S, Hause B, Hajirezaei M (2014) Transcriptomic analysis reveals ethylene asstimulator and auxin as Regulator of adventitious root formation in petunia cuttings. Front Plant SCI 5:494
Sieberer T, Leyser O (2006) Plant science––auxin transport, but in which direction? Science 312:858–860
Sauer M, Balla J, Luschnig C, Wisniewska J, Reinöhl V, Friml J (2006) Canalization of auxin flow by Aux/IAA-ARF-dependent feedback regulation of PIN polarity. Genes Dev 20:2902–2911
Xu M, Zhu L, Shou HX, WuP (2005) A PIN1 Family Gene, OsPIN1, involved in auxin-dependent adventitious root emergence and tillering in rice. Plant Cell Physiol 46:1674–1681
Marchant A, Bhalerao R, Casimiro I, Eklöf J, Casero PJ, Bennett M, Sandberg G (2002) AUX1 promotes lateral root formation by facilitating indole-3-acetic acid distribution between sink and source tissues in the Arabidopsis seedling. Plant Cell 14:589–597
Kong WW, Li Y, Zhang MM, Jin F, Li J (2014) A novel Arabidopsis microRNA promotes IAA biosynthesis via the indole-3-acetaldoxime pathway by suppressing SUPERROOT1. Plant Cell Physiol 56:715–726
Bian HW, Xie YK, Guo F, Han N, Ma SY, Zeng ZH, Wang JH, Yang YN, Zhu MY (2012) Distinctive expression patterns and roles of the miRNA393/TIR1 homolog module in regulating flag leaf inclination and primary and crown root growth in rice (Oryza sativa). New Phytol 196:149–161
Nakamura A, Fujioka S, Sunohara H, Kamiya N, Hong Z, Inukai Y, Miura K, Takatsuto S, Yoshida S Ueguchi-Tanaka M, Hasegawa Y, Kitano H, Matsuoka M (2006) The role of OsBRI1 and its homologous genes, OsBRL1 and OsBRL3, in rice. Plant Physiol 140: 580–590
Liu W, Xu ZH, Luo D, Xue HW (2003) Roles of OsCKI1, a rice casein kinase I, in root development and plant hormone sensitivity. Plant J 36:189–202
Maharjan PM, Dilkes BP, Fuka S, Fujiokajio S, Pěník A, Ljung K, Burow M, Halkier BA, Choe S (2014) Arabidopsis gulliver1/SUPERROOT2-7 identifies a metabolic basis for auxin and brassinosteroid synergy. Plant J 80:797–808
Li GJ, Meng XZ, Wang G, Mao GH, Han L, Liu YD, Zhang Q (2012) Dual-level regulation of ACC synthase activity by MPK3/MPK6 cascade and it downstream WRKY transcription factor during ethylene induction in Arabidopsis. PLoS Genet 8:e1002767
Zhang P, Luo Q, Wang RL, Xu J (2017) Hydrogen sulfide toxicity inhibits primary root growth through the ROS-NO pathway. Scientific Rep 7:868
Pagnussat GC, Lanteri ML, Lombardo MC, Lamattina L (2004) Nitric oxide mediates the indole acetic acid induction activation of a mitogen activated protein kinase cascade involved in adventitious root development. Plant Physiol 135:279–286
Vartapetian BB, Jackson MB (1997) Plant adaptations to anaerobic stress. Ann Bot 79:3–30
Ishizawa A, Murakami S, Kawakami Y, Kuramochi H (1999) Growth and energy status of arrowhead tubers, pondweed turions and rice seeding under anoxic conditions. Plant Cell Environ 22:505–514
Jung SH, Lee JY, Lee DH (2003) Use of SAGE technology to reveal changes in gene expression in Arabidopsis leaves undergoing cold stress. Plant Mol Biol 52:553–567
Geigenberger P, Stitt M, Fernie AR (2004) Metabolic control analysis and regulation of the conversion of sucrose to starch in growing potato tubers. Plant Cell Environ 27:655–673
Gietl C (1992) Malate dehydrogenase isoenzymes: cellular locations and role in the flow of metabolites between the cytoplasm and cell organelles. Biochem Biophys Acta 1100:217–234
Scheibe R (2004) Malate valves to balance cellular energy supply. Physiol Plant 120:21–26
Hameister S, Becker B, Holtgreffe S, Strodtkotter I, Linke V, Backhausen JE, Scheibe R (2007) Transcriptional regulation of the NADP dependent malate dehydrogenase: comparative genetics and identification of DNA-binding proteins. J Mol Evol 65:437–455
Becker B, Holtgrefe S, Jung S, Wunrau C, Kandlbinder A, Baier M, Dietz KJ, Backhausen JE, Scheibe R (2006) Influence of the photoperiod on redox regulation and stress responses in Arabidopsis thaliana L. (Heynh.) plants under long- and short-day conditions. Planta 224:380–393
Cushman JC (1993) Molecular cloning and expression of chloroplast NADP-malate dehydrogenase during Crassulacean acid metabolism induction by salt stress. Photosynth Res 35:15–27
Sahi C, Singh A, Kumar K, Blumwald E, Grover A (2006) Salt stress response in rice: genetics, molecular bilogy, and comparative genomics. Func Integr Genom 6:263–284
Xu XW, Ji J, Xu Q, Qi XH, Weng YQ, Chen XH (2018) The major-effect quantitative trait locus CsARN6.1 encodes an AAA ATPase domain-containing protein that is associated with waterlogging stress tolerance by promoting adventitious root formation. Plant J 93:917–930
Acknowledgements
This work was supported by Jiangsu Agriculture Science and Technology Innovation Fund (Grant No. CX (18) 3066), and the modern agriculture of Yangzhou (Grant No. YZ2017044). We extend our thanks to some members of BIG for their cooperation in obtaining the data during ARs formation of the lotus by RNA-seq technique. The authors also thank Editage Ltd for their editorial assistance.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2019_4749_MOESM4_ESM.xls
Additional File 1 All differentially expressed known and novel miRNAs in C1/C0 libraries and C2/C1 libraries (XLS 155 KB)
Rights and permissions
About this article

Cite this article
Libao, C., Huiying, L., Yuyan, H. et al. Transcriptome analysis of miRNAs expression reveals novel insights into adventitious root formation in lotus (Nelumbo nucifera Gaertn.). Mol Biol Rep 46, 2893–2905 (2019). https://doi.org/10.1007/s11033-019-04749-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-04749-z