
Abstract
The gene lpmo1 encoding Penicillium verruculosum lytic polysaccharide monooxygenase (PvLPMO9A) was sequenced and homologously overexpressed in P. verruculosum B1-537 (ΔniaD) auxotrophic strain under the control of the cbh1 gene promoter in combination with either the cbh1 signal sequence (sCBH1-X series of samples) or the native lpmo1 signal sequence (sLPMO1-X series). Three enzyme samples of the sCBH1-X series were characterized by a lower overall content of cellobiohydrolases (CBHs: 26–45%) but slightly higher content of endoglucanases (EGs: 17–23%) relative to the reference B1-537 preparation (60% of CBHs and 14% of EGs), while the PvLPMO9A content in them made up 9–21% of the total secreted protein. The PvLPMO9A content in four enzyme preparations of the sLPMO1-X series was much higher (30–57%), however the portion of CBHs in most of them (except for sLPMO1-8) decreased even to a greater extent (to 21–42%) than in the samples of the sCBH1-X series. Two enzyme preparations (sCBH1-8 and sLPMO1-8), in which the content of cellulases was substantially retained and the portion of PvLPMO9A was 9–30%, demonstrated the increased yields of reducing sugars in 48-h saccharification of Avicel and milled aspen wood: 19–31 and 11–26%, respectively, compared to the reference cellulase cocktail.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The biotechnological production of second-generation biofuels from renewable lignocellulosic biomass has been implemented on an industrial scale in the last few years in Italy, USA and Brazil [1, 2]. The most laborious stage of this technology is enzymatic saccharification of cellulose, which is catalyzed by a multienzyme system of hydrolytic cellulases, including endo-1,4-β-glucanases (EGs), exo-cellobiohydrolases (CBHs) and β-glucosidases (BGLs) [3]. Rather recently, metal-dependent chitin- and cellulose-oxidizing enzymes, called lytic polysaccharide monooxygenases (LPMOs), have been discovered [4,5,6]. LPMOs act as auxiliary activities (AA) to hydrolytic cellulases, enhancing the rate of cellulose destruction and boosting the yield of sugars in the enzymatic reaction [7,8,9,10,11]. For this reason, LPMOs have been introduced into modern commercial cellulase preparations produced by leading enzyme manufacturing companies [12].
Penicillium verruculosum is an efficient producer of highly-active extracellular cellulase multienzyme cocktails [13, 14]. LPMOs from Thielavia terrestris and Trichoderma reesei have previously been cloned and heterologously expressed in P. verruculosum in order to enhance the hydrolytic performance of its cellulases [9, 15]. At the same time, our earlier attempts to isolate a native LPMO from this fungus failed, although some experimental data indicated the presence of a minor LPMO activity in P. verruculosum multienzyme cocktails [9]. It should be noted that none of LPMOs from Penicillium species has been characterized up date, although the genes and translated amino acid sequences of one LPMO from P. oxalicum and four LPMOs from P. rubens are present in the CAZy database (http://www.cazy.org/AA9_all.html).
In this paper, a gene encoding the P. verruculosum LPMO I (PvLPMO9A) was sequenced and homologously overexpressed in this fungal host using different constructions of the expression plasmids, and the performance of the secreted crude multienzyme preparations in saccharification of cellulosic materials was investigated.
Materials and methods
Microorganisms and the growth conditions
Penicillium verruculosum B1-537 strain [16, 17] was used as a host strain for transformation and as a source for genomic DNA preparation. This host strain was deficient by the nitrate reductase gene (ΔniaD) for auxotrophic selection of recombinants. The Escherichia coli MachI strain (Thermo Fisher Scientific Inc., Waltman, MA, USA) was used for bacterial transformation and isolation of new plasmid constructs containing target genes.
The medium for screening of fungal clones contained 40 g/l cellulose, 15 g/l KH2PO4, 10 g/l of wheat bran (pH 5.0).
Cloning of the lpmo1 gene
The nucleotide sequence corresponding to the full-length lpmo1 gene was amplified by polymerase chain reaction (PCR). In order to amplify the lpmo1 gene, two pairs of oligonucleotide primers (CBH1-LPMO1-LIC5/CBH1-LPMO1-LIC3 and LPMO1-LIC5/LPMO1-LIC3) were designed:
- CBH1-LPMO1-LIC5:
-
GGCAACAGCAGGAGCTCATGGTTTTGTGCAAAACATCATTATTGAC
- CBH1-LPMO1-LIC3:
-
AGAGGAGGGCGACACAGTTAAAAGACAGTAGTGGTGATGACGGTAGTC
- LPMO1-LIC5:
-
CAAACAGAAGCAACCGACACAATGCCTTCTACTAAAGTCGCTGCCCT
- LPMO1-LIC3:
-
GAGGAGAAGCCCGGTTAAAAGACAGTAGTGGTGATGACGGTAGTC
The PCR was carried out using a freshly prepared genomic DNA of the fungus P. verruculosum (Dneasy® Plant Mini Kit, QIAGEN, Valencia, CA, USA), the derived primers, Long polymerase mix (LPM, Thermo Fisher Scientific Inc., Waltham, MA, USA) and MyCycler equipment (Bio-Rad Laboratories, Hercules, CA, USA) according to the following protocol: 3 min at 95 °C, followed by 25 cycles of 1.5 min at 95 °C, 1 min at 50 °C, 2 min at 68 °C, 10 min at 4 °C. The resulting PCR product was purified from agarose gel using QiAquick Gel Extraction Kit (QIAGEN, Valencia, CA, USA). The PCR products were sequenced, and identified as the lpmo1 gene of P. verruculosum.
Construction of expression plasmids and generation of mutants
A full lpmo1 gene (1040 bp) encoding the PvLPMO9A (LPMO I) was cloned into a modified pUC19 vector, pCBHI, containing the promoter and terminator regions of the cbh1 gene of P. verruculosum, by Ligation Independent Cloning method (LIC-method) as previously described [17, 18]. As an alternative, the polynucleotide sequence of the lpmo1 gene without a signal sequence (977 bp) was cloned into the vector pCBHI-SS, that is, pCBHI [17] with an additive sequence encoding the CBH I signal sequence (SS) fused to the 3′-end of the cbh1 promoter region (see Fig. 1).

Construction of pCBHI and pCBHI-SS vectors for cloning the lpmo1 gene into P. verruculosum B1-537 recipient strain under the control of the cbh1 gene promoter in combination with either the native lpmo1 signal sequence (a) or the cbh1 signal sequence (b)
The PCR-products (1040 bp or 977 bp) and linearized pCBHI or pCBHI-SS vectors were treated with T4 DNA polymerase (Thermo Fisher Scientific Inc., Waltman, MA, USA) in the presence dATP and dTTP (Thermo Fisher Scientific Inc., Waltman, MA, USA), respectively. The treated inserts were ligated into the treated vectors by mixing 50 ng of vector with 150 ng of insert. The mixture was incubated for 30 min at 22 °C and then transformed into E. coli MachI competent cells using a standard transformation protocol [19]. Thus, plasmids pLPMOI and pCBHI-LPMOI, containing the complete lpmo1 gene and lpmo1 gene fused to the cbh1 signal sequence were obtained. The absence of mutations, additional insertions or deletions in the cassettes containing the lpmo1 gene was confirmed by sequencing in both directions by the method described by Sanger et al. [20].
Obtaining enzyme preparations
The P. verruculosum B1-537 (ΔniaD) host strain was co-transformed with the obtained pLPMOI or pCBHI-LPMOI plasmid together with pSTA10 plasmid taken at a standard ratio (10:1, µg), using the transformation protocol described elsewhere [21]. The pSTA10 plasmid contained a nitrate reductase gene, allowing carrying out the selection of transformants in the medium with 10 mM NaNO3.
Screening of transformants was carried out in shake flasks (total volume 500 ml, fermentation volume 100 ml). The P. verruculosum B1-537 (ΔniaD) host strain was used as a control. As a result of primary screening, several recombinant P. verruculosum strains of the sCBH1-X and sLPMO1-X series secreting the PvLPMO9A, visible as a remarkable band on the SDS-PAGE gel, were selected. The enzyme identification in the protein bands was carried out by MALDI-TOF mass spectrometry (MS) peptide fingerprinting using the online FindPept tool (https://web.expasy.org/findpept/) as described elsewhere [13, 22].
Supernatants of the culture filtrates were freeze-dried on a VirTis BenchTop 2K ES freeze dryer (SP Scientific,Warminster, PA, USA).
Enzyme activity assays
Enzyme activities against carboxymethylcellulose (CMC) and birchwood xylan (Sigma, St. Louis, MO, USA) were determined by analyzing reducing sugars (RS) released from 5 g/l substrates after 5 min (CMC) or 10 min (xylan) of the enzymatic reaction at pH 5.0 and 50 °C as described elsewhere [23]. Avicelase activity was assayed by analyzing RS released after 60 min of the enzymatic reaction with 5 g/l Avicel (microcrystalline cellulose from Vitek Company, Russia) at pH 5.0 and 40 °C as described elsewhere [24]. Reducing sugars were analyzed by the Nelson-Somogyi method [25]. The standard calibration plot was made using different concentrations of glucose (Helicon, Russia) in the range of 0.01–0.2 g/l. The enzyme quantity that catalyzed the formation of 1 µmol RS (in glucose equivalents) per 1 min was defined as the enzyme activity unit. Protein concentration was determined by the modified Lowry method [26].
Analysis of the enzyme component composition
Analysis of the component composition of multienzyme cocktails produced by recombinant P. verruculosum strains overexpressing PvLPMO9A was carried out essentially as described elsewhere [16, 27]. In brief, HPLC fractionation of enzymes was carried out on a FPLC Pharmacia system using Source 15Q and MonoQ HR 5/5 columns (Pharmacia, Uppsala, Sweden). Each protein fraction was analyzed for different enzyme activities and protein content using the Lowry assay [26], as described in the preceding section, and it was also characterized by SDS-PAGE in a 12% gel using a Mini Protein Cell system (Bio-Rad Laboratories, Hercules, CA, USA).The enzyme components in fractions were identified based on the obtained activities and MALDI-TOF MS peptide fingerprinting of the corresponding protein bands from a gel [13, 22]. Identified proteins having purity higher than 90% in the respective fractions, according to the SDS-PAGE data, were considered as individual enzymes, and the content of the corresponding enzymes in the initial crude multienzyme preparation was calculated as the ratio of the protein content in those fractions to the total protein content in the initial preparation. Some protein fractions contained more than one enzyme (purity less than 90%) identified by its substrate specificity and MALDI-TOF MS. In such case, we first estimated the enzyme share in a fraction based on the intensity of its band on the electrophoretic gel, and then, knowing the protein content in this fraction and the enzyme share, its content in the initial preparation was calculated.
Cellulose saccharification
Avicel (Vitek Company, Russia) and aspen wood, pretreated by milling on an impeller mill Mikrosilema IM-450 (Monolitstroy, Russia) as described elsewhere [28], were treated in 2-ml test tubes at pH 5.0 and 40 °C by composite multienzyme cocktails containing 85% of the enzyme preparation under study and 15% of F10 preparation as a source of BGL as described elsewhere [9]. The F10 preparation contained the Aspergillus niger BGL (~ 80% of the total protein) heterologously expressed in P. verruculosum B1-537 host strain, while the rest of proteins (~ 20%) represented cellulases of P. verruculosum [16, 29]. In all cases, the substrate concentration was 100 g/l, and the total protein dosage in the reaction mixture was 5 mg/g substrate. In Avicel saccharification experiments, 5 mM gallic acid was added to the reaction system as an electron donor to LPMO. After 6, 24 and 48 h of the enzymatic reaction, 0.1-ml aliquots of reaction mixture were taken, centrifuged, and then the RS concentration was determined in the supernatant by the Nelson-Somogyi method [25]. The experiments were carried out in triplicate.
Results and discussion
PvLPMO9A expression
The lpmo1 gene encoding LPMO I was sequenced and homologously overexpressed in P. verruculosum B1-537 auxotrophic strain using two constructions of the expression plasmids, one of which used the cbh1 gene promoter and the full-sized lpmo1 gene and the other used a native cbh1 signal sequence instead of the lpmo1 signal sequence. The native cbh1 signal sequence was fused directly upstream the lpmo1 gene encoding a mature LPMO I (PvLPMO9A) protein (Fig. 1).
The lpmo1 gene consists of 1040 bp and contains one intron. The nucleotide sequence of the gene was deposited in the GenBank with an accession number MK158950. A translated amino acid sequence contains 328 residues. The first 21 residues (MPSTKVAALSAVLALASTVAG) represent a signal peptide; the N-terminal sequence of the mature protein starts from a histidine residue (HGYVQN...), like in other LPMOs [6, 10]. The alternative signal peptide sequence from CBH I, used for the expression of PvLPMO9A (Fig. 1), was MSALNSFNMYKSALILGSLLATAGA [30].
Three clones of the sCBH1-X series (sCBH1-8, sCBH1-14, sCBH1-17) and four clones of the sLPMO1-X series (sLPMO1-1, sLPMO1-4, sLPMO1-8, sLPMO1-9) of fungal transformants were selected for further analysis and testing (sCBH1- and sLPMO1—denote the respective signal sequences used in the expression plasmids). SDS-PAGE data for the respective multienzyme preparations are shown in Fig. 2. Bands on the SDS-PAGE image marked by an arrow correspond to the secreted PvLPMO9A, as MALDI-TOF MS peptide fingerprinting showed. The MS analysis also confirmed the N-terminal peptide sequence of the mature PvLMPO9A for both constructions of the expression plasmids. Peaks in the mass spectra of the enzyme tryptic digests with m/z of 6393 and 6464 matched the specific tryptic peptide 1 HGY...CHK 58 of the mature protein; the second mass corresponded to the peptide modified by acrylamide at Cys56 residue, a typical CYS_PAM modification in such kind of MALDI-TOF MS analysis. Moreover, the observed peaks with m/z 6407 and 6478, differed by 14 Da from the above mentioned peaks, matched the same N-terminal peptide (without and with CYS_PAM modification), in which the N-terminal histidine residue is methylated. So, both recombinant forms of PvLPMO9A expressed with different signal peptides were methylated incompletely. The methylation of the N-terminal histidine that coordinates the copper ion in the enzyme active site is rather typical for cellulose-active LPMOs, however the role of this modification is unclear [6, 10]. It should be noted that we have previously observed similar partial His1 methylation in the case of recombinant LPMO from T. terrestris, also expressed in P. verruculosum [9], while the native LPMO from Myceliophthora thermophila studied in the same work was found to exist only in the methylated form, since the N-terminal peptide with unmodified His1 residue was not observed in the mass spectrum of the enzyme tryptic digest. Thus, incomplete methylation of N-terminal histidines in recombinant LPMOs, homologously or heterologously expressed by P. verruculosum, seems to be a typical feature of this fungal host.

SDS-PAGE of crude enzyme preparations based on recombinant strains of P. verruculosum overexpressing the lpmo1 gene. M molecular markers (shown in kDa); 1 B1-537 (control); 2 sCBH1-8; 3 sCBH1-14; 4 sCBH1-17; 5 sLPMO1-1, 6 sLPMO1-4; 7 sLPMO1-8; 8 sLPMO1-9
The specific activities of the enzyme preparations under study against cellulosic substrates (Avicel, CMC) and xylan are shown in Table 1. Using a previously developed method for assaying the component composition of cellulase multienzyme cocktails [16, 27], the content of major enzymes was estimated in each sample (Table 2). It should be noted that CBH I, CBH II and EG I of P. verruculosum are usually found in a culture liquid in two forms, a high-molecular-weight and low-molecular-weight form, the second one (without a cellulose-binding module) being formed as a result of limited proteolysis of the enzyme linker [13]. For simplicity, the total content of both forms for each of the mentioned enzymes is given in Table 2.
The enzyme preparations based on sCBH1-X series of transformants were generally characterized by lower overall content of CBHs (26–45%) but slightly higher content of EGs (17–23%) relative to the control B1-537 sample, in which CBHs and EGs represented 60 and 14% of the total secreted protein, respectively (Table 2). For sLPMO1-X series, the content of CBHs decreased even to a greater extent (to 21–42%), while the content of EGs (7–14%) was closer to that in the control preparation. The content of BGL and Xyl I for both series of transformants also varied compared to the reference B1-537 sample. The specific activities of the preparations against Avicel, CMC and xylan (Table 1) fairly well correlated with a total content of CBHs, EGs and Xyl I (Table 2) in them; the coefficient of determination R2, calculated with the assumption of a linear dependence between the parameters, was found to be 0.975, 0.779 and 0.955, respectively. The lower value of R2 for the pair “CMCase activity—total content of EGs” was the result of different specific activities of the four major endoglucanases [13] and their different shares in the P. verruculosum multienzyme cocktails (Table 2).
As regarding to the PvLPMO9A content in the samples, it was much higher in the sLPMO1-X series of the enzyme preparations (30–57%) compared to the sCBH1-X series (9–21%); this is also clearly visible on the electrophoregrams (Fig. 2). Thus, using a native signal sequence of the lpmo1 gene for the expression of PvLPMO9A has proven to be more effective than using the cbh1 signal sequence. It should be noted that P. canescens xylanase A, P. verruculosum EG IIa and P. funiculosum dextranase have previously been expressed in P. verruculosum B1-537 strain under the control of a strong cbh1 gene promoter using the native signal sequences of the enzymes mentioned; the secretion of the target proteins reached 31–36% of the total protein in culture broths [17, 31, 32]. However, Gouka et al. [33] reported that using the glaA signal sequence for the expression of foreign proteins in Aspergillus species under the control of the Aspergillus glaA gene promoter provides better results than using the native signal sequences of those proteins because of their improved translocation and folding. Therefore, in this work we used both possible variants of PvLPMO9A expression, that is, with and without a native signal sequence of the lpmo1 gene, in the latter case replacing it by a signal sequence of the cbh1 gene. Regarding the influence of the signal peptide structure on the expression of other LPMOs, systematic studies on this subject are rather scarce. The codon-optimized native signal sequence of T. reesei LPMO9A (Cel61A) was found to provide better secretion of this enzyme by Pichia pastoris than the more commonly used signal sequence of the alpha-mating factor (α-MF) from Saccharomyces cerevisiae [34]. In another work, twelve signal peptides with reported high secretion efficiency were screened to assess the extracellular production of the chitin-active LPMO10A (CBP21) from Serratia marcescens in E. coli [35]. Only four signal peptides, including those from PelB, CBHI, SacB and XCs, increased the protein production level of CBP21, the PelB being the most efficient, while two another signal peptides improved the release of the recombinant chitinase Chi92 from Aeromonas veronii [35]. The reported data confirm a paradigm that a suitable signal peptide for one protein may not be efficient for another protein [36].
Enzymatic saccharification of cellulosic substrates
Avicel and milled aspen wood were hydrolyzed by the enzyme preparations under study with the addition of F10 preparation enriched by a BGL activity [16, 29]. The ratio between cellulase samples and F10 preparation was 85:15 by protein, as that optimized in our previous studies [16, 37], while maintaining the total protein dosage in the reaction system at the level of 5 mg/g substrate.
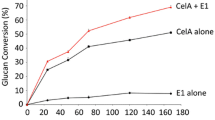
The results of Avicel hydrolysis are shown in Fig. 3. In the presence of 5 mM gallic acid as an electron donor to PvLPMO9A, the enzyme preparations sCBH1-8 and sLPMO1-8 demonstrated superior performance over the control B1-537 sample at each time point, the RS concentration after 48 h being higher by 19 and 31%, respectively, relative to the control. The sCBH1-14 sample exhibited the performance close to that of the reference sample only in the first period of the reaction (6 and 24 h), while the final RS yield was lower than in the control. The rest of samples containing homologously overexpressed PvLPMO9A could not compete with the reference B1-537 multienzyme cocktail by the saccharification efficiency. To identify the role of LPMO activity in improving the efficiency of cellulose saccharification, the hydrolysis of Avicel was also carried out by the sLPMO1-8 preparation in the absence of gallic acid. The removal of the electron donor led to a decrease in the final RS concentration by 19% (see bars above the sLPMO1-8* legend in Fig. 3). These results show that the PvLPMO9A was indeed active and the increase in the sugar yields for the best preparations containing this enzyme was substantially due to its presence in the cocktails.

Yields of reducing sugars (RS) in hydrolysis of Avicel (100 g/l) by composite multienzyme cocktails containing 85% of the enzyme preparations produced by recombinant P. verruculosum strains overexpressing PvLPMO9A and 15% of F10 preparation as a source of BGL, pH 5.0, 40 °C, 5 mM gallic acid, enzyme loading 5 mg protein/g substrate. B1-537, control (reference sample consisting of 85% of B1-537 preparation and 15% of F10 preparation); sLPMO1-8*, data for sLPMO1-8 sample in the absence of gallic acid
In hydrolysis of milled aspen wood (Fig. 4), the picture was rather similar to that observed with Avicel as a substrate, except the superiority of sCBH1-8 and sLPMO1-8 samples over the reference preparation was less pronounced: by 11 and 26% considering the final RS concentration.

Yields of reducing sugars (RS) in hydrolysis of milled aspen wood (100 g/l) by composite multienzyme cocktails containing 85% of the enzyme preparations produced by recombinant P. verruculosum strains overexpressing PvLPMO9A and 15% of F10 preparation as a source of BGL, pH 5.0, 40 °C, enzyme loading 5 mg protein/g substrate. B1-537, control (reference sample consisting of 85% of B1-537 preparation and 15% of F10 preparation)
Data shown in Figs. 3 and 4, together with those presented in Table 2, demonstrate that sCBH1-8 and sLPMO1-8 multienzyme preparations based on recombinant P. verruculosum strains, in which the secretion of major cellulases was substantially retained (at the level of 58–71% of the total protein) relative to the reference strain (77%) and which additionally secrete PvLPMO9A at a moderate level (9–30%), provide the highest yields of RS in hydrolysis of both cellulosic and lignocellulosic substrate. The positive effects observed for sCBH1-8 and sLPMO1-8 samples in comparison with the reference B1-537 preparation were largely the result of the synergistic cooperation between cellulolytic enzymes and PvLPMO9A, the latter acting as an auxiliary activity to hydrolytic cellulases, as already reported in other publications [8, 9, 11, 38, 39]. At the same time, the enzyme preparations, in which the total content of cellulases was < 50%, exhibited a poor performance in saccharification of both cellulosic substrates despite the high content of PvLPMO9A in them, that is, the extra high PvLPMO9A secretion by sLPMO1-1, sLPMO1-4 and sLPMO1-9 strains caused a detrimental effect on the secretion of cellulases, resulting in a poor hydrolytic performance of the respective multienzyme cocktails. Nevertheless, the latter mentioned samples with PvLPMO9A content of ~ 50% or higher may be used as blends to the traditional cellulase preparations not having their own LMPO. Their using as the blends has at least one advantage: it allows the optimization of the ratio between cellulase and LPMO activities. Hu et al. [11] showed that the optimal ratio between cellulase enzymes and LPMO depends on the cellulosic feedstock and the loading of solids in the reaction system. In particular, for steam pretreated poplar and corn stover the optimal LPMO content in the multienzyme cocktail decreased from 20 to 3% and from 6.7 to 2%, respectively, with increasing the solids’ loading from 2 to 20% [11]. Thus, a variety of obtained in this work cellulase preparations, containing the PvLPMO9A, opens up broad prospects for further optimization of multienzyme cocktails for saccharification of lignocellulosic materials.
Conclusions
In the present work, a variety of recombinant P. verruculosum strains secreting cellulase multienzyme cocktails containing the homologously overexpressed PvLPMO9A were obtained using two different constructions of the expression plasmids, based on the cbh1 gene promoter in combination with either the cbh1 signal sequence (sCBH1-X series of samples) or the native lpmo1 signal sequence (sLPMO1-X series). Three enzyme samples of the sCBH1-X series were characterized by lower overall content of CBHs but slightly higher content of EGs relative to the reference P. verruculosum B1-537 preparation, while the PvLPMO9A content in them made up 9–21% of the total secreted protein. The PvLPMO9A content in four enzyme preparations of the sLPMO1-X series was much higher (30–57%), however the portion of CBHs in most of them (except for sLPMO1-8) decreased even to a greater extent than in the samples of the sCBH1-X series. At least two new promising recombinant strains of P. verruculosum overexpressing the native PvLPMO9A were developed. The secreted multienzyme cocktails based of these strains (sCBH1-8 and sLPMO1-8), in which the content of major cellulases was substantially retained and the portion of PvLPMO9A was 9–30%, demonstrated the increased yields of reducing sugars in saccharification of Avicel and milled aspen wood: by 19–31 and 11–26%, respectively, compared to the reference cellulase cocktail.
References
Gusakov AV (2013) Cellulases and hemicellulases in the 21st century race for cellulosic ethanol. Biofuels 4:567–569
dos Santos LV, de Barros Grassi MC, Gallardo JCM, Pirolla RAS, Calderón LL, de Carvalho-Netto OV, Parreiras LS, Camargo ELO, Drezza AL, Missawa SK, Teixeira GS, Lunardi I, Bressiani J, Pereira GAG (2016) Second-generation ethanol: the need is becoming a reality. Ind Biotechnol 12:40–57
Payne CM, Knott BC, Mayes HB, Hansson H, Himmel ME, Sandgren M, Ståhlberg J, Beckham GT (2015) Fungal cellulases. Chem Rev 115:1308–1448
Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, Eijsink VGH (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330:219–222
Phillips CM, Beeson WT, Cate JH, Marletta MA (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem Biol 6:1399–1408
Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VGH (2012) Novel enzymes for the degradation of cellulose. Biotechnol Biofuels 5:45
Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels 6:41
Müller G, Várnai A, Johansen KS, Eijsink VGH, Horn SJ (2015) Harnessing the potential of LPMO-containing cellulase cocktails poses new demands on processing conditions. Biotechnol Biofuels 8:187
Bulakhov AG, Gusakov AV, Chekushina AV, Satrutdinov AD, Koshelev AV, Matys VY, Sinitsyn AP (2016) Isolation of homogeneous polysaccharide monooxygenases from fungal sources and investigation of their synergism with cellulases when acting on cellulose. Biochemistry 81:530–537
Hemsworth GR, Johnston EM, Davies GJ, Walton PH (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol 33:747–761
Hu J, Chandra R, Arantes V, Gourlay K, van Dyk JS, Saddler J (2015) The addition of accessory enzymes enhances the hydrolytic performance of cellulase enzymes at high solid loadings. Biores Technol 186:149–153
Cannella D, Jørgensen H (2014) Do new cellulolytic enzyme preparations affect the industrial strategies for high solids lignocellulosic ethanol production? Biotechnol Bioeng 111:59–68
Morozova VV, Gusakov AV, Andrianov RM, Pravilnikov AG, Osipov DO, Sinitsyn AP (2010) Cellulases of Penicillium verruculosum. Biotechnol J 5:871–880
Gusakov AV, Sinitsyn AP (2012) Cellulases from Penicillium species for producing fuels from biomass. Biofuels 3:463–477
Bulakhov AG, Volkov PV, Rozhkova AM, Gusakov AV, Nemashkalov VA, Sinitsyn AP (2017) Using an inducible promoter of a gene encoding Penicillium verruculosum glucoamylase for production of enzyme preparations with enhanced cellulase performance. PLoS ONE 12:e0170404
Dotsenko GS, Gusakov AV, Rozhkova AM, Korotkova OG, Sinitsyn AP (2015) Heterologous β-glucosidase in a fungal cellulase system: comparison of different methods for development of multienzyme cocktails. Process Biochem 50:1258–1263
Denisenko YA, Gusakov AV, Rozhkova AM, Osipov DO, Zorov IN, Matys VY, Uporov IU, Sinitsyn AP (2017) Site-directed mutagenesis of GH10 xylanase A from Penicillium canescens for determining factors affecting the enzyme thermostability. Int J Biol Macromol 104:665–671
Aslanidis C, de Jong PJ (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucl Acids Res 18:6069–6074
Sambrook J, Russell D (2001) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Sanger F, Nicklen S, Chase AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Aleksenko A, Makarova N, Nikolaev I, Clutterbuck A (1995) Integrative and replicative transformation of Penicillium canescens with a heterologous nitrate-reductase gene. Curr Genet 28:474–478
Gusakov AV, Semenova MV, Sinitsyn AP (2010) Mass spectrometry in the study of extracellular enzymes produced by filamentous fungi. J Anal Chem 65:1446–1461
Sinitsyna OA, Bukhtoyarov FE, Gusakov AV, Okunev ON, Bekkarevitch AO, Vinetsky YP, Sinitsyn AP (2003) Isolation and properties of major components of Penicillium canescens extracellular enzyme complex. Biochemistry 68:1200–1209
Gusakov AV, Sinitsyn AP, Salanovich TN, Bukhtojarov FE, Markov AV, Ustinov BB, van Zeijl C, Punt P, Burlingame R (2005) Purification, cloning and characterisation of two forms of thermostable and highly active cellobiohydrolase I (Cel7A) produced by the industrial strain of Chrysosporium lucknowense. Enzyme Microbiol Technol 36:57–69
Nelson N (1944) A photometric adaptation of the Somogyi method for the determination of sugars. J Biol Chem 153:375–379
Peterson GL (1979) Review of the Folin phenol protein quantitation method of Lowry, Rosebrough, Farr and Randall. Anal Biochem 100:201–220
Markov AV, Gusakov AV, Kondratyeva EG, Okunev ON, Bekkarevich AO, Sinitsyn AP (2005) New effective method for analysis of the component composition of enzyme complexes from Trichoderma reesei. Biochemistry 70:657–663
Volkov PV, Rozhkova AM, Gusakov AV, Sinitsyn AP (2014) Homologous cloning, purification and characterization of highly active cellobiohydrolase I (Cel7A) from Penicillium canescens. Prot Expr Purif 103:1–7
Chekushina AV, Dotsenko GS, Kondratieva EG, Sinitsyn AP (2013) Enzyme preparations from Penicillium verruculosum for bioconversion of plant raw materials is an alternative to commercial preparations obtained using Trichoderma fungi species. Biotekhnologiya 3:69–80
Dotsenko AS, Gusakov AV, Volkov PV, Rozhkova AM, Sinitsyn AP (2016) N-linked glycosylation of recombinant cellobiohydrolase I (Cel7A) from Penicillium verruculosum and its effect on the enzyme activity. Biotechnol Bioeng 113:283–291
Merzlov DA, Zorov IN, Dotsenko GS, Denisenko YA, Rozhkova AM, Satrutdinov AD, Rubtsova EA, Kondratieva EG, Sinitsyn AP (2015) Properties of enzyme preparations and homogeneous enzymes—endoglucanases EG2 Penicillium verruculosum and LAM Myceliophthora thermophila. Biochemistry 80:473–482
Volkov PV, Gusakov AV, Rubtsova EA, Rozhkova AM, Matys VY, Nemashkalov VA, Sinitsyn AP (2019) Properties of a recombinant GH49 family dextranase heterologously expressed in two recipient strains of Penicillium species. Biochimie 157:123–130
Gouka RJ, Punt PJ, van den Hondel CA (1997) Efficient production of secreted proteins by Aspergillus: progress, limitations and prospects. Appl Microbiol Biotechnol 47:1–11
Tanghe M, Danneels B, Camattari A, Glieder A, Vandenberghe I, Devreese B, Stals I, Desmet T (2015) Recombinant expression of Trichoderma reesei Cel61A in Pichia pastoris: optimizing yield and N-terminal processing. Mol Biotechnol 57:1010–1017
Yang Y, Li J, Liu X, Pan X, Hou J, Ran C, Zhou Z (2017) Improving extracellular production of Serratia marcescens lytic polysaccharide monooxygenase CBP21 and Aeromonas veronii B565 chitinase Chi92 in Escherichia coli and other synergism. AMB Expr 7:170
Low KO, Muhammad MN, Illias RM (2013) Optimization of signal peptide for recombinant protein secretion in bacterial hosts. Appl Microbiol Biotechnol 97:3811–3826
Martin C, Volkov PV, Rozhkova AM, Puls J, Sinitsyn AP (2015) Comparative study of the enzymatic convertibility of glycerol- and dilute acid-pretreated sugarcane bagasse using Penicillium- and Trichoderma-based cellulase preparations. Ind Crops Prod 77:382–390
Guo Z, Duquesne S, Bozonnet S, Nicaud J-M, Marty A, O’Donohue MJ (2017) Expressing accessory proteins in cellulolytic Yarrowia lipolytica to improve the conversion yield of recalcitrant cellulose. Biotechnol Biofuels 10:298
Chylenski P, Forsberg Z, Ståhlberg J, Várnai A, Lersch M, Bengtsson O, Sæbø S, Horn SJ, Eijsink VGH (2017) Development of minimal enzyme cocktails for hydrolysis of sulfite-pulped lignocellulosic biomass. J Biotechnol 246:16–23
Acknowledgements
This work was supported by the fundamental research program of the Presidium of the Russian Academy of Sciences No. 33 “Carbon energy: chemical aspects”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Semenova, M.V., Gusakov, A.V., Volkov, P.V. et al. Enhancement of the enzymatic cellulose saccharification by Penicillium verruculosum multienzyme cocktails containing homologously overexpressed lytic polysaccharide monooxygenase. Mol Biol Rep 46, 2363–2370 (2019). https://doi.org/10.1007/s11033-019-04693-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-04693-y