
Abstract
The auxiliary activity family 9 (AA9, formerly GH61) harbors a recently discovered group of oxidative enzymes that boost cellulose degradation. Indeed, these lytic polysaccharide monooxygenases (LPMOs) are able to disrupt the crystalline structure of cellulose, thereby facilitating the work of hydrolytic enzymes involved in biomass degradation. Since these enzymes require an N-terminal histidine residue for activity, their recombinant production as secreted protein is not straightforward. We here report the expression optimization of Trichoderma reesei Cel61A (TrCel61A) in the host Pichia pastoris. The use of the native TrCel61A secretion signal instead of the alpha-mating factor from Saccharomyces cerevisiae was found to be crucial, not only to obtain high protein yields (>400 mg/L during fermentation) but also to enable the correct processing of the N-terminus. Furthermore, the LPMO activity of the enzyme is demonstrated here for the first time, based on its degradation profile of a cellulosic substrate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lytic polysaccharide monooxygenases (LPMOs) are a recently identified group of enzymes that assist in the breakdown of carbohydrate polymers like cellulose, chitin, and starch by oxidative cleavage [1]. Although the first representatives were assigned to glycoside hydrolase (GH) families, they have recently been reclassified in auxiliary activity (AA) families [2–4]. Family AA9 (formerly GH61) only contains eukaryotic LPMOs with activity toward cellulose as a substrate [5–7]. By disrupting the crystalline structure of cellulose, they facilitate the enzymatic cleavage of classical cellulases, what is expected to significantly boost the production efficiency of second-generation ethanol [8, 9]. The production of several AA9 members has already been described, either by their native hosts [7, 10] or using Pichia pastoris for heterologous expression [5, 11–13]. Nevertheless, every protein needs optimization of its own expression conditions [14].
All LPMOs have a similar active site architecture, where an N-terminal histidine is involved in binding a copper ion. The correct processing of the protein’s N-terminus is therefore crucial to obtain an active enzyme [7, 15]. In particular, the choice of the secretion signal may have a big influence, as this leader sequence needs to be cleaved off after the pre-protein has been targeted to the secretory pathway. In Pichia pastoris, the most commonly used secretion signal is the alpha-mating factor (alpha-MF) originating from Saccharomyces cerevisiae. However, many alternatives exist and whether or not the protein can be correctly processed depends on the compatibility of the secretion signal used and machinery of the host cell [16].
The filamentous fungus Trichoderma reesei (Hypocrea jecorina) is one of the best-studied organisms in the field of biomass valorization [17–19]. Besides a whole array of hydrolytic enzymes, it also produces two LPMOs of family AA9, namely TrCel61A and TrCel61B. The former consists of a catalytic domain connected to a cellulose-binding domain (CBM1) by a flexible linker, whereas the latter only harbors a catalytic domain, which is 49 % identical to that of Cel61A [10]. Before the discovery of LPMO as a new enzyme class, Cel61A was thought to have a very weak endoglucanase (EG) activity and was therefore referred to as EGIV. The enzyme has already been both produced in a heterologous host (S. cerevisiae, 1997 [20]) and overexpressed from the native host (T. reesei, 2001 [21]). However, its LPMO activity has not yet been demonstrated.
In this paper, the recombinant expression of TrCel61A in the yeast Pichia pastoris is described for the first time, resulting in the highest yields that have been reported so far. Crucially, several secretion signals have been compared with respect to N-terminal processing, to ensure the generation of a catalytically active enzyme. Finally, this allowed us to confirm that TrCel61A indeed is a polysaccharide monooxygenase that generates oxidized oligosaccharides from cellulose substrate.
Materials and Methods
Biological Materials
The cDNA coding for Trichoderma reesei Cel61A (GenBank Y1113) was isolated from T. reesei QM6A (MUCL 44908) via total RNA extraction (RNeasy Mini kit, Qiagen) followed by cDNA production (Superscript III first strand synthesis kit, Invitrogen). This gene was cloned in pJET1.2 plasmid (Life Technologies) for further use. A codon-optimized secretion signal and gene sequence were ordered from GeneArt Gene Synthesis (Thermo Fisher Scientific). Pichia pastoris strain CBS7435 and all plasmids (pPpT4 plasmid variants, described by Näätsaari [22]) were provided by the institute of Molecular Biotechnology at TU Graz, Austria.
Molecular Work
The molecular constructs were completed in E. coli cells (Transformax™ EC100™ Electrocompetent E. coli cells from Epicentre). The required primers were ordered from Integrated DNA Technologies (IDT). In all cases, the constructs were integrated in the pPpT4 vectors downstream of the promoter (GAP or AOX1) and included an N-terminal secretion signal followed by Trcel61A gene with a His6 tag directly attached to its C-terminus (Fig. 1). The constructs described in the section ‘Optimizing the yield of secreted protein’ were cloned by restriction ligation using XhoI, EcoRI, and NotI, using Fast digest enzymes (Life Technologies). All other variations of the secretion signal were constructed by various molecular techniques such as the method developed by Sanchis to eliminate a few base pairs [23] and Gibson assembly [24] to insert TrCel61A preceded by another secretion signal in the pPpT4 backbone. More detailed information on the molecular constructs can be found in Supplementary material.

Molecular background of cloning strategies. a Representation of the vector with the relative positions of promoter, secretion signal, and coding sequence of TrCel61A. All evaluated options are indicated in a list. b Short schematic overview of the different steps going from DNA sequence till mature active protein
After confirming its correct sequence (LGC Genomics) in E. coli, the resulting plasmid was linearized and transferred into freshly prepared competent Pichia pastoris CBS7435 cells [25]. Positive transformants were selected by incubating the transformation mixture at 30 °C for 48 h on YPD agar plates containing 100 µg/mL Zeocin. At least 5 colonies per transformation were grown on microscale (96-deep-well plate, see further), and the supernatant was analyzed via SDS-PAGE.
Media and Growth Methods
LB medium was used for growing E. coli cultures containing 2 % (w/v) agar, if required, and 25 µg/mL Zeocin for selection. The cultures were grown overnight at 37 °C while shaking at 200 rpm.
The standard medium for P. pastoris was YPD medium (1 % (w/v) yeast extract, 2 % (w/v) tryptone, and 2 % (w/v) glucose) containing 2 % (w/v) agar, if required, and 100 µg/mL Zeocin for selection. For growth experiments, a buffered minimal medium was used. The basic composition of the medium consisted of 200 mM potassium phosphate buffer (pH 6), 1.34 % yeast nitrogen base without amino acids, and 4*10−5 % biotin. For initial growth, BMD medium was completed by adding 1 % glucose while induction media were formed by adding 1 % (BMM2) or 5 % (BMM10) methanol (v/v) to the basic medium.
P. pastoris strains were grown in 250-mL (unbaffled) shake flasks at 30 °C by 200 rpm shaking. Initially, the culture was started in 45 mL BMD medium, followed by induction after 48-60 h by adding 5 mL BMM10 medium. Subsequently, methanol supply was kept at 2 shots of 0.5 mL methanol a day. After 3 to 5 days of induction, cultures were harvested by centrifuging at 1500 rpm for 15 min (4 °C). This method can be scaled to different volumes. Alternatively, microscale cultivation [26] was performed in 96-deep-well plates (8 × 8 × 40 mm with round bottom, Enzyscreen) that were sealed with a low-evaporation sandwich cover (Enzyscreen). Plates were incubated at 28 °C, tilted under an angle of 25°, shaking at 300 rpm. A high methanol supply was applied at a rate of 2 shots of 50 µL BMM10 medium a day.
Fermentation and Purification
TrCel61A was fermented in a 2 L fermentation vessel (Biostat B, B. Braun Biotech) starting with one liter BSM culture medium, as described by De Winter et al. [27]. Methanol feed was initiated at a cell wet weight volume of 160 g/L (30 h batch phase) and was gradually increased to 2 g/(L h) for 6 h and kept at this rate for 90 h. After 60 h of induction, the pO2 feed was increased to maintain a dissolved oxygen percentage of 50 %. The protein was obtained by centrifuging the fermentation broth for 10 min at 10.000 rpm. The resulting supernatant was filtered using depth filtration, followed by cross-flow filtration and buffer exchange for 50 mM sodium acetate buffer pH 5.2 (Vivaflow 200, Sartorius).
Protein Analysis and Concentration
Culture supernatant and purified proteins were analyzed on 12 % SDS-PAGE gels to confirm the presence of the correct protein [28]. The proteins were stained with Coomassie Brilliant Blue (R-250). Apart from a reference standard protein ladder (Pageruler Prestained Protein Ladder, Thermo Fischer Scientific), each gel was also provided with a reference protein, 0.1 mg/mL BSA, to estimate the protein concentration. The intensity of the desired bands was measured and compared by digital imaging [29], using ImageJ (Image Processing and Analysis in Java, available at http://imagej.nih.gov/ij/). Samples were diluted to fit in the linear quantifiable range (0.050–0.250 mg/mL protein).
Activity Testing
To measure activity, 37 µg of TrCel61A was mixed with 1 mM ascorbic acid and 1.2 % Phosphoric Acid Swollen Cellulose (PASC), prepared from Avicel PH-101 (Sigma-Aldrich) following instructions from Wood [30]. The volume was adjusted to 1 mL with 10 mM sodium acetate buffer pH 5.2 in Eppendorf tubes. The tubes were incubated overnight at 50 °C while shaking at 1400 rpm in an Eppendorf Thermomixer. The enzyme was heat-inactivated by incubating the tubes at 95 °C for 10 min and the samples analyzed by HPAEC-PAD using the method described by Forsberg et al. [31].
Protein Sequencing
The N-terminus of the proteins was determined by Edman degradation. The proteins were blotted on a PVDF membrane, and the desired bands were excised from the blot. Subsequently, the samples were analyzed using a 494 Procise protein sequencer (Applied Biosystems).
Results and Discussion
Optimizing the Yield of Secreted Protein
When using Pichia pastoris as an expression host, many factors can influence the yield of secreted recombinant proteins [14]. In this work, the evaluation was focused on (1) the type of promoter, (2) the codon usage, and (3) the secretion signal. To that end, 6 different constructs were prepared and their expression yields were compared by SDS-PAGE analysis (Table 1). In the compared analysis of at least three biologic replicates per construct, the TrCel61A protein was detected at a molecular mass of about 60 kDa (Figure S1), which is higher than its calculated mass of 35 kDa due to glycosylation. Although the methanol-regulated AOX1 promoter and the constitutive GAP promoter are supposed to be equally strong [32], only a very low expression level could be detected for the GAP constructs in our case. This could be an indication that either the secretion pathway was not followed correctly or the proteins ended in the ER-associated protein degradation (ERAD) pathway as described earlier in literature [33]. A further increase in expression yield was achieved by modifying the codon usage toward P. pastoris (Figure S2), as has been reported previously for the recombinant expression of cellobiohydrolase and mannanase [34]. Interestingly, the codon-optimized native secretion signal of Trichoderma reesei Cel61A in combination with the codon-optimized gene was found to give better results than the more commonly used alpha-mating factor from Saccharomyces cerevisiae (α-MF). At the end of the optimization process, the optimized construct yielded 70 ± 2 mg/L of secreted protein on microscale, which is about 4 times higher than the starting construct (Table 1).
Optimizing N-terminal Processing
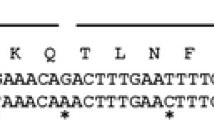
The correct removal of the secretion signal is absolutely essential in case of LPMO expression, as the enzyme requires an N-terminal histidine for activity. So far, several LPMOs have been produced using the α-MF for secretion purposes in Pichia pastoris, but little attention has been paid to its processing [5, 35]. Therefore, N-terminal sequence analysis has now been performed on several constructs (Table 2). The α-MF sequence ends with the amino acid sequence EKR, which is recognized by the protease Kex2 in the Golgi apparatus. To increase the cleavage efficiency of Kex2, the signal peptide can be extended with 2 EA repeats, which are subsequently cleaved off by another Golgi protease Ste13 [36]. However, neither of these constructs resulted in a correct N-terminus for TrCel61A (Fig. 2). The former was not recognized at all by Kex2, resulting in the entire pro-sequence still present in the protein. Although the addition of the EA repeats indeed increased the activity of Kex2, their removal by Ste13 was highly inefficient. As an alternative to the EA repeats, an enterokinase cleavable sequence (DDDDR [11]) was tested, to check whether an artificially introduced cleavage site could help circumvent an inefficient biologic post-translational processing. Although this complicates the process by the introduction of an extra step, the result was satisfactory (Fig. 2), as was also reported for the LPMO from Phanerochaete chrysosporium GH61D by Westereng et al. [11].

Effect of secretion signal on yield and processing. The expression level of the secreted protein (white) and corresponding share of correctly processed form (black) is given. (αMF is indicated with its different amino acids in the end, DDDK protein = secretion signal of DDDK protein, PcGH61A-SS = Phanerochaete chrysosporium GH61D native secretion signal, TrCel61A-SS = Trichoderma reesei Cel61A secretion signal)
As a comparison, two related native LPMO secretion signals have also been evaluated, i.e., that of TrCel61A and that of Phanerochaete chrysosporium GH61D (PcGH61D). Interestingly and despite that both secretion signals are equally foreign for Pichia pastoris, the former is completely and correctly removed by the host, and thus is the preferred option in terms of both protein yield and N-terminal processing. In contrast, the latter only had a removal efficiency of 85 % and also resulted in a product with a twofold lower expression yield. Finally, inserting an endogenous secretion signal from P. pastoris has also been tried, as one would expect that the cellular machinery is optimized to recognize this signal. For example, the DDDK protein signal sequence has been proposed as an alternative for the α-MF [37]. Unfortunately, the N-terminus was found to be processed incorrectly in our case and more specifically by cleaving at position His3 in the protein.
Scale-up to Fermentation and Demonstration of LPMO Activity
In order to get a better insight into the expression capabilities for our optimized construct (AOX1 promoter, codon-optimized TrCel61A secretion signal and gene, and native TrCel61A secretion signal), the production process was scaled-up to a 2L fed-batch fermentation. Since there is no direct, fast, and quantitative activity measurement (yet) available for LPMO, the protein concentration was monitored during fermentation. To that end, four different methods were compared: (1) OD280 (Nanodrop 2000c), (2) Bradford protein assay, (3) Bicinchoninic acid (BCA) protein assay, and (4) quantitative SDS-PAGE. The first three suffered from a high background, high variation, and/or a low response to increases in protein content, most likely due to interference by the culture medium or glycosylation of the protein [12]. In contrast, quantification of the protein band on SDS-PAGE by digital imaging was found to be a more reliable method. After an induction phase of 96 h, a protein concentration of 447 mg/L was obtained (Fig. 3), which outperforms production in the natural host Trichoderma reesei by almost a factor four [21] and represents the highest expression level for TrCel61A reported so far. Although the enzyme was already expressed in S. cerevisiae, comparison was not possible since no yields were reported [20]. As a comparison, expression of 4 Neurospora crassa PMOs in Pichia pastoris yielded 340 mg per liter medium.

Fermentation parameters. Protein concentration (g/L) and cell wet weight (g/L) monitored and given for different time points. The methanol feed was initiated at 30 h (first vertical line). A second increase in cell wet weight was found after 90 h (second vertical line) due to pressure increase
More importantly, TrCel61A was shown to be active as LPMO by incubating the enzyme with phosphoric acid swollen cellulose (PASC) as a substrate and ascorbic acid as an electron donor. HPAEC-PAD analysis showed the formation of cello-oligosaccharides in their neutral and oxidized form (Fig. 4). Interestingly, the even-numbered aldonic acids (cellobionic acid and cellotetraonic acid) have a higher prevalence than the odd-numbered counterparts, which might suggest a preference in the cleavage mechanism for this enzyme. Furthermore, the release of cellobiose, cellotriose, and their oxidized counterparts cellobionic acid and cellotrionic acid keeps increasing over time, while larger products start to decrease. This suggests that TrCel61A is also active on short soluble oligos as described earlier for NcLPMO9C [38]. TrCel61A would need a substrate of minimally 4 glucose units in order to be active. Moreover, small peaks appear in the region for C4-oxidized and double (C1- and C4-) oxidized products, what classifies TrCel61A as a type-3 LPMO [39].

Enzymatic activity of recombinant TrCel61A. a HPAEC-PAD profile of TrCel61A activity on PASC at 50 °C. Samples were analyzed after 0, 2, 8, 19, and 44 h of reaction. a Enzyme reaction with the addition of TrCel61A. b Control reaction performed without the addition of enzyme. Cellobionic acid (GlcGlcA) and cellotrionic acid (Glc2GlcA) were used as standard whereas the nature of the other oxidized products was inferred from the literature [40]
Conclusion
The yeast Pichia pastoris was found to be a suitable expression host for the LPMO Cel61A from Trichoderma reesei, with a protein yield during fermentation of >400 mg/L when using the AOX1 promoter, a codon-optimized gene and the protein’s native secretion signal. Considering the importance of an N-terminal histidine residue, we here report that the processing of the native secretion signal is much more accurate than that of the more commonly used alpha-mating factor from Saccharomyces cerevisiae. Furthermore, our results demonstrate for the first time that TrCel61A is an active LPMO that generates both neutral and oxidized cello-oligosaccharides from PASC as a substrate.
References
Vu, V. V., Beeson, W. T., Span, E. A., et al. (2014). A family of starch-active polysaccharide monooxygenases. Proc Natl Acad Sci USA, 111, 13822–13827.
Levasseur, A., Drula, E., Lombard, V., et al. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels, 6, 41.
Lo Leggio, L., Simmons, T. J., Poulsen, J.-C. N., et al. (2015). Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat Commun, 6, 5961.
Horn, S. J., Vaaje-Kolstad, G., Westereng, B., et al. (2012). Novel enzymes for the degradation of cellulose. Biotechnol Biofuels, 5, 45.
Dimarogona, M., Topakas, E., Olsson, L., et al. (2012). Lignin boosts the cellulase performance of a GH-61 enzyme from Sporotrichum thermophile. Bioresour Technol, 110, 480–487.
Beeson, W. T., Phillips, C. M., Cate, J. H. D., et al. (2012). Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Am Chem Soc, 134, 890–892.
Li, X., Beeson, W. T., Phillips, C. M., et al. (2012). Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure, 20, 1051–1061.
Ekwe, E., Morgenstern, I., Tsang, A., et al. (2013). Non-hydrolytic cellulose active proteins: research progress and potential application in biorefineries. Ind Biotechnol, 9, 123–131.
Dimarogona, M., Topakas, E., & Christakopoulos, P. (2013). Recalcitrant polysaccharide degradation by novel oxidative biocatalysts. Appl Microbiol Biotechnol, 97, 8455–8465.
Karkehabadi, S., Hansson, H., Kim, S., et al. (2008). The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6 A resolution. J Mol Biol, 383, 144–154.
Westereng, B., Ishida, T., Vaaje-Kolstad, G., et al. (2011). The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS ONE, 6, e27807.
Kittl, R., Kracher, D., Burgstaller, D., et al. (2012). Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol Biofuels, 5, 79.
Bey, M., Zhou, S., Poidevin, L., et al. (2013). Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (family GH61) from Podospora anserina. Appl Environ Microbiol, 79, 488–496.
Daly, R., & Hearn, M. T. W. (2005). Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit, 18, 119–138.
Quinlan, R. J., Sweeney, M. D., Lo Leggio, L., et al. (2011). Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA, 108, 15079–15084.
Hegde, R. S., & Bernstein, H. D. (2006). The surprising complexity of signal sequences. Trends Biochem Sci, 31, 563–571.
Merino, S. T., Cherry, J., & Ave, D. (2007). Progress and challenges in enzyme development for biomass utilization. Adv Biochem Eng Biotechnol, 108, 95–120.
Wilson, D. B. (2009). Cellulases and biofuels. Curr Opin Biotechnol, 20, 295–299.
Kubicek, C. P., Mikus, M., Schuster, A., et al. (2009). Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol Biofuels, 2, 19.
Saloheimo, M., Nakari-Setälä, T., Tenkanen, M., et al. (1997). cDNA cloning of a Trichoderma reesei cellulase and demonstration of endoglucanase activity by expression in yeast. Eur J Biochem, 249, 584–591.
Karlsson, J., Saloheimo, M., Siika-Aho, M., et al. (2001). Homologous expression and characterization of Cel61A (EG IV) of Trichoderma reesei. Eur J Biochem, 268, 6498–6507.
Näätsaari, L., Mistlberger, B., Ruth, C., et al. (2012). Deletion of the pichia pastoris ku70 homologue facilitates platform strain generation for gene expression and synthetic biology. PLoS ONE, 7, e39720.
Sanchis, J., Fernández, L., Carballeira, J. D., et al. (2008). Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: application to difficult-to-amplify templates. Appl Microbiol Biotechnol, 81, 387–397.
Gibson, D. G., Young, L., Chuang, R., et al. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases., 6, 12–16.
Lin-Cereghino, J., Wong, W. W., Xiong, S., et al. (2005). Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. Biotechniques, 38, 44–48.
Weis, R., Luiten, R., Skranc, W., et al. (2004). Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res, 5, 179–189.
De Winter, K., Šimčíková, D., Schalck, B., et al. (2013). Chemoenzymatic synthesis of α-l-rhamnosides using recombinant α-l-rhamnosidase from Aspergillus terreus. Bioresour Technol, 147, 640–644.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Sun, L. W., Zhao, Y., Niu, L. P., et al. (2011). A rapid method for determining the concentration of recombinant protein secreted from Pichia pastoris. J Phys: Conf Ser, 276, 012144.
Wood, T. M. (1988). Preparation of crystalline, amorphous, and dyed cellulase substrates. Methods Enzymol, 160, 19–25.
Forsberg, Z., Vaaje-Kolstad, G., Westereng, B., et al. (2011). Cleavage of cellulose by a CBM33 protein. Protein Sci, 20, 1479–1483.
Cregg, J. M., Cereghino, J. L., Shi, J., et al. (2000). Recombinant protein expression in Pichia pastoris. Mol Biotechnol, 16, 23–52.
Hohenblum, H., Gasser, B., Maurer, M., et al. (2004). Effects of gene dosage, promoters, and substrates on unfolded protein stress of recombinant Pichia pastoris. Biotechnol Bioeng, 85, 367–375.
Mellitzer, A., Weis, R., Glieder, A., et al. (2012). Expression of lignocellulolytic enzymes in Pichia pastoris. Microb Cell Fact, 11, 61.
Sygmund, C., Kracher, D., Scheiblbrandner, S., et al. (2012). Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl Environ Microbiol, 78, 6161–6171.
Cereghino, J. L., & Cregg, J. M. (2000). Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev, 24, 45–66.
Govindappa, N., Hanumanthappa, M., Venkatarangaiah, K., et al. (2014). A new signal sequence for recombinant protein secretion in Pichia pastoris. J Microbiol Biotechnol, 24, 337–345.
Isaksen, T., Westereng, B., Aachmann, F. L., et al. (2014). A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J Biol Chem, 289, 2632–2642.
Vu, V. V., Beeson, W. T., Phillips, C. M., et al. (2014). Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J Am Chem Soc, 136, 562–565.
Forsberg, Z., Mackenzie, A. K., Sorlie, M., et al. (2014). Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc Natl Acad Sci, 111, 8446–8451.
Ahmad, M., Hirz, M., Pichler, H., et al. (2014). Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol, 98, 5301–5317.
Acknowledgments
The authors wish to thank the Agency for Innovation by Science and Technology (IWT) Flanders for financial support (Ph.D.-grant to M.T.). The study was carried out in the frame of the MRP Project “Ghent Bio-economy” granted by Ghent University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tanghe, M., Danneels, B., Camattari, A. et al. Recombinant Expression of Trichoderma reesei Cel61A in Pichia pastoris: Optimizing Yield and N-terminal Processing. Mol Biotechnol 57, 1010–1017 (2015). https://doi.org/10.1007/s12033-015-9887-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-015-9887-9