
Abstract
N-ethyl-N-nitrosourea (ENU) is highly used in rodent models of tumerogenesis/carcinogenesis. Xenografting human-cancer tissues/cells with estradiol (E2) treatment is also used to generate rodent-models of gynaecological cancers. The altered metabolic-redox environment leading to establishment of pre-tumorigenesis condition and their mechanism are less studied. Here, female Wister rats were treated with these drugs at their pre-tumerogenic dosage (one group ENU single intra-peritoneal dose of 90 mg/kg b.w. and another group were implanted with human breast tumor (stage-IIIB) and fed with 2.5 mg of 17β-estradiol once in a week for 4 months). After 4 months, animals were sacrificed; their serum and liver tissues were tested. A brief comparison was made with a rat model (regarded as positive control) of toxicity induced by mutagenic environmental pollutant arsenic (0.6 ppm daily/4 weeks). The increase in serum alkaline phosphatase and glutamate-pyruvate transaminase suggests the possible organ toxicity is favoured by the increase in hepatic/systemic free radicals and oxidative stress in all drug application models. But the increase in the serum E2 level as noted in the ELISA data with impairment in the hepatic estrogen sulfotransferase (SULT1E1) protein expression (immuno-blot data) were noticed with interfered hepatic free-thiols only in ENU and xenograft-E2 group compared to arsenic group. It is also evident in the in vitro result from E2/GSH/NAC added hepatic slices with altered antioxidant regulations. Moreover, impairment in hepatic SOD1, catalase and glutathiole peroxidase activities (PAGEzymographic data), especially in the ENU-treated group makes them more vulnerable to the oxidative threat in creating pre-tumerogenic microenvironment. This is evident in the result of their higher DNA-damage and histological abnormalities. The Bioinformatics study revealed an important role of rSULT1E1 in the regulations of E2 metabolism. This study is important for the exploration of the pre-tumerogenic condition by ENU and E2 by impairing SULT1E1 expression and E2 regulations via oxidant-stress signalling. The finding may help to find new therapeutic-targets to treat gynaecological-cancers more effectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Different gynaecological cancers are leading cause of deaths worldwide. Drug-induced mutations, oncogenesis and steroid deregulations are the main determinants of these cancers. Moreover, liver being the main metabolic path of entry for any endo- or exo-genous compounds it experiences primary threat from the toxicity and tumerogenesis initiated by these compounds. So, hepatocellular carcinoma (HCC) also accounts for an important cause of cancer-related deaths, globally. During chemical processing of both endo- and exo-genous compounds, toxic metabolites are sometimes produced that might be injurious to the liver [1]. So the mechanism of tumorigenesis/carcinogenesis induction by some compound and its other toxicity may be one of the important objectives for the study on cancer biology. The toxic potentials of any proposed compounds and their nature are also important during any anti-cancer therapeutic study.
For the generation of experimental animal models for tumorigenesis/carcinogenesis different exo- and endo-genous drugs are used in the laboratory. N-ethyl-N-nitrosourea (C3H7N3O2; ENU) is an important carcinogen used in generating animal cancer models mainly different types of gynaecological cancers (breast, uterus and endometrium) [2]. ENU is also used to induce mammary tumors and liver cancers [3]. This indirect alkylating agent produces DNA-ethyl adducts [4]. ENU is an alkylating agent which promotes A to T base transversions, AT to GC transitions, and also some GC to AT transitions [5, 6]. The ENU is a structurally diverse group of DNA damaging compounds which form adducts at ring nitrogen (N) and extra cyclic-oxygen (O) atoms of DNA bases [7, 8]. These mutations are of major importance in induction of cancers. ENU has been traditionally characterised as a severely potent trans-placental teratogen and carcinogen in rodents [9, 10].
The endogenous compounds like steroids are responsible for several pathological conditions especially in females. It is widely known that many initiators and promoters of carcinogenesis include estradiol and others, those produce reactive oxygen species (ROS) [11]. The ROS causes oxidative damage to a variety of molecules, including DNA and protein structure leading to dysregulation of its function [11]. So, the role of free radicals and oxidant mechanisms during the cancer pathogenesis in patients or in experimental animal models or during anti-cancer therapy are of great importance and should be taken into account during the investigation. In animal model of cancer, xenografting i.e. implantation of human tumor cells adjoined/beneath tissues/organs (under the mammary/breast tissues of rodents) is a routine laboratory work. This is conducted with or without estradiol application in mostly nude rodents (athymic or whole body γ-irradiated or drug-induced immuno-compromised). During this procedure a significant level of toxicity, oxidative stress may also be occurring [12]. The features of the general level of toxicity, stress and metabolic dysregulations might be accountable in immuno-uncompromised condition in the presence of drug application. In anti-cancer therapeutic studies the evaluation of basal levels of toxicity is of great importance. Antioxidant depletion, oxidative stress and metabolic damages are reported in rodent models after E2 and ENU application. Studies demonstrate and support the correlation between lipid peroxidation, systemic stress and hepato-carcinogenicity [12].
Estradiol (E2) is considered as a carcinogen causing dysregulation of pathways involved in cell proliferation and tissue growth. In some cases this steroid is prescribed for the post menopausal hormone replacement therapy in some women. But, some time the unwanted level of E2 has been reported to be carcinogenic. The mechanisms involved in carcinogenic effects of estradiol are stimulation of cell proliferation and differentiation via estrogen receptor (ER)-mediated pathway [13, 14]. ROS generation during a cytochrome P450 (CYP)-mediated estrogen metabolism leads to genotoxic effects like mutation and chromosome abnormalities, and may also regulate enzyme activity or transcription factors through redox regulation [15, 16]. Indeed, non-transformed breast epithelial cells experience variable amount of production of ROS by peroxisomal β-oxidation, cytochrome P450 and other phase I/II xenobiotic metabolism [17]. As for example, UDP-glucuronosyltransferase (UGT) is also an important participant. In terms of steroid and specially E2 metabolism there have been significant association amongst SULT1E1 and other drug metabolizing enzymes. Thus, estradiol being a vital molecule in maintenance of normal cellular proliferation and growth is accountable for different physiological as well as pathological conditions. Abundance of estradiol in postmenopausal state was found to be correlated with different gynaecological cancers [17, 18]. This molecule induces malignant transformation or rapid neoplastic growth via impaired mitochondrial oxidative-phosphorylation (BOX-2), oxidative-stress and develops breast cancer. It is unclear that other than direct DNA damage or impaired cellular signalling how oxidative-stress influences the tumor biology via endocrine regulation that promote ER-positive breast cancer [19].
Since, estradiol being considered as a carcinogen, it makes us inquisitive about estradiols effect on liver in terms of oxidative stress, toxicity induction and its effect on estradiol metabolizing enzyme, estrogen sulfotransferase (SULT1E1). Estrogen sulfotransferase a phase-2 drug metabolizing enzyme, sulfonates estrogen at nM concentration and produces estradiol sulphate (E2S) or estrone sulphate (E1S) which are biologically inactive and unable to bind to ERα or ERβ. Thereby, those are unable to propagate its receptor associated function [18, 20]. E2S and E1S are more susceptible to elimination. Previous studies have reported redox regulation of SULT1E1 [19, 21] and a few studies have reported reduced SULT1E1 expression and activity in MCF7 cell line inferring that may be redox regulation is the reason for the reduced activity of this in cancer cells [21, 22]. Oxidative stress has a vital role in E2 regulation and can structurally modify or alter the expression level of E2 metabolizing proteins. As such, estrogen receptors loose their DNA binding ability and thus impairing transcription. Estradiol binding site gets blocked in estrogen sulfotransferase under oxidative stress [19, 21].
Previous studies have suggested a possible role for oxidative stress in nitrosoamine-induced carcinogenesis [23]. Earlier studies have correlated lipid peroxidation as oxidative stress marker with carcinogenicity. Therefore, the ENU-induced tumor/cancer model gives a scope to study the corresponding changes in oxidative stress, or its physiological consequences, such as changes in redox-sensitive signalling. The purpose of this study was to establish the profile of ENU-induced and E2-xenografting induced oxidative stress and toxicity/necrosis/carcinogenesis in the liver of female rat. This toxicity model has been compared to one positive control animal group of toxicity induced by arsenic in rat. Arsenic is an environmental contaminant and exerts its carcinogenic and genotoxic effects in different human organs mainly in the liver. Arsenic-induced hyperkeratosis, gangrene; hepatic tumors are well documented [24].
Materials and methods
Ethical clearance and fulfilments of other regulatory affairs
This is to state that the present study was carried out in accordance with the National Institutes of Health, USA guidelines and the institutional ethical concerns, relevant guidelines were maintained throughout the investigation. The Breast cancer tissue samples were collected from the Oncology and Radiology Dept. Midnapore Medical College and Hospital, West Bengal. This is to confirm that all experimental protocols were approved by the institutional (Oriental Institute of Science and Technology) Ethics Committee. This is also to state that informed consent was obtained from all participant patients who were at their post-menopausal age.
Animal selection and treatment
Female albino rats of Wister strain ageing 3–4 weeks were acclimatized for 10 days at 12-h light–dark cycle, 32 °C ± 2 °C temperature, 50–70% humidity in the institutional animal resource facility. Those were fed with a standard pellet diet (Hindustan Lever Ltd, Mumbai, India) and water ad libitum. Studies were carried out in accordance with the National Institutes of Health, USA guidelines and the institutional (Oriental Institute of Science and Technology) ethical concerns were maintained throughout the investigation. Rats were randomly distributed in three groups having 5–9 in each as mentioned in the specified method/figure section. Animals of Group-II were implanted in the inguinal mammary fat pad area with a total of 400 mg tumor-tissue (stage-IIIB)/rat from a common stock of single cell preparation [25, 26]. Those animals were fed by gavages with 17β-estradiol (E8875 SIGMA) 2.5 mg/in 500 µl of distilled water once in a week for 4 months. The E2 administration by this way yielded the best physiological responses which has been demonstrated several times [27,28,29]. Animals in Group-III were administered by N-ethyl-N-nitrosourea (ENU), intra-peritoneal at a dose of 90 mg/kg body weight and were regularly observed for 4 months. After conducting several dose–response studies and reviewing some relevant work the present treatment schedule for both estradiol and ENU were employed [29,30,31,32]. This dose schedule fulfilled the purpose of the establishment of pre-tumorigenic condition. Remaining Group I animal as control was supplemented with the same amount of drinking water once in a week for 4 months. For arsenic toxicity experiment, rats of Group-IV and Group-V (six rats in each group) were fed with 0.5 ml drinking water (control) and the same amount of drinking water containing sodium arsenite at a concentration of 0.6 ppm/100 g b.w./day for 28 days, respectively. After conducting several dose–response studies the present treatment schedule was employed and published elsewhere [33]. The present dose schedule usually does not cause animal mortality but exposure for a moderate time period (> 3 weeks), increase liver and kidney toxicity marker and other clinical marker suggesting significant level of cellular and metabolic impairment.
On the day of sacrifice, animals were exposed to light anaesthesia (by ether) and blood was collected using a disposable syringe (21-gauge needle), serum was separated from the collected blood samples. The liver tissue was carefully collected and stored at − 20 °C for experimental purposes.
Evaluation of general toxicity
Serum glutamate pyruvate transaminase (SGPT), serum glutamate oxaloacetate transaminase (SGOT) alkaline phosphatase (ALP), albumin, bilirubin, urea, uric acid and creatinine were measured from the rats by standard protocol with the assay kits (Ranbaxy, India or other reputed company). Total protein (serum) was measured following Lowry method using standard protocol [34].
Estimation of malondialdehyde (MDA) Levels
MDA was estimated both from tissue and serum samples. Tissue was homogenized (10% w/v) in the ice-cold phosphate buffer (0.1 mol/L, pH 7.4) and the homogenate was centrifuged at 10,000 rpm at 4 °C for 10 min. The MDA assay was conducted using the supernatant following the protocol [35]. To chelate iron and reduce its interference in peroxidation reaction of unsaturated fatty acid, 1 mM EDTA was used in the reaction mixture. To reduce the interference caused by a yellow–orange colour produced by some carbohydrates, the reaction mixture was heated at 80 °C instead of 100 °C. Finally, the MDA was measured and calculated utilizing the molar extinction coefficient of MDA (1.56 × 105 cm2/mmol).
Estimation of non protein soluble thiol (NPSH)
The NPSH in serum and liver tissue homogenates (prepared in 0.1 M phosphate buffer, pH 7.4) were determined by the standard DTNB (5,5′-dithiobis-2-nitrobenzoic acid) method with a slight modification [36]. In brief, the protein was precipitated by trichloroacetic acid and clear cytosol was added to 0.1 M sodium phosphate buffer containing 5 µM DTNB. The level of NPSH was determined against a GSH standard curve.
Assay of super oxide dismutase (Cu–Zn, SOD1) in PAGE-zymography technique
A tablet of nitro blue tetrazolium (NBT) was dissolved in 30 mL water. The non-denaturing (10%) acrylamide electrophoresed (for 3 h at 40 mA at 4 °C) gel carrying all the samples (125 µg of protein) with the desired bands was soaked in it for 30 min with shaking. The gel was shaken in 40 mL superoxide dismutase (SOD1) solution [0.028 M tetra methyl ethylene diamine (TEMED), 2.8 × 10−5 M riboflavin, and 0.036 M potassium phosphate at pH 7.8] for 15 min. The soaked gel was placed on a clean acetate sheet and illuminated for 5–15 min. The gel became purple except at the position containing SOD1. The gel was scanned when the maximum contrast between the band and background has been achieved. An identical gel was stained with Coomassie brilliant blue to verify the liver protein of the corresponding SOD1 protein bands in different group of animals [37,38,39].
Catalase activity in PAGE-zymography technique
A non-denaturing gel (8%) was loaded with a 25 µg of cytosolic and electrophoresed for 3 h at 40 mA at 4 °C. The gel was washed 3 × 10 min in distilled water. protein and was incubated in 0.003% H2O2 for 10 min followed by staining with 2% ferric chloride and 2% potassium ferricyanide. In this critical step two reagents were not mixed prior to staining rather poured of together directly on top of the gel. The gel became greenish blue except at the position containing the catalase. The gel was rinsed with distilled water and then scanned when maximum contrast between the band and the background was obtained [38, 39].
Glutathione peroxidase assay PAGE-zymography technique
A non-denaturing gel (8%) was loaded with a 150 µg of protein. The gel was run for 3 h at 40 mA at 4 °C. The gel was removed from the glass plates and placed into a glass staining dish. The gel was washed 3 × 10 min in distilled water containing 1 mM GSH using about 50 ml per wash. In this critical step washing the gels with GSH solution allows the gel to absorb this substrate needed for GPx to function.The gel was incubated in 100 ml ddH2O containing 0.008% cumenehydroperoxide for 10 min and then rinsed twice with distilled water. Two solutions were prepared in separate vials. These are 1% ferric chloride (w/v, 0.3 gm in 30 ml ddH2O containing GSH) in one tube and 1% potassium ferricyanide (w/v, 0.3 gm in 30 ml ddH2O containing GSH) in other tubes. The gel was incubated with the prepared stains. Importantly, two reagents were poured together directly on top of the gel. When achromatic bands begin to form (5–15 min), the stain was poured off and the gel was washed extensively with distilled water. The image of the bands demonstrated GPx activity was evaluated. The cumenehydroperoxides instead of H2O2 performed as a good substrate and the GPx (contains a selenium in the active site) can utilize this hydroperoxide to determine total peroxidase activity according to the established protocol as described by [38].
In vitro SOD and catalase activity assay
Liver tissue slices from control rats were incubated in Krebs ringer buffer with different reducing agents i.e. N-acetyl cysteine (NAC, 1 mM) or reduced glutathione (GSH, 1 mM) or in combination and 10−7 M estradiol for 0, 6 and 12 h and then its cytosolic fraction was used for SOD and catalase activity [38, 39] .
DNA Fragmentation Analysis
The liver tissue was lysed with 500 µl of lysis buffer (50 mM Tris pH 8.0, 20 mM EDTA, 10 mM NaCl, 1% SDS, 0.5 mg/ml proteinase K) for 20 min on ice (4 °C) and centrifuged at 12,000×g for 30 min at 4 °C. The supernatant was extracted with 1:1 mixture of phenol: chloroform with gentle agitation for 5 min followed by centrifugation and precipitated in two equivalence of cold ethanol and one-tenth equivalence of sodium acetate. After spinning down and decanting, the precipitate was re-suspended in 30 µl of deionised water-RNase solution [0.4 ml water + 5 µl of RNase] and 5 µl of loading buffer for 30 min at 37 °C. The 0.8% agarose gel with ethidium bromide was run at 5 V for 10 min before increasing to 100 V and documented in gel documentation system [35].
E2 assay in rat serum and liver tissue by ELISA method
Estradiol (E2) was assayed in serum and liver tissue of rats by the ELISA Kit (LILAC-ACCUBIND, Mumbai, India) method based on the principle of a solid phase enzyme-linked immunosorbent assay. Absorbance was measured spectrophotometrically at 450 nm.
Studies on expression of estrogen sulfotransferase protein by immuno-blotting
Cytosol proteins from liver (10 µg) were separated on a 12% (w/v) polyacrylamide gel in an electrophoresis system (BIORAD, USA). After running at 200 V, the protein bands were transferred overnight at 40 V onto a nitrocellulose membrane. All membranes were blocked in TBST (50 mM Tris, pH 7.5, 150 mM NaCl, and 0.05% (v/v) Tween 20) containing 5% (w/v) dried milk for 1 h on a shaker at room temperature. Membranes were incubated with rabbit anti-(rat)SULT1E1 (1:500) in TBST containing 5% (w/v) dry milk for 2 h on a shaker at room temperature. After incubation, membranes were washed with TBST for 4 × 15 min and incubated with secondary antibody (horseradish peroxidase-conjugated Immuno-Puregoat anti-rabbit IgG; H + L) at 1:5000 dilutions in the same buffer for 2 h. The membranes were washed with TBST for 4 × 15 min and then with trisbuffered saline (TBS) 3 × 5 min. The bands were developed with 1 ml of substrate containing 3,3′-Diaminobenzidine (DAB) Liquid Substrate System tetrahydrochloride. The metal enhanced DAB substrate utilizes cobalt chloride and nickel chloride in a special formulation to produce a dark brown/black precipitate in the presence of horseradish peroxidase (HRP). The SULT1E1 bands on the membrane were scanned and the densitometry analysis was performed with ImageJ software in a Gel Documentation and Analysis System from Advanced American Biotechnology (Fullerton, CA). We sincerely thank Dr. Guangping Chen of Department of Physiological Sciences of Oklahoma State University for kindly providing us the primary antibody against rSULT1E1.
Histological studies
Liver tissues of control group, ENU treated, arsenic treated and estradiol + xenografted animals were processed according to the standard protocol and were embedded in paraffin, serially sectioned at 5 µM. The slides were then stained with eosin and hematoxylin (Harris), and were observed under a microscope (Nikon, Eclipse LV100, magnification × 10 and × 100) to study the tissue histo-architecture.
Bioinformatics analysis of possible interactions of SULT1E1 with other proteins
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) is a biological database and web resource of known and predicted protein–protein interactions [40, 41]. String is a precomputed database derived from experimental data, literature mining, analysis of coexpressed genes etc. String applies a unique scoring method based on the different types of associations against a common reference set and produces a single confidence score per prediction [42]. Evidence based interactive or interrelated pattern has been deduced by this online resource/analytical software (http://string-db.org/).
Statistical analysis
The statistical analyses were done by using the SPSS for Windows statistical software package (SPSS Inc., Chicago, IL, USA, 2010). Normally distributed data were tested by Kolmogorov–Smirnov test. Baseline continuous-variables and outcome-measures were compared by Students t’ test analysis.
Results
rSULT1E1 protein expression and E2 level
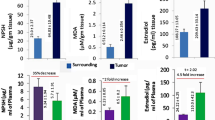
The rSULT1E1 expression was found to be reduced in the liver tissues of ENU-administered group (p < 0.01) and in estradiol-treated group (p < 0.001) compared to the control group. The average serum estradiol was found to be significantly increased in rats of ENU-treated group (p < 0.01) in comparison to that of control group. In E2-administered group this value increased by 100% (p < 0.001). The average tissue estradiol was found to be significantly increased (2.2 fold p < 0.001) in rats of E2-treated group (Fig. 1). Generally highly expressed SULT1E1 will increase sulfoconjugation of E2 resulting in less tissue E2. Our results shows less SULT1E1 expression and hence elevated E2. This result signifies that amount of E2 and SULT1E1 expressions are inverse in nature.

The rSULT1E1 and E2 regulations are presented. a Immunoblotting of rat estrogen sulfotransferase (rSULT1E1). Lane distribution, 1–3—control; 4–6—ENU; 7–9—estradiol. b Densitometric analysis of rSULT1E1 protein band and their statistical analysis (n = 3 in each group). c Serum and d liver E2 level in control, ENU treated and xenograft-E2 treated rats (n = 9 in each group). (Data in the bar diagram graph represent mean ± SE. Level of significances of differences are shown between the treated and control group; (a) p < 0.05, (b) p < 0.01, (c) p < 0.001)
Assay of super oxide dismutase (SOD1) and catalase activities; in vivo and in vitro
The SOD activity was found reduced in ENU administered group as compared to the control group (p < 0.05) whereas the activity was found to be increased in estradiol-fed group (p < 0.01) (Fig. 2). The catalase activity was reduced in the ENU administered group as compared to the control group whereas slightly increased in the estradiol-fed group (Fig. 2). Both ENU and E2 impose threats like oxidative stress via different ways. ENU may cause mutations in the DNA leading to structural impairment in SOD and catalase resulting in their low activities. On the other hand E2 causes increased SOD activities though E2 also induces oxidative stress via estrogen metabolites such as Catecholamine, Quinone’s and semiquinones. For better evaluation we did some in-vitro experiments using NAC, GSH and E2.

Regulations of antioxidant-enzyme activities are shown by PAGE zymography. The samples were prepared from female rat liver in response to different drugs. a Left panel—SOD1, 100 µg protein was loaded, Lane distribution; 1–4—control, 5–8—ENU treated, 9, 10—E2 treated in xenograft rats. Catalase: 50 µg of protein was loaded, Lane distribution; 1–4—control, 5–8—ENU treated, 9, 10—E2 treated in xenograft rats. GPx: 100 µg protein was loaded, lane distribution; 1–4—control, 5–8—ENU treated, 9, 10—E2 treated in xenograft rats. Corresponding densitometric analysis data are shown in the bar diagram (mean ± SE). Level of significances of differences are shown between the means of treated and control group; a, p < 0.05, b, p < 0.01, c, p < 0.001). b Right panel—liver tissue slices were incubated with different reducing agents or with estrdiol for 0, 6 or 12 h and then its cytosolic fraction were tested to determine antioxidant enzyme activities in gel zymogram study. Lane distribution; 1—1 mM NAC for 6 h, 2—1 mM GSH for 6 h, 3—1 mM NAC + 1 mM GSH, 4—10−7 M estradiol, 5—incubated control, 6—1 mM NAC for 12 h, 7—1 mM GSH for 12 h, 8—1 mM NAC + 1 mM GSH for 12 h, 9—10−7 M estradiol for 12 h, 10—incubated control for 12 h, 11—unincubated control, 12—old liver sample
The SOD activity was found to be increased most in 1 mM NAC followed by 1 mM GSH and a combination of both 1 mM GSH and NAC but was found to be decreased in 10–7 M estradiol, as compared to control at 6 h of incubation. The SOD activity in all of them was found to be further decreased at 12 h of incubation compared to 6 h. The catalase activity was found to be slightly increased in 10–7 M E2 treated group (Fig. 2). This explains E2 alone may decrease SOD activity due to increased oxidative stress but may increase activity via GSH induction. This is also satisfied by in vivo experiment where E2 treated animals showed elevated NPSH (Fig. 3).

Toxicity parameters in the serum/liver of control, ENU treated and xenograft-E2 treated female rats are represented here (data in the graph represent mean ± SE). NPSH and MDA levels in serum and liver tissue of control, ENU treated and xenograft-E2 treated female rats are also shown (data in the graph represent mean ± SE). Level of significances of differences are shown between the means of treated and control group; (a) p < 0.05, (b) p < 0.01, (c) p < 0.001). (Data in the bar diagram graph represent mean ± SE. level of significances of differences are shown between the treated and control group; (a) p < 0.05, (b) p < 0.01, (c) p < 0.001)
General toxicity
In the present investigation the mean SGPT level in control group was 11.89 U/L (± 3.49), in ENU administered group was 16.76 U/L (± 3.96), and in xenograft- E2 group (p < 0.05) was 20.865 U/L (± 0.751) (Fig. 3). The SGPT was found to be significantly increased (p < 0.001) in E2-treated group and SGOT increased (p < 0.01) in ENU-treated group. No significant increase was noticed in serum albumin level in either drug group but bilirubin increased in ENU-treated (p < 0.01) and E2-tretaed group (p < 0.05) with comparison to that of control. The mean ALP activity in control group was 8.9 KA units (± 0.303), in ENU administered group ~ 85–100% activity was increased (15.173 ± 1.092 KA, p < 0.01), and in xenograft-E2 group (17.13 ± 5.65 KA, 96% higher) (Fig. 3). A notable increase (55%) in mean serum urea level, an important kidney toxicity marker was noticed only in ENU-treated group (Fig. 3). Both the ENU and E2 were toxic for renal and hepatic system.
The NPSH acts as a soluble anti-oxidant which maintains the cellular redox environment. The mean serum NPSH level did not change in any treated group with comparison to that of control (Fig. 3). The mean tissue NPSH level increased ~ 47% in ENU treated and 250% in E2 treated group (p < 0.001). The lipid peroxidation product and strong anti-oxidant MDA increased in serum by 2–2.5 times (p < 0.01) in the drug treated groups (Fig. 3). The mean tissue MDA level in control group was 4.89 ± 0.56 µM/g and in ENU administered group it was 8.97 ± 1.11 µM/g (higher p < 0.01), and in xenograft-E2 group the value was 3.53 ± 1.368 µM/gm (Fig. 3). This concludes that E2 might have both pro-oxidant and antioxidant property.
Arsenic toxicity results
Present result suggests that arsenic can induce hepatotoxicity. This is evident from the result of increased serum ALP (p < 0.05) and SGPT activities (p < 0.01). Increased MDA and decreased NPSH levels (p < 0.05) also suggest the possible incidence free radical associated lipid damage and depletion of cellular antioxidants. Uric acid regarded as a dependable antioxidant molecule which was noticed to be depleted by 50% in arsenic treated rat group. In the current study, unlike ENU and E2 group rat estrogen sulfotransferase (rSULT1E1) did not alter in response to arsenic treatment (Fig. 4). Liver and serum E2 levels were also remained unchallenged (data not shown). But the oxidative stress and free radicals was found to induce DNA and tissue damage in arsenic treated rats. Arsenic ingestion with the present dose and duration resulted in hepatocyte disarrangement with lobular degeneration.

Effect of arsenic toxicity is demonstrated on different biochemical, molecular and histological parameters in experimental rat model (n = 6 in each group). (Data in the bar diagram graph represent mean ± SE. level of significances of differences are shown between the treated and control group; (a) p < 0.05, (b) p < 0.01, (c) p < 0.001)
Histology and DNA fragmentation results
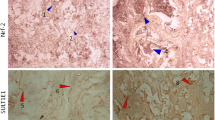
Low density DNA bands and DNA laddering was observed in the ENU administered group as compared to the control and the estradiol administered group. Central veins are quite prominent in the control rat livers (Fig. 5) as compared to the ENU and E2 treated groups. Deeply eosinophilic cytoplasm was noticed both in the E2 and the ENU (Fig. 5) treated animals. The control group indicates cord-like arrangement of hepatocytes with clear nucleoli. The arrows in the Fig. 5 shows distorted and elongated central vein. The nucleus seems to be darkly stained in the E2 group in (yellow arrow) Fig. 5, representing nuclear condensation and indicates swollen hepatocytes. The arrow in the Fig. 5 indicates infiltration of the inflammatory cells along with vascular congestion in the ENU treated group. Disarranged hepatocytes were noticed in both E2 and ENU treated group.

Changes are demonstrated in histo-architecture and DNA stability in liver tissue of female rats in response to different drugs. Left panel- Histological evaluations: a control group (× 10); b control group (× 100); c estradiol (× 10); d estradiol treated group (× 100); e ENU treated group (× 10); f ENU treated group (× 100). The control group indicates cord-like arrangement of hepatocytes with clear nucleoli. The arrows shows distorted and elongated central vein. The nucleus seems to be darkly stained in the E2 group in (yellow arrow). Right panel—DNA fragmentation analysis: lane 1–3—control, 4–6—ENU treated, 7, 8—estradiol
Bioinformatics results
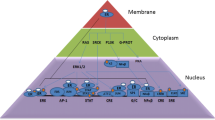
Our present bioinformatics result suggests that rSULT1E1 interacts with several enzyme proteins as described in Fig. 6, showing a strongest interacting score with phase I Cyp450 group of enzymes (Cyp19 and Cyp3). The rSULT1E1 is also highly interactive with UDP glycosyltransferase 2 family. Like rSULT1E1 this enzyme is a phase II drug metabolizing enzyme. In all cases the intensity score for the interaction was > 0.900. The predicted specific action was a strong dependence among phase I and phase II enzymes. It concludes that rSULT1E1 is a novel interacting proteins for steroid specially E2 metabolism (Fig. 6).

Interactive role of rSULT1E1 is represented. The STRING-10, efficient protein interacting software was used to prepare this diagram. The rSULT1E1 is an important protein member belongs to Phase II drug metabolizing enzyme which exerts direct influence to HSD-17β, several UDP-glucaronosyltransferase and cyp450 group of enzymes which is discussed in the text. Neighborhood, gene-fusion, concurrence, co-expressions, experiments, databases, test-mining and homology modeling were the default option in the software to generate this interactive diagram
Discussion and conclusion
The ENU is extensively used to generate animal cancer models due to its potent carcinogenic nature [4, 6]. Immuno-compromised rodents, xenografted with breast cancer cells or patient-derived cancer-cells and treated with estradiol have been used to generate animal models of breast cancer [43,44,45]. In the present study we evaluated the patho-physiology, toxicity and oxidative-stress of humanized-rat initiated by human-tumor xenografting and E2 feeding to the animal. In another group of rats, same parameters were screened after ENU treatment. The use of disease initiation and prognostic markers for breast cancer may help therapeutic strategies to be defined more efficiently. In the current study the role of SOD, catalase and GPx has been studied in relation to the pre-tumorigenic disease condition. The role of SULT1E1 and E2 has also been studied in vitro and in vivo experiments.
Initiation of carcinogenesis has involvement of several biochemical, genetic or proteomic events occurring together or separately. In both cases alterations of SULT1E1 and E2 were noticed. But the result in arsenic treated group was little different. This supports our present finding. It is evident that arsenic-induced liver damages like necrosis and degeneration was the result from oxidative stress and free radical attack [42]. This is manifested by the liver function test and histoarchitecture data. SULT1E1 did not change in this drug group suggesting no alterations of E2 regulations via this pathway as noticed in ENU and E2 groups. Oxidative stress is one of the biochemical events that may initiate carcinogenesis [46]. Indeed, ROS are important regulators of growth-factor receptor signalling [47]. Moreover these are involved in estrogen-inducible cancer cell proliferation [48, 49].
Carcinogenesis induced by ENU at genomic level has been reported. Mainly several oncogene and/or tumor suppressor gene are shown to be involved. As for example, miRNA-mediated BRCA1 and P53 signalling has been identified as important pathways for the tumorigenesis/carcinogenesis [50]. In accordance with the present study, oxidative-stress initiated by both ENU and E2 is evident and indicates an enhanced lipid-peroxidation and other stress-parameters [46], along with histoarchitectural abnormalities in the treated group compared to the control (Fig. 5). The GSH is accepted as a free radical scavenger and associated in maintaining cellular reducing environment and normal physiological state. The cysteine or non-protein soluble thiol (NPSH) is required for the synthesis of GSH. Not only during disease initiation or progression, but also during therapeutic approaches against disease, GPx and GSH have exerted their valuable role. The increase of glutathione (GSH) level and GPx activity in tumor cells has been noted as a determinant of disease prognosis and response to cytotoxic treatments. Doxorubicin treatment was able to eliminate tumor cells and at the same time suppressed the expression of the GPx gene. High levels of GPx may be related to the development of resistance during breast cancer chemotherapy [51].This suggests that antioxidant protection by GPx should be abrogated in tumor cells. However, in our study impairment of GPx after ENU treatment was helpful in the initiation of the pre-tumorigenic condition. Not only in breast cancer, has GPx been shown to be a tumor suppressor gene in pancreatic cancer also. Delivery of the GPx gene alone or in combination with the MnSOD gene may prove beneficial for the treatment of pancreatic cancer [52]. In the current study, increase of NPSH in ENU or estradiol treated group suggest that NPSH and GSH synthesizing machinery in liver as a protective factor, but not sufficient in the ENU group to suppress the entire stress generated. Eventually, it results in enhanced lipid-peroxidation and MDA level. A moderate level of MDA in hepatocyte but higher level in serum was noticed in rats of xenograft-E2 group. Report suggests that MDA, advanced oxidation protein product (AOPP) and SOD activities were significantly higher in patients with breast cancer. These have been pointed as useful biomarkers in breast cancer diagnosis and treatment [53]. Influences on GPx and SOD activity in our experiment may be noted as important factor prior to the disease initiation process. Report reveals that antioxidant enzyme; SOD and catalase activities significantly increased in liver of ENU-treated rodents with an elevated level of tissue MDA [54]. In the present positive control experiment the elevated levels of serum transaminases and alkaline phosphatase (indicator of liver functions) were observed in arsenic treated rat and this may be related to the extensive alterations in the liver histology indicating liver damage [55]. Present possibilities may be corroborated with our evidence on the increase in oxidant stress response initiated by MDA and resulting from the impaired antioxidant capacity, that is, NPSH and uric acid level. Altered redox environment may be vulnerable to proper SOD structure/ activity and DNA stability [39]. Proteomic analyses identified that inactivation of SOD is related to the redox modification of important cysteines [39]. These altered regulations might have been without the influences of SULT1E1 expressions (Fig. 2).
Serum NPSH level in rats of ENU or estradiol treated group was similar to that of control. Earlier studies reported that ENU induces increase in gamma-glutamyl-transpeptidase (GGT) [56, 57], which provides cysteine for the synthesis of GSH by γ-glutamyl cysteine synthetase [57]. It results in the decrease in extra-cellular reducing component [58]. A high increase is noticed in liver thiol in rats of E2-treated group (Fig. 3). A moderate increase is also observed in ENU-treated group (Fig. 3). This may support the upregulation of both the enzymes for GSH synthesis in liver. Thus, oxidative stress may increase GGT to constitute an adaptation to stress, but high GGT also induces preneoplastic lesions [56]. Estradiol infusion has been shown to increase GSH synthesis by activating the γ-glutamyl cysteine synthetase in liver but not in breast tissues [59]. Impairment of SOD activity in ENU-treated group and activation in estradiol-treated group were noticed in the current study. The result from animal model is also supported by the present in vitro experiment (Fig. 2). Low catalase activity in ENU-treated rats strongly justifies the correlation between oxidative stress and DNA damage in this group [24, 60]. Our finding on SOD dysregulation in pre-disease condition is substantial and this may be therapeutically targeted during even post-disease period. SOD/catalase ‘mimetic’ (EUK 134) has been shown to effectively attenuate via inhibition of H2O2 signalling the viability, proliferation, clonal expansion, adhesion, and migration of human breast cancer cells; MCF-7. This ‘mimetic’ has been shown to act by TNFα-induced reduction of NF-κB function [61]. This suggests that the inflammation associated transcriptional regulation of certain gene may be a target of redox impairment. This also supports our proposal on redox regulation of E2 signalling by the modification of E2 metabolizing proteins. In case of positive control, arsenic exposure also exhibited oxidative stress through significant reduction of GSH in liver, cultured lung epithelial cells and discrete brain areas [62, 63]. But unlike ENU/E2 treated groups these interferences were not related to the regulations of SULT1E1 expressions (Fig. 4).
ROS ultimately results in mutagenic DNA breakage [64]. It is evident in ENU-treated group where possible alteration in gene-expression might result in pre-malignant tissue lesions [6] as visible in our histology results. It indicates central vein distortion and nuclear condensation was found in the E2 group (Fig. 4) supported by earlier studies which concluded that E2 induced a dose-dependent increase in DNA fragmentation and nuclear condensation [65]. Infiltration of inflammatory cells in the central vein of the ENU treated group was found may be due to the induction of necrosis [66]. The apoptotic-degenerative signalling might have been generated via mitochondrial-membrane instability followed by cytochrome-c release, caspases, and BAD/Bcl-2 activation [67] resulting in DNA damage. ENU was found to induce liver toxicity resulting in increased serum markers (Fig. 3). Increased serum and tissue MDA indicates direct relation between ROS and oxidative stress and liver-kidney toxicity in both E2 and ENU groups. Estradiol was also found to increase the liver toxicity with reduced lipid peroxidation in this tissue but, overall increase in serum indicating two possibilities. Either estradiol acts as an antioxidant in liver by increasing GSH and reducing MDA at production level or augmenting transportation of MDA to serum (Fig. 3). Estrogen is an endogenous and normal physiological biomolecule. This may indicate the protective or detrimental role of estrogen is associated to its dose and exposure schedule. Report reveals that estrogen plus progestin increase superoxide dismutase and total antioxidant capacity in postmenopausal women [65]. The removal of E2 by ovariectomy decreases the activity of the antioxidant enzymes and increases lipid peroxidation in the female rats [66].
The role of estrogen nuclear-receptor is important for the genomic regulations by estrogen. During reproductive functioning receptor-estrogen complex influences several associated genes [67]. Not only reproductive functions, an important role of estrogen receptor α (ERα) results in estrogen-mediated transcriptional signalling and overexpression of the important drug-metabolizing enzyme, glutathione S-transferase P1-1 (GSTP). This enzyme utilizes GSH for the conjugation reactions in tumors. GSTP overexpression has been correlated to the poor prognosis in breast cancer [68]. The interdependence of GSTP-expression and ERα signalling apparently indicate an association between E2 and GSH function. In the current study E2 associated GSH increase has been indicative in this regard. Interestingly, production of GSH-E2 conjugate has been evident, but several fold lower than the amount of catechol-estrogen in cell. So, the accumulation of estrogenquinones is higher (than the GSH-E2 conjugation) which participate in DNA damaging events in the E2 dependant breast carcinogenesis [69]. The regulation of E2 metabolism is also great importance for the metabolites it generates. SULT1E1, HSD17 and aromatase are important enzymes in this regard. Our present bioinformatics study shows the significant association amongst these enzymes in term of their protein–protein interaction. This might have a significant impact on the concentration of active E2.
Regulation of E2 function is significantly influenced by the redox-regulated enzyme estrogen sulfotransferase (SULT1E1). This enzyme is responsible for sulfoconjugation followed by the inactivation of estrogen and estradiol-mediated signalling [70]. The protein expression of SULT1E1 was found to be reduced in ENU or estradiol-administered group in the present study. ENU and E2 thus interfere either with transcriptional or translational machinery involved in SULT1E1 expression. This needs further confirmation. Estradiol level was increased in liver of both test groups as compared to the control. Low SULT1E1 expression in the xenograft-E2 group (Fig. 1) supports the outcome of E2 level far exceeding than that of the control rats. It is considered that high E2 level that is exceeding even the highest level of E2 in the oestrus cycle may influence the SULT1E1 expression in the liver tissue where GSH level was also found to be much higher than compared to that of control [70]. High E2 level along with low SULT1E1 expression (Fig. 1) determines that E2 controls its abundance through SULT1E1 expression/regulation. And this control is conferred by the altered GSH-regulation [70]. Earlier studies supports that E2 induces GSH synthesis in the rat liver. In vitro, treatment of normal human breast epithelial cells or breast tissue with estradiol and progesterone did not alter GSH levels. But the exposure of hepatocytes to this steroid increased the cellular content of gamma-glutamylcysteinesynthetase followed by increase in GSH level [59]. This is evident in our current study. Beside synthesis, report reveals that estradiol also increase consumption of plasma GSH to the erythrocytes during the regular reproductive cycle of women [71].
GSH generates a reducing environment which may not be favourable for the expression of SULT1E1, since this enzyme is an oxidative stress induced protein via Nrf2 signalling [72]. Noticeably, tocopherol a strong oxidant-scavenger has been shown to stimulate Nrf2-dependent antioxidant response in the mammary glands of E2-treated rats. Inductions of mRNA levels of Nrf2 along with antioxidant enzymes; superoxide dismutase, catalase, and glutathione peroxidase have been demonstrated and this anti-oxidative induction diminished E2-induced mammary hyperplasia [73]. Stronger but differential involvement of GPx has been shown in breast cancer patients. Studies indicated a role for GPx-1 allelic identity and loss of heterozygosity as factors of significance to breast cancer development [74].
In the present study also, strong antioxidative potentiations have been noticed in terms of SOD and catalase activities (Fig. 2). E2-regulated GSH synthesis in liver may activate SULT1E1 which in turn deactivate E2 by forming E2-sulfate. Thus, E2 can auto-regulate at least partially, the level of its active form via GSH in liver. The cellular protection of E2 is conferred by its physiological/pharmacological relevant dose via the non-receptor mediated hence, non-genomic pathway. And the potency of E2 found to be few 100 folds higher in the presence of glutathione [75]. The paucity of cytoplasm and the abundance of nuclei is visible in HE stained slides from the arsenic treated group Arsenic and ROS-induced elevations of c-reactive proteins have been noticed in vitro in human hepatocarcinoma HepG2 cell lines [76]. Evidence suggests both ROS and CRP can activate the transcription factor NF-κ-β [76] and several mitogen-activated protein kinases (MAPKs) [77]. In contrast to the MAPKs regulations during arsenic-related mutagenesis, redox regulation act via inverse Akt regulated phosphorylation in breast cancer cells MCF-7 which are oxidative stress resistant. On the other hand, only MCF-7 cells (oxidative stress sensitive) are shown to be responsive and dependent on PI3K/Akt/mTOR signalling [78]. This suggests that classical transcriptional regulations works in different ways in arsenic carcinogenesis and breast carcinogenesis model, though both are regulated by the cellular redox environment.
In support to our rat experiments, we also noticed an increase of SOD activity in all in-vitro treatment (6 h) groups, which include N-acetyl cysteine (NAC) and GSH, and in the E2-administered group. In 6 h group cell remains more alive and metabolically active than that of 12 h group which favours transportation of the biomolecule from the external environment to the internal milieu of the cell. It is reported that E2 at its highest concentration is a stress inducer that favours the adaptive responses in terms of the increase of intracellular thiol [59] by activating synthesizing machineries. This is noticed in our in vivo experiments. In addition, the enhanced thiol activated hepatocytes SOD (Fig. 2). Being isolated, the in-vitro system was not capable to activate all steps up to the thiol synthesis, so no change was found. In case of NAC being either the direct stimulator of the thiol or thiol itself, can stimulate the gamma glutamyl cysteine synthetase mediated GSH synthesis [79], and GSH itself being thiol, these molecules were able to activate SOD better than estradiol. Evidence suggests that for one-electron oxidation of estradiol to its reactive phenoxyl radical intermediate is possible. The hydrogen is abstracted by the phenoxyl-radical metabolite from reduced glutathione. This generates the glutathione thiyl radical. Moreover, the estradiolphenoxyl radical also can abstract hydrogen from reduced NADH which generates the NAD·radical [80]. The superoxide generated may spontaneously dismutate to form hydrogen peroxide. It may also react with another NADH to form NAD·, thus propagating a cascade reaction leading to oxygen consumption and hydrogen peroxide accumulation. The accumulation of intracellular hydrogen peroxide could explain the hydroxyl radical-induced DNA base lesions recently reported for female breast cancer tissue [80, 81].
In case of catalase we did not get that much changes within groups. Catalase is a terminal antioxidant enzyme which utilizes its substrate H2O2 which comes from the different metabolic pathways. So, high level of H2O2 induces for an optimum and sufficient levels of catalase activation. This may be the reason that catalase activity was found to be consistently high to ensure the possible oxidative damage from H2O2. Due to this initial high level, no sufficiently significant change was noticed. On the other hand superoxide anion (O2.−) is the substrate of an initial or early stage-antioxidant enzyme like SOD. The control value of SOD itself is lower which keep space for its modification depending on factor that’s why we get changes in different groups. The lower value of SOD in the 12 h control group with corresponding to the 6 h group suggests that cell and tissue become metabolically less responsive. A study shows inactivation of SULT1E1 under oxidative stress. Thus, E2 either inducing a reducing environment or an oxidising environment differentially respond in cellular metabolism. Report reveals that co-treatment with tamoxifen decreased glutathione peroxidase and catalase levels. But, it did not completely inhibit E2-mediated oxidative DNA damage, cell death and breast carcinogenesis.
Present result suggests that ENU or E2 applications differentially induce oxidative stress. E2 can be regarded as a protective or detrimental bio-molecule depending on its dose and exposure time/schedule. Unlike arsenic treated group, SULT1E1 and E2-mediated GSH regulation and how the redox environments influence the SULT1E1 or other gene/proteins need to be explored further. The inter-dependence of SULT1E1 and other proteins as noticed in our bioinformatics study infers the role of direct or indirect role of these proteins on active E2 metabolism and its functioning. The understanding of toxicity and oxidative-stress induction and tumorigenesis mechanism is very important stepping stone for any study of anti-cancer therapy. The present study is very much helpful in this regard.
References
Dietrich CG, Rau M, Jahn D, Geier A (2017) Changes in drug transport and metabolism and their clinical implications in non-alcoholic fatty liver disease. Expert Opin Drug MetabToxicol 13(6):625–640
Tsubura A, Lai YC, Miki H, Sasaki T, Uehara N, Yuri T, Yoshizawa K (2011) Animal models of N-methyl-N-nitrosourea-induced mammary cancer and retinal degeneration with special emphasis on therapeutic trials. In Vivo 25(1):11–22
Gurley KE, Moser RD, Kemp CJ (2015) Induction of liver tumors in mice with N-ethyl-N-nitrosourea or N-nitrosodiethylamine. Cold Spring Harb Protoc 2015(10):941–942
Stottmann R, Beier D (2014) ENU mutagenesis in the mouse. Curr Protoc Mouse Biol 4(2):25–35
Coghill EL, Hugill A, Parkinson N, Davison C, Glenister P, Clements S, Hunter J, Cox RD, Brown SD (2002) A gene-driven approach to the identification of ENU mutants in the mouse. Nat Genet 30(3):255–256
Barbaric IWS, Russ A. Dear TN (2007) Spectrum of ENU-induced mutations in phenotype-driven and gene-driven screens in the mouse. Environ Mol Mutagen 48:124–142
Shrivastav N, Li D, Essigmann JM (2010) Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis 31:59–70
Engelbergs J, Thomale J, Rajewsky MF (2000) Role of DNA repair in carcinogen-induced ras mutation. Mutat Res 450:139–153
Thordarson G, Lee AV, McCarty M, Van Horn K, Chu O, Chou YC, Yang J, Guzman RC, Nandi S, Talamantes F (2001) Growth and characterization of N-methyl-N-nitrosourea-induced mammary tumors in intact and ovariectomized rats. Carcinogenesis 22(12):2039–2047
Katayama K, Ueno M, Takai H, Ejiri N, Uetsuka K, Nakayama H, Doi K (2002) Ethylnitrosourea induces apoptosis and growth arrest in the trophoblastic cells of rat placenta. Biol Reprod 67(2):431–435
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5):479–496
Sanchez-Perez Y, Carrasco-Legleu C, Garcia-Cuellar C, Pérez-Carreon J, Hernández-Garcia S, Salcido-Neyoy M, Aleman-Lazarini L, Villa-Trevino S (2005) Oxidative stress in carcinogenesis. Correlation between lipid peroxidation and induction of preneoplastic lesions in rat hepatocarcinogenesis. Cancer Lett 217(1):25–32
Yager JD, Davidson N (2006) Estrogen carcinogenesis in breast cancer. Engl J Med 354:270–282
Deblois G, Giguère V (2011) Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim Biophys Acta 1812(8):1032–1040
Russo J, Russo IH (2006) The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol 102:89–96
Boukari K, Ciampi ML, Guiochon MA, Young J, Lombes M, Meduri G (2007) Human fetal testis: source of estrogen and target of estrogen action. Hum Reprod 7:1885–1892
Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78(2):547–581
Nelson HD, Zakher B, Cantor A, Fu R, Griffin JO, Meara ES, Buist DS, Kerlikowske K, van Ravesteyn NT, Trentham-Dietz A, Mandelblatt JS, Miglioretti DL (2012) Risk factors for breast cancer for women aged 40 to 49 years: a systematic review and meta-analysis. Ann Intern Med 156(9), 635–648
Benz CC, Yau C (2008) Ageing, oxidative stress and cancer: paradigms in parallax. Nat Rev Cancer 8(11):875–879
Maria BR, Elaine R, Nancy D, Maureen KW, Charles EW (2012) Genomics of estradiol-3-sulfate action in the ovine fetal hypothalamus. Physiol Genom 44(13):669–677
Maiti S, Zhang J, Chen G (2007) Redox regulation of human estrogen sulfotransferase (hSULT1E1). Biochem Pharmacol 73(9):1474–1481
Pasqualini JR, Chetrite GS (2007) Correlation of estrogen sulfotransferase activity and proliferation in normal and carcinomatous human breast. A hypothesis. Anticancer Res 27(5A):3219–3225
Santarelli RL, Pierre F, Corpet DE (2008) Processed meat and colorectal cancer: a review of epidemiologic and experimental evidence. Nutr Cancer 60(2):131–144
Maiti S, Chattopadhyay S, Deb B, Samanta T, Maji G, Pan B, Ghosh A, Ghosh D (2012) Antioxidant and metabolic impairment result in DNA damage in arsenic-exposed individuals with severe dermatological manifestations in Eastern India. Environ Toxicol 27(6):342–350
Wu YJ, Muldoon LL, Dickey DT, Lewin SJ, Varallyay CG, Neuwelt EA (2009) Cyclophosphamide enhances human tumor growth in nude rat xenografted tumor Models. Neoplasia 11(2):187–195
Ligresti A, Moriello AS, Starowicz K, Matias I, Pisanti S, De Petrocellis L, Laezza C, Portella G, Bifulco M, Di Marzo V (2006) Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther 318(3):1375–1387
Gordon MN, Osterburg HH, May PC, Finch CE (1986) Effective oral administration of 17 beta-estradiol to female C57BL/6J mice through the drinking water. Biol Rep 35(5):1088–1095
Levin-Allerhand JA, Sokol K, Smith JD (2003) Safe and effective method for chronic 17beta-estradiol administration to mice. Contemp Top Lab Anim Sci 42(6):33–35
Mense SM, Remotti F, Bhan A, Singh B, El-Tamer M, Hei TK, Bhat HK (2008) Estrogen-induced breast cancer: alterations in breast morphology and oxidative stress as a function of estrogen exposure. Toxicol Appl Pharmacol 232:78–85
Stoica G, Koestner A, Capen CC (1984) Neoplasms induced with high single doses of N-ethyl-N-nitrosourea in 30-day-old Sprague-Dawley rats, with special emphasis on mammary neoplasia. Anticancer Res 4(1–2):5–12
Pacheco-Martínez MM, Cortés-Barberena E, Cervantes-Ríos E, Del Carmen García-Rodríguez M, Rodríguez-Cruz L, Ortiz-Muñiz R (2016) Moderate malnutrition in rats induces somatic gene mutations. Mutat Res 789:26–32
Zaizen Y, Higuchi Y, Matsuo N, Shirabe K, Tokuda H, Takeshita M (2000) Antitumor effects of soybean hypocotyls and soybeans on the mammary tumor induction by N-methyl-n-nitrosourea in F344 rats. Anticancer Res 20(3A):1439–1444
Acharyya N, Sajed Ali S, Deb B, Chattopadhyay S, Maiti S (2015) Green tea (Camellia sinensis) alleviates arsenic-induced damages to DNA and intestinal tissues in rat and in situ intestinal loop by reinforcing antioxidant system. Environ Toxicol 30(9):1033–1044
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Methods Enzymol 52:302–310
Forman HJ (2009) Critical methods in free radical biology & medicine. Free Radic Biol Med 47:S207
Gamero-Sandemetrio E, Gómez-Pastor R, Matallana E (2017) Zymography methods to simultaneously analyze superoxide dismutase and catalase activities: novel application for yeast species identification. Methods Mol Biol 1626:189–198
Christine JW, Joseph JC (2010) Measurement of superoxide dismutase, catalase, and glutathione peroxidase in cultured cells and tissue. Nat Protoc 5(1):51–66
Acharyya N, Chattopadhyay S, Maiti S (2014) Chemoprevention against arsenic-induced mutagenic DNA breakage and apoptotic liver damage in rat via antioxidant and SOD1 upregulation by green tea (Camellia sinensis) which recovers broken DNA resulted from arsenic-H2O2 related in vitro oxidant stress. J Environ Sci Health C 32(4):338–361
Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovi M, Bork P, von Mering C (2009) STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37:412–416
Taboada B, Verde C, Merino E (2010) High accuracy operon prediction method based on STRING database scores. Nucleic Acids Res 38:130
Von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P (2005) STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res 33:D433–D437
Roomi MW, Roomi NW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M (2005) Modulation of N-methyl-N-nitrosourea induced mammary tumors in Sprague-Dawley rats by combination of lysine, proline, arginine, ascorbic acid and green tea extract. Breast Cancer Res 7(3):R291–R295
Morton CL, Houghton PJ (2007) Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc 2(2):247–250
Morton JJ, Bird G, Refaeli Y, Jimeno A (2016) Humanized mouse xenograft models: narrowing the tumor-microenvironment gap. Cancer Res 76(21):6153–6158
Yesennia SP, Claudia CL, Claudia GC, Julio PC, Sergio HG, Martha SN, Leticia AL, Saul VT (2005) Correlation between lipid peroxidation and induction of preneoplastic lesions in rat hepatocarcinogenesis. Oxidative stress in carcinogenesis. Cancer Lett 217(1):25–32
Lander HM (1997) An essential role for free radicals and derived species in signal transduction. FASEB J 11:118–124
Felty Q, Singh KP, Roy D (2005) Estrogen-induced G1/S transition of G0-arrested estrogen-dependent breast cancer cells is regulated by mitochondrial oxidant signaling. Oncogene 24:4883–4893
Qiu Y, Waters CE, Lewis AE, Langman MJ, Eggo MC (2002) Oestrogen-induced apoptosis in colonocytes expressing oestrogen receptor beta. J Endocrinol 174(3):369–377
Luan Y, Qi X, Xu L, Ren J, Chen T (2014) Absence of mature microRNAs inactivates the response of gene expression to carcinogenesis induced by N-ethyl-N-nitrosourea in mouse liver. J Appl Toxicol 34(12):1409–1417
Jardim BV, Moschetta MG, Leonel C, Gelaleti GB, Regiani VR, Ferreira LC, Lopes JR, Zuccari DA (2013) Glutathione and glutathione peroxidase expression in breast cancer: an immunohistochemical and molecular study. Oncol Rep 30(3):1119–1128
Liu J, Hinkhouse MM, Sun W, Weydert CJ, Ritchie JM, Oberley LW, Cullen JJ (2004) Redox regulation of pancreatic cancer cell growth: role of glutathione peroxidase in the suppression of the malignant phenotype. Hum Gene Ther 15(3):239–250
Kilic N, Yavuz Taslipinar M, Guney Y, Tekin E, Onuk E (2014) An investigation into the serum thioredoxin, superoxide dismutase, malondialdehyde, and advanced oxidation protein products in patients with breast cancer. Ann Surg Oncol 21(13):4139–4143
Silig Y, Ozdemir O, Atalay A (2003) Liver and colon pro- and anti-oxidant enzyme activities in rats after long-term ethylnitrosourea exposure. Indian J Biochem Biophys 40(2):136–138
Chattopadhyay S, Deb B, Maiti S (2012) Hepatoprotective role of vitamin B(12) and folic acid in arsenic intoxicated rats. Drug Chem Toxicol 35(1):81–88
Stark AA, Russell JJ, Langenbach R, Pagano DA, Zeiger E, Huberman E (1994) Localization of oxidative damage by a glutathione-gamma-glutamyltranspeptidase system in preneoplastic lesions in sections of livers from carcinogen-treated rats. Carcinogenesis 15(2):343–348
Zhang H, Forman HJ, Choi J (2005) Gamma glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol 401:468–483
Hanigan MH (2014) Gamma-glutamyl transpeptidase: redox regulation and drug resistance. Adv Cancer Res 122:103–141
Dabrosin C, Ollinger K (2004) Variability of glutathione during the menstrual cycle-due to estrogen effects on hepatocytes? Free Radic Biol Med 36(2):145–151
Calatayud M, Devesa V, Velez D (2013) Differential toxicity and gene expression in Caco-2 cells exposed to arsenic species. Toxicol Lett 218:70–80
Shah MH, Liu GS, Thompson EW, Dusting GJ, Peshavariya HM (2015) Differential effects of superoxide dismutase and superoxide dismutase/catalase mimetics on human breast cancer cells. Breast Cancer Res Treat 150(3):523–534
Li M, Cai JF, Chiu JF (2002) Arsenic induces oxidative stress and activates stress gene expressions in cultured lung epithelial cells. J Cell Biochem 87:29–38
Chattopadhyay S, Maiti S, Maji G, Deb B, Pan B, Ghosh D (2011) Protective role of Moringa oleifera (Sajina) seed on arsenic-induced hepatocellular degeneration in female albino rats. Biol Trace Elem Res 142(2):200–212
Lantz RC, Hays AM (2006) Role of oxidative stress in arsenic-induced toxicity. Drug Metab Rev 38(4):791–804
Unfer TC, Figueiredo CG. Zanchi MM, Maurer LH, Kemerich DM, Duarte MM, Konopka CK, Emanuelli T (2015) Estrogen plus progestin increase superoxide dismutase and total antioxidant capacity in postmenopausal women. Climacteric 18(3):379–388
Guerra RC, Zuniga-Munoz A, Guarner Lans V, Diaz-Diaz E, Tena Betancourt CA, Pérez-Torres I (2014) Modulation of the activities of catalase, Cu-Zn, Mn superoxide dismutase, and glutathione peroxidase in adipocyte from ovariectomised female rats with metabolic syndrome. Int J Endocrinol. https://doi.org/10.1155/2014/175080
Singh B, Bhat NK, Bhat HK (2011) Partial inhibition of estrogen-induced mammary carcinogenesis in rats by tamoxifen: balance between oxidant stress and estrogen responsiveness. PLoS ONE 6(9):25125
Liu X, An BH, Kim MJ, Park JH, Kang YS, Chang M (2014) Human glutathione S-transferase P1-1 functions as an estrogen receptor α signaling modulator. Biochem Biophys Res Commun 452(3):840–844
Hachey DL, Dawling S, Roodi N, Parl FF (2003) Sequential action of phase I and II enzymes cytochrome p450 1B1 and glutathione S-transferase P1 in mammary estrogen metabolism. Cancer Res 63(23):8492–8499
Mauvais-Jarvis F (2012) Estrogen sulfotransferase: intracrinology meets metabolic diseases. Diabetes 61(6):1353–1354
Sheng-Huang C, Chieh-Hsin C, Mu-Chun Y, Wen-Tung H, Chia-Ying H, Ya-Ting H, Wan-Ling SU. Jiuan-Jen S, Chih-Yang H, Jer-Yuh L (2015) Effects of estrogen on glutathione and catalase levels in human erythrocyte during menstrual cycle. Biomed Rep 3(2):266–268
Guo Y, Hu B, Huang H, Tsung A, Gaikwad NW, Xu M, Jiang M, Ren S, Fan J, Billiar TR, Huang M, Xie W (2015) Estrogen sulfotransferase is an oxidative stress-responsive gene that gender-specifically affects liver ischemia/reperfusion injury. J Biol Chem 290(23):14754–14764
Das Gupta S, So JY, Wall B, Wahler J, Smolarek AK, Sae-Tan S, Soewono KY, Yu H, Lee MJ, Thomas PE, Yang CS, Suh N (2015) Tocopherols inhibit oxidative and nitrosative stress in estrogen-induced early mammary hyperplasia in ACI rats. Mol Carcinog 54(9):916–925
Hu YJ, Diamond AM (2003) Role of glutathione peroxidase 1 in breast cancer: loss of heterozygosity and allelic differences in the response to selenium. Cancer Res 63(12):3347–3351
Green PS, Gridley KE, Simpkins JW (1998) Nuclear estrogen receptor-independent neuroprotection by estratrienes: a novel interaction with glutathione. Neuroscience 84(1):7–10
Druwe IL, Sollome JJ, Sanchez-Soria P, Hardwick RN, Camenisch TD, Vaillancourt RR (2012) Arsenite activates NFκB through induction of C-reactive protein. Toxicol Appl Pharmacol 261(3):263–270
Xu Y, Li Y, Li H, Pang Y, Zhao Y, Jiang R, Shen L, Zhou J, Wang X, Liu Q (2013) The accumulations of HIF-1α and HIF-2 α by JNK and ERK are involved in biphasic effects induced by different levels of arsenite in human bronchial epithelial cells. Toxicol Appl Pharmacol 266(2):187–197
Glorieux C, Auquier J, Dejeans N, Sid B, Demoulin JB, Bertrand L, Verrax J, Calderon PB (2014) Catalase expression in MCF-7 breast cancer cells is mainly controlled by PI3K/Akt/mTor signaling pathway. Biochem Pharmacol 89(2):217–223
Manjula RT (2015) Depletion of glutathione during oxidative stress and efficacy of N-acetyl cysteine: an old drug with new approaches. Med Chem 5(1):037–039
Sipe HJ Jr, Jordan SJ, Hanna PM, Mason RP (1994) The metabolism of 17 beta-estradiol by lactoperoxidase: a possible source of oxidative stress in breast cancer. Carcinogenesis 15(11):2637–2643
Pullela PK, Alby K, Varela H, Sem DS (2006) Effect of thiol reductants on estrogen and estrogen receptor: revisiting previous assays. FASEB J 20:A74
Acknowledgements
University Grants Commission, New Delhi provided JRF and SRF to AN who is a Ph.D. students working in the Post Graduate Department of Biochemistry, OIST.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicting interest
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
Rights and permissions
About this article

Cite this article
Nazmeen, A., Maiti, S. Oxidant stress induction and signalling in xenografted (human breast cancer-tissues) plus estradiol treated or N-ethyl-N-nitrosourea treated female rats via altered estrogen sulfotransferase (rSULT1E1) expressions and SOD1/catalase regulations. Mol Biol Rep 45, 2571–2584 (2018). https://doi.org/10.1007/s11033-018-4425-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-4425-z