
Abstract
Human estrogen sulfotransferase (SULT1E1) and nuclear factor erythroid 2-related factor 2 (Nrf-2) expression influences each other in advanced human breast carcinogenesis. The difference in the metabolism of estradiol (E2) in pre- and post-menopausal women remains to be connected with post-menopausal breast cancer. A synergism between ROS production and E2 generation has been demonstrated. No definite mechanism for simultaneous functions of Nrf2, oxidative stress E2 regulating enzymes (SULT1E1) has been yet clarified. Our present review demonstrates that ROS dependent regulation of Nrf-2 is one of the most important determinants of E2 regulation by altering SULT1E1 expression. This study also focuses the idea that estrogen receptor cased subtypes of cancer may have different molecular environments which has an impact on the therapeutic efficacy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The aerobic life system in the mammalians has extensive cellular protection machinery. Oxidative stress affects the cells and exhibits a coordinated expression of genes and their corresponding proteins having antioxidant capacity and different phases of drug metabolizing efficacy [1,2,3]. Carcinogenesis is a multi-step and multi-factorial disease. Oxidative stress is one of the steering factors in the initiation and pathogenesis of cancer. Modifiable risk factors are related to breast cancer pathogenesis and severity [4]. Even the obesity associated risk of breast cancer differs on the basis of estrogen receptor (ER) status in pre- and postmenopausal women [5]. Estrogen and estrogen receptor influences several factors that may cause breast cancer. An animal study confirms the effect of estrogen (mainly estradiol) on adipose tissue. Some studies also reports that visceral adipose tissue (VAT) is a major site of estrogen production. Obesity can induce pulmonary hypertension and causes changes in estrogen metabolism. This condition results in an increased production of 16α-hydroxyestrone (16αOHE1) instead of normal estrogen from visceral adipose tissue (VAT) and 16α-OHE1 contributes to oxidative stress [6]. These reports suggest influence of estrogen and its metabolite directly or indirectly on the occurrence of breast cancer.
The Sulfotransferase family 1E member 1 (SULT1E1) disrupts estrogen homeostasis by sulfo-conjugation/deactivation of estrogen and may control tumorigenesis and or progression of breast and endometrial cancers. The protein levels of SULT1E1 were found to be reduced in breast cancer cell line (MCF-7, T47D and MDA-MB-435 cells).Whereas the same study have also shown that inducing SULT1E1 overexpression were able to inhibit the growth of breast cancer cell, induced apoptosis and arrested cell cycle progression [7]. Studies also suggest that an impairment of SULT1E1 activity occurs via redox modulation [8]. Oxidative stress takes regulatory actions on estrogen metabolizing proteins. Studies suggested that Nrf2 induces the expression of SULT1E1, which subsequently increases sulfation of estrogen leading to its deactivation. This sequentially is able to limit the estrogen-mediated activation of Nrf2 [9]. Hence, there seems to have an existence of a fulcrum which balances E2 and Nrf2 or making them interdependent. This review aimed to conduct an in-depth review based on our earlier studies and few results, in order to find the link between Nrf2 and SULT1E1 expressions in breast cancer patients. There are other sulfotransferases which also plays vital role in estrogen metabolism and metabolism of xenobiotics. These SULTS are known to metabolize drugs used in breast cancer treatments and thus they pose important effect during breast cancer treatment. Rat hydroxysteroid sulfotransferase a (STa) is reported to metabolize alpha-OH-TAM [10]. Methotrexate (MTX) treatment induces mRNA expressions of aryl sulfotransferase (AST-IV) and hydroxysteroid sulfotransferase (SULT1A1) in liver and intestine of rats [11]. Ethanol induces hSULT1A1/hSULT1E1 protein expressions along with their enzymatic-activities [12]. Sulfotransferases were found to be induced at transcriptional level by retinoic acid [13]. These studies suggest that oxidative stress in disease and reactive oxygen species (ROS) producing drugs influence SULTs expressions. This may have long course effects on disease outcome, drug metabolisms, drug-drug interactions and even in the normal physiological processes.
Overview of the function of SULT1E1, Nrf2 and their impact on each other
Oxidants and antioxidants are fundamentals of a balanced physiological state. Redox imbalance is known to be the core cause of several metabolic disorders and plays a vital role in carcinogenesis too. A number of transcriptional regulatory components have evolved to be ROS responsive and have formed a complex network. Nrf2 seems to be a dominant regulator, and guards cells from oxidative and electrophilic stress. Elevated amount of ROS activates tyrosine kinases to dissociate Nrf2: Keap1 complex, nuclear import of Nrf2 and coordinated activation of cytoprotective gene expression [14]. Hence, ROS is one of the known factors responsible as an inducer or supporter of metabolic disorder and cancer. Role of Nrf2 in oxidative stress maintenance may also hold a role in breast cancer.ROS such as superoxide, hydroxyl radical, and peroxyl radicals are metabolic by-products leaking from the complexes I and III of the mitochondrial respiratory chain [15]. Thymidine phosphorylase enzyme which is highly expressed in most breast cell carcinoma plays an important role in the production and enhancement of ROS in cancer cells. Rise in gene expression of thymidine phosphorylase in breast tumour cells is one of the causes of oxidative stress in these patients [16]. Eventually, there are several ways in which ROS are accumulated in the cancer tissue, thus elevating oxidative stress.
Increased oxidative stress in terms of malondialdehyde (MDA) an end product of lipid peroxidation was found in the breast tumour samples as compared to their surrounding [17]. ROS generation may occur via inflammatory pathways also. Inflammation, a process in a variety of cancers, also takes place in breast cancer and involves immune cells, including macrophages and neutrophils, in the immune response. Therefore, breast tumors are susceptible to macrophage penetration. Macrophages through NADPH oxidase cause ROS to increase in these cells. Studies also reveal that normal adjacent tissue from patients with breast tumours (cancer) exhibited significantly higher levels of the putative MDA adducts (MDA-deoxyguanosine [DG] and MDA-deoxyadenosine [DA]) than their corresponding tumors [18]. As a result of elevated MDA in tumours [17], MDA-adducts would probably be higher in surrounding tissue as reported earlier. This also suggests that lipid peroxidation increases in breast tumours and its product form DNA adducts which accumulate in adjacent breast tissues in breast cancer patients [18]. If the cellular repair system is unable to mend MDA adducts in transcribed genes, underlies the worsening of the patients pathological state, because MDA-DG adducts in repetitive CpG sequences causes frame shift mutation [19]. Thus, lipid peroxidation and MDA adducts may contribute to the progression and or upgradation of these disease. Generation of high ROS levels is detrimental for the cells as it can lead to DNA damage and oxidation of proteins and lipids changing their functions. Accumulating evidence indicates that apart from their harmful effects ROS act as second messenger signaling molecules regulating numerous pathways including cell cycle, autophagy, apoptosis, endoplasmic reticulum stress and cellular energy metabolism [20].
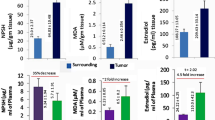
The breast tumour tissue also exhibited elevated E2 concentrations as compared to the surrounding [17]. Nrf2 expression was intensely increased in breast tumour tissue as compared to the corresponding surrounding (Fig. 1). In the tumour tissue of an established breast cancer patient who have undergone whole mastectomy, it was found that there persists a scenario with elevated E2 that may be responsible for carcinogenesis, elevated Nrf2 (inducer of antioxidant genes), along with an elevated SULT1E1 [21], Here is the difference between patients tumour cell and cell lines like MCF-7, Since studies report reduced SULT1E1 expression in MCF-7 cells [22]. An earlier study revealed Nrf2 as an inducer of SULT1E1 [9], so an elevated Nrf2 is directly responsible for an elevated SULT1E1. Since, Nrf2 is an oxidative stress responsive transcription protein and so SULT1E1 may also be considered as a stress responsive protein, as Nrf2 is an inducer of SULT1E1. Thus, regulating E2 via E2 metabolizing enzyme may physiologically be considered as stress regulation. However elevated SULT1E1 did not led to low E2 in breast tumours, tumours had elevated E2, Suggesting that SULUT1E1 is either non-functional or not sufficiently available. Earlier studies revealed that SULT1E1 may be unable to bind to E2 due to oxidation of a cysteine in the E2 binding site [23] and ultimately leading to no sulfation and no E2 inactivation under oxidative stress environment.

Expression of Nrf2 and SULT1E1 protein in tumor tissue and adjacent surrounding breast tissue in breast cancer patients. Demarcated arrows are (1) Adipose tissue, (2) Low Nrf2 expression in stromal and cellular regions, (3) Highly expressed Nrf2, (4) Stromal region, (5) Adipose tissue, (6) Stromal region with low expression of SULT1E1, (7) Adipose tissue, (8) Increased expression of SULT1E1 in the stromal region
Earlier study shows that ENU treated rat livers represents an elevated MDA with no significant change in E2 level [24] and the current study shows low or negligible increase in Nrf2 levels in the ENU treated group. Nrf2 in ENU group was more aggregated and localized instead of being distributed. This infers that the existing Nrf2 in the cytosol are forced to get activated and enter the nucleus or to a particular location in the cell. This immunohistochemistry result reveals a direct relationship between increased oxidative stress and translocation of Nrf2. A small or negligible quantity of Nrf2has been induced in response to the ROS threat in ENU group (which altogether seems to encircle nucleus). On the other hand the animal group treated with E2 alone shows a significant increase in Nrf2 expression and distribution (Fig. 2), whereas according to the earlier studies the E2 treated group shows an elevated MDA [24]. A comparison between these results declares that Nrf2 is activated to move and function due to oxidative stress but induction of Nrf2 expression at a larger scale is clearly influenced by E2 in this case and denotes that Nrf2 expression is dependent on some other pathway accompanied with oxidative stress (Fig. 3). This study also infers that only an extensive oxidative stress is not enough to induce Nrf2, evident from ENU group of animals where there is elevated MDA but low distributed Nrf2 as compared to control and E2 group. However, Nrf2 is noticed to be increased in E2 group despite of low oxidative stress in this group. It is evident from low MDA i.e., lipid peroxidation (marker of oxidative stress). Hence, E2 group shows a reductive stress and however E2 seems to be directly proportional to Nrf2 according to both ENU and E2 group [24]. This suggests that E2 is anyhow extremely involved in the Nrf2 regulation along with a stress that might be an oxidative or other stress. Oxidative stress alone may effectively cause Nrf2 to detach from keap1 and translocate to the nucleus where it heterodimerizes with musculoaponeurotic fibrosarcoma (Maf), the Nrf2-Maf heterodimer binds to ARE to induce the expression of antioxidant and metabolic genes [25]. However, induction of Nrf2 synthesis depends on some other factor also (or pathway) accompanied with oxidative stress. The ROS alone may not be the core reason of Nrf2 expression, but is the most important factor in Nrf2 activation. As evident from the ENU and E2 animal experiment, the elevated Nrf2 in breast tumor may be because of elevated E2 along with Oxidative stress. Either an E2 associated pathway or E2 induced oxidative stress play some significant role in Nrf2 expression.

Immunohistochemistry of rat liver tissues. Tumor shows an increased expression of Nrf-2 as compared to their corresponding surrounding tissue. The red arrows are pointing the central vein

Stress activation of Nrf2 and HNF α mediated induction of SULT1E1 and its role in Estradiol regulation via P13K pathway
The biphasic activity of ROS or oxidative stress may probably depend on the grade and environmental requirements of the cancer cells. After the attainment of a certain cellular properties a cancer cell becomes enormously intelligent and this study hypothesize that E2 dependent cancers are more intelligent, tolerant, resistant which makes cancer cells pathologically more severe. Nrf2 in non E2 dependent cancer cases may get activated by oxidative stress in order to protect against oxidative stress. Nrf2 may not be expressed or synthesized, if the Nrf2 expressing factors or pathway is not activated, as evident from our studies. This study infers that probably oxidative stress can only activate Nrf2 and translocates it to nucleus, but E2 is capable of inducing Nrf2 expression [24]. Thus E2 may be one among the several Nrf2 inducing (via synthesis) factor.
Oxidative stress is the link between SULT1E1 and Nrf2 expression
Basic biological response towards oxidative stress is initiated by a group of molecules that is keap1, Nrf2, Maf and delivered via expression of antioxidant response element (ARE). On requirement, Nrf2 escapes degradation by Keap1 and translocate to the nucleus, and activates gene expression of antioxidant response element and induces enzymes such as c-glutamyl cysteine synthetase, heme oxygenase-1 (HO-1), glutathione peroxidase 1, NADPH dehydrogenase, quinone 1 (NQO1), glutathionine S transferase (GST), and glutathione reductase (GR) [26]. These proteins are antioxidative and cytoprotective in nature. In recent study SULT1E1 was also shown to be a transcriptional target of Nrf2 [9]. SULT1E1 deactivates estradiol and estrone by adding a sulphate group and forming estrogen sulphate which has least addiction towards ER and unknown functional contributions, rather more prone to be eliminated from the tissue.
Transcriptional signaling and inflammatory responses
Nrf, Maf and antioxidant response element (ARE) complex executes its function in terms of anti-inflammatory responses, antioxidative responses, detoxification of toxins, autophagic activity, and proteasome actions and contributes vital physiological role. Breakage in the Nrf2 and Keap1 interaction triggers the initiation of the Nrf2 pathway. As in the case of p21Cip/WAF1 which successfully competes Keap1 to bind to the DLG motif (low-affinity binding site) of Nrf2 protecting it from ubiquitination [26]. The first selected autophagy protein, p62 also competes with Nrf2for binding to Keap1. p62–Keap1 complex enables the ubiquitinated aggregate formation and creating a positive feedback loop with Nrf2 [27,28,29,30]. Thus, autophagy and p62 both have a grip on Nrf2 activation. p62 is degraded through autophagy under normal conditions.post-translational autophagic degradation regulates the intracellular level of p62. Oxidative stress modulates the transcription of p62 via Nrf2 by the Ras/MAPK pathway, the JNK/c-Jun pathway. Resveratrol is an inducer of autophagy [31]. P62 protein mediates aggresome formation and triggers the activation of selective autophagic degradation [32]. In addition, protein kinase C, mitogen-activated protein kinases, and phosphotidylinositol 3-kinase (PI3K) have been implicated in the regulation of Nrf2/ARE signaling [33].
Estrogen and autophagy share deep Crosstalk signaling between them. Estrogenic effectors affect autophagy. Autophagy-targeted transcription factors (TFs) and translocation of estrogen receptors, as well as histone modifications is regulated by E2. Estrogen is able to delay the apoptosis in breast cancer patients allowing longer survivals of the cells via ESRs [34]. Antagonizing estrogen is the most important action in treatment of estrogen- induced or -dependent cancers. Antagonizing estrogen may disrupt cell balance provided by it via regulation of apoptosis and autophagy which might be a protective effect of estrogen towards cancer growth. There are reports and cases which reveals that E2 promotes autophagy [35,36,37], but a few studies also report that when hypoxia, lipopolysaccharides (LPS), or ovariectomy stimulated cellular autophagy, presence of E2 restricts gene expression of some autophagy proteins [38, 39]. Thus, E2 mediated inhibition of autophagy via hypoxia induced oxidative stress may be one of the pathways for Nrf2 overexpression and activation in the breast cancer tissue as reported in the current study (Fig. 1). Earlier study found an elevated MDA along with over-expressed Nrf2 in Breast tumor tissue and the surrounding tissue represented comparatively less MDA and Nrf2 expression [17]. This implies that cellular environment plays a crucial role in the modulation of molecular pathways.
Nrf2-mediated expression of genes on ARE is dependent on Small Maf proteins. Among all the domains and motifs on Nrf2, NESzip motif (nuclear export signal co-localized with the leucine zipper (ZIP) domain) is one of the most important one. Nrf2 heterodimerizes with MafG via ZIP–ZIP binding which enhances retention of Nrf2 in the nucleus. Nrf2/Maf heterodimer binds specifically to antioxidant response element which is a cis-acting enhancer stimulating transcription of a series of genes which possess antioxidant and detoxification properties [40]. The Musculoaponeurotic fibrosarcoma (Maf) oncogene or protein has three homologs that Maf F, Maf G, and Maf K. Maf G is acetylated by cAMP-response element-binding protein (CBP) in erythroid cells. The transcriptional activity of the heterodimer Nrf2/MafG is reported to be enhanced due to acetylation [41]. This suggests an important role of CBP in delivering the Nrf2/Maf G heterodimers transcriptional activity. Inhibitor of DNA binding protein (Id1/3) inhibition reduces the expression of small MafF, MafG and MafK transcription factors [42]. The overexpression of SULT1E1 inhibits proliferation in vitro and tumorigenesis in vivo [7]. Thus, modulation of SULT1E1 expression and activation is crucial stepin maintenance of a healthy physiology or occurrence of disease.
Earlier studies report that bile acid induced by cholestasis activates farnesoid X receptor (FXR). Now FXR tends to compete with CBP for binding hepatocyte nuclear factor 4α (HNF4α). HNF4α is the transcription factor involved in the transactivation and a determination of gender-specific expression and activation of signaling pathways important in the regulation of phase II enzymes and transporters in hepatocytes. The competition between CBP and FXR tends to decrease acetylation of Hepatocytes nuclear factor 4α (HNF4α) and its nuclear retention, which sequentially repressed HNF4α-dependent SULT1E1 gene transcription [43].
Both Nrf2 and pregnane X receptor (PXR) actively participated in hepatic signaling in HNF4α Null mice. HNF4α deficiency markedly alters hepatic mRNA expression of a large number of phase II enzymes and transporters [44]. It’s a good example of how molecular pathways act differently on the basis of cellular environment as well as type. Nrf2 is an inducer of SULT1E1 in breast tissue and some other tissue also and HNF4α is a major inducer of SULT1E1 in liver. Earlier studies also indicate that hepatic Nrf2 signaling is enhanced by of HNF4a [44]. It seems like HNF4α does not utilizes Nrf2 for SULT1E1 expression since Nrf2 is active in liver when HNF4α is absent and vice versa. Rather, we can also assume presence of HNF4α may have some inhibitory role of Nrf2 in certain environments. An earlier study from our lab has shown that MDA level was much lower in the liver tissue of E2 administered group whereas MDA was highly increased in the ENU administered animal group [23]. The same animal group with E2 administration has shown an increased serum MDA level; this infers that E2 acts in different ways and via different pathways in different organs depending on the organs cellular environment .E2 may also induce Nrf2 in breast cancer tissue by activation of P13K pathways along with the inactivation of GSK3B, via increasing the phosphorylation of both the proteins.
Two points can be explained in relation to the SULT1E1/Nrf-2 expression and disease condition. First, it may be clarified that cancer is a stepwise process that is initiated and influenced by several factors. Breast cancer pathogenesis usually occurs in months to years with different degrees of severity and designated as stages of the diseases. On the course of time, when severity is increases, disease causing factor dominates over the adaptive factors. Now, SULT1E1 is an adaptive factor that decreases active E2 level which restricts E2 dependent breast carcinogenesis. The SULT1E1 expressions vary in relation to disease types and disease stages. If 1E1 expression is high so E2 is low and disease is less severe and when 1E1 is low E2 is high and the disease is at a progression stage. The second point is little paradoxical in relation to the oxidative burden in the internal milieu of the cells. When oxidative stress increased in the cells the reducing environment (NADPH and GSH) is depleted. In this environment the 1E1 remain in inactive form though the protein expression remain high. So, to get more activity tissue express more amount of 1E1 protein. So, higher expression does not always impart sufficient activity. As a result, disese progression is not restricted. So, to judge the state we need to consider two states, that is 1E1 expression and intracellular redox environment. Though in our result SULT1E1 is highly expressed but it may be interpreted to be active or inactive.
Conclusion
These data indicates that E2 associated cancer may be more severe when in collaboration with ROS or oxidative stress. Where, Oxidative stress have an ability to turn normal pathway into a carcinogenic one, as well as switch factors on and off differently in separate organs. Cancers might be able to resist against oxidative stress more strongly in comparison to E2 independent cancers. There are several therapies which utilizes oxidative or ROS as the therapy to kill or destruct cancer cells. In the cases of cancers where E2 is high, the E2 mediated Nrf2 expression and activation (induction) may make cancer cells resistant to therapeutically used oxidative stress than those cases where Nrf2 is only activated but not expressed by oxidative stress. Oxidative stress may regulate Nrf2 and thereby SULT1E1 by modulating organ specific transcription factors like HNF4α in liver and directly Nrf2 in other tissue. In certain cases once the stock Nrf2 is utilized and oxidative stress overrules the antioxidant status, cell starts to be victim of excessive oxidative stress. There is yet no specific marker to identify the level of oxidative stress (OS) that is working in favour of tumorigenesis or the level of OS that may inhibit the disease, but here our study brings forward the possibilities of inter-regulations among Oxidative stress responsive genes such as Nrf2, proliferative molecules responsible for disease severity that is E2 and oxidative-stress itself. Future research works may clarify these facts more clearly.
Data availability
The datasets used and/or analysed during the current study available on request.
Abbreviations
- SULT1E1:
-
Estrogen sulfotransferase
- Nrf-2:
-
Nuclear factor erythroid 2-related factor 2
- VAT:
-
Visceral adipose tissue
- 16αOHE1:
-
16α-Hydroxyestrone
- STa:
-
Hydroxysteroid sulfotransferase
- MTX:
-
Methotrexate
- AST-IV:
-
Aryl sulfotransferase IV
- MDA:
-
Malondialdehyde
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate hydrogen
- MDA DG:
-
Deoxyguanosine malondialdehyde
- MDA DA:
-
Deoxyadenosine malondialdehyde
- ENU:
-
Ethyl nitroso urea
- ARE:
-
Antioxidant response element
- HO-1:
-
Heme oxygenase-1
- GPX:
-
Glutathione peroxidase 1
- NQO1:
-
NADPH dehydrogenase, quinone 1
- GST:
-
Glutathionine S transferase
- SOD:
-
Superoxide dismutase
- Keap1:
-
Kelch-like ECH-associated protein 1
- DLG:
-
Low-affinity binding site
- LPS:
-
Lipopolysaccharides
- CBP:
-
Cyclic-AMP response element binding protein
- MafF:
-
Musculoaponeurotic fibrosarcoma homolog F
- MafG:
-
Musculoaponeurotic fibrosarcoma homolog G
- MafK:
-
Musculoaponeurotic fibrosarcoma homolog K
- HNF4α:
-
Hepatocyte nuclear factor 4α
- FXR:
-
Farnesoid X receptor
- PXR:
-
Pregnane X receptor
References
Motohashi H, Yamamoto M (2004) Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 10:549–557
Klaassen CD, Slitt AL (2005) Regulation of hepatic transporters by xenobiotic receptors. Curr Drug Metab 6:309–328
Mandlekar S, Hong JL, Kong AN (2006) Modulation of metabolic enzymes by dietary phytochemicals: a review of mechanisms underlying beneficial versus unfavorable effects. Curr Drug Metab 7:661–675
Fares MY, Salhab HA, Khachfe HH, Khachfe HM (2019) Breast cancer epidemiology among Lebanese women: an 11-year analysis. Medicina (Kaunas) 10(55):8
Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM (2017) Obesity and adverse breast cancer risk and outcome: mechanistic insights and strategies for intervention. CA Cancer J Clin 67(5):378–397
Sowers M, McConnell D, Jannausch ML, Randolph JF, Brook R, Gold EB, Crawford S, Lasley B (2008) Oestrogen metabolites in relation to isoprostanes as a measure of oxidative stress. Clin Endocrinol (Oxf) 68(5):806–813
Xu Y, Liu X, Guo F, Ning Y, Zhi X, Wang X, Chen S, Yin L, Li X (2012) Effect of estrogen sulfation by SULT1E1 and PAPSS on the development of estrogen-dependent cancers. Cancer Sci 103:1000–1009
Maiti S, Nazmeen A (2019) Impaired redox regulation of estrogen metabolizing proteins is important determinant of human breast cancer. Cancer Cell Int 19:111
Guo Y, Hu B, Huang H, Tsung A, Gaikwad NW, Xu M, Jiang M, Ren S, Fan J, Billiar TR, Huang M, Xie W (2015) Estrogen sulfotransferase is an oxidative stress-responsive gene that gender-specifically affects liver ischemia/reperfusion injury. J Biol Chem 290(23):14754–14764
Chen G, Yin S, Maiti S, Shao X (2002) 4-Hydroxytamoxifen sulfation metabolism. J Biochem Mol Toxicol 16(6):279–285
Maiti S, Chen G (2003) Methotrexate is a novel inducer of rat liver and intestinal sulfotransferases. Arch Biochem Biophys 418(2):161–168
Maiti S, Chen G (2015) Ethanol up-regulates phenol sulfotransferase (SULT1A1) and hydroxysteroid sulfotransferase (SULT2A1) in rat liver and intestine. Arch Physiol Biochem 121(2):68–74
Maiti S, Chen X, Chen G (2005) All-trans retinoic acid induction of sulfotransferases. Basic Clin Pharmacol Toxicol 96(1):44–53
Sajadimajd S, Khazaei M (2018) Oxidative stress and cancer: the role of Nrf2. Curr Cancer Drug Targets 18(6):538–557
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94(3):909–950
Brown NS, Bicknell R (2001) Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res 3(5):323–327
Nazmeen A, Chen G, Ghosh TK, Maiti S (2020) Breast cancer pathogenesis is linked to the intra-tumoral estrogen sulfotransferase (hSULT1E1) expressions regulated by cellular redox dependent Nrf-2/NFκβ interplay. Cancer Cell Int 4(20):70
Wang M, Dhingra K, Hittelman WN, Liehr JG, de Andrade M, Li D (1996) Lipid peroxidation-induced putative malondialdehyde-DNA adducts in human breast tissues. Cancer Epidemiol Biomark Prev 5(9):705–710
Vander Veen LA, Hashim MF, Shyr Y, Marnett LJ (2003) Induction of frameshift and base pair substitution mutations by the major DNA adduct of the endogenous carcinogen malondialdehyde. Proc Natl Acad Sci USA 100(24):14247–14252
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24(10):R453–R462
Lena S, Svoboda M, Klameth L et al (2013) The sulfatase pathway for estrogen formation: targets for the treatment and diagnosis of hormone-associated tumors. J Drug Deliv 95:7605
Yali Xu, Lin X, Jiawen Xu, Jing H, Qin Y, Li Y (2018) SULT1E1 inhibits cell proliferation and invasion by activating PPARγ in breast cancer. J Cancer 9(6):1078–1087
Maiti S, Zhang J, Chen G (2007) Redox regulation of human estrogen sulfotransferase (hSULT1E1). Biochem Pharmacol 73(9):1474–1481
Nazmeen A, Maiti S (2018) Oxidant stress induction and signalling in xenografted (human breast cancer-tissues) plus estradiol treated or N-ethyl-N-nitrosourea treated female rats via altered estrogen sulfotransferase (rSULT1E1) expressions and SOD1/catalase regulations. Mol Biol Rep 45(6):2571–2584
Li W, Yu S, Liu T, Kim JH, Blank V, Li H, Kong AN (2008) Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim Biophys Acta 1783(10):1847–1856
Li W (2009) Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog 48(2):91–104
Liu WJ, Ye L, Huang WF (2016) p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 21:29
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145
Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614
Puissant A, Fenouille N, Auberger P (2012) When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2:397–413
Zheng Q, Su H, Ranek MJ, Wang X (2011) Autophagy and p62 in cardiac proteinopathy. Circ Res 109:296–330
Jixiang Z, Wang X, Vikash V (2016) ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev 435:965
Petrovski G, Zahuczky G, Katona K (2007) Clearance of dying autophagic cells of different origin by professional and non-professional phagocytes. Cell Death Differ 14:1117–1128
Totta P, Busonero C, Leone S (2016) Dynamin II is required for 17β-estradiol signaling and autophagy-based ERα degradation. Sci Res 6:23727
Lahm T, Petrache I (2012) LC3 as a potential therapeutic target in hypoxia-induced pulmonary hypertension. Autophagy 8:1146–1147
Guido C, Panza S, Santoro M (2012) Estrogen receptor beta (ERβ) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle 11:2911–2921
Hsieh DJ, Kuo W-W, Lai Y-P (2015) 17β-Estradiol and/or estrogen receptor β attenuate the autophagic and apoptotic effects induced by prolonged hypoxia through HIF-1α-mediated BNIP3 and IGFBP-3 signaling blockage. Cell Physiol Biochem 36:274–284
Yang Y, Zheng X, Li B (2014) Increased activity of osteocyte autophagy in ovariectomized rats and its correlation with oxidative stress status and bone loss. Biochem Biophys Res Commun 451:86–92
Li W, Siwang Yu, Liu T, Kim J-H, Blank V, Hong Li A-N, Kong T (2008) Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip Motif. Biochim Biophys Acta 1783(10):1847–1856
Hung HL, Kim AY, Hong W, Rakowski C, Blobel GA (2001) Stimulation of NF-E2 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J Biol Chem 276(14):10715–10721
Bensellam M, Montgomery MK, Luzuriaga J, Chan JY, Laybutt DR (2015) Inhibitor of differentiation proteins protect against oxidative stress by regulating the antioxidant-mitochondrial response in mouse beta cells. Diabetologia 58(4):758–770
Liu X, Xue R, Yang C, Gu J, Chen S, Zhang S (2018) Cholestasis-induced bile acid elevates estrogen level via farnesoid X receptor-mediated suppression of the estrogen sulfotransferase SULT1E1. J Biol Chem 293(33):12759–12769
Hong Lu, Gonzalez FJ, Klaassen C (2010) Alterations in hepatic mRNA expression of phase II enzymes and xenobiotic transporters after targeted disruption of hepatocyte nuclear factor 4 alpha. Toxicol Sci 118(2):380–390
Acknowledgements
University Grants Commission, New Delhi provided JRF and SRF to AN who is a Ph.D. students working in the Post Graduate Department of Biochemistry, OIST.
Funding
Institutional, no external funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nazmeen, A., Chen, G. & Maiti, S. Dependence between estrogen sulfotransferase (SULT1E1) and nuclear transcription factor Nrf-2 regulations via oxidative stress in breast cancer. Mol Biol Rep 47, 4691–4698 (2020). https://doi.org/10.1007/s11033-020-05518-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05518-z