
Abstract
The immunomodulatory property of mesenchymal stem cells (MSCs) has been previously reported. Still it is unclear if this property can be affected by the cell origin and cell quality. Using primary MSCs expanded from bone marrow (BM-MSCs) and adipose tissue (AD-MSCs) of mice, we investigated whether the immunomodulatory property of MSCs varied with cell origin and cell quality (early- vs. late-passaged BM-MSCs). BM-MSCs (p1) and AD-MSCs (p1) had a typical spindle shape, but morphological changes were observed in late-passaged BM-MSCs (p6). A pathway-focused array showed that the expression of chemokine/cytokine genes varied with different cell origins and qualities. By co-culturing with spleen mononuclear cells (MNC) for 3 days, the expression of CD4 was suppressed by all types of MSCs. By contrast, the expression of CD8 was suppressed by BM-MSCs and increased by AD-MSCs. The expression ratio of CD206 to CD86 was at a comparable level after co-culture with AD-MSCs and BM-MSCs, but was lower with late-passaged BM-MSCs. AD-MSCs highly induced the release of IL6, IL-10 and TGF-β in culture medium. Compared with early-passaged BM-MSCs (p1), late-passaged BM-MSCs (p6) released less TGF-β. Our data suggests that the immunomodulatory properties of MSCs vary with cell origin and cell quality and that BM-MSCs of good quality are likely the optimal source of immunomodulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Mesenchymal stem cells (MSCs) are a population of undifferentiated cells that were first isolated from human bone marrow and defined by their ability to adhere to plastic surfaces when cultured under standard conditions [1]. Because of their immunomodulatory properties [2, 3], scientists have used MSCs not only for tissue/organ regeneration but also for the treatment of autoimmune diseases. Studies on the secretory profile of human MSCs have confirmed the secretion of many cytokines, including TGFβ, IL6, VEGF, IDO, HGF and IL10 [4,5,6,7,8,9].
MSCs have been isolated from many species, including rats and mice, with different success rates [10, 11]. BM-MSCs isolation is a challenging process due to low yield, contamination by haemopoietic cells and the tendency of the cells to differentiate in the absence of differentiation culture conditions [10, 12]. MSCs have been isolated from other tissues, including adipose tissue, dental pulp, heart and brain [13,14,15,16], but adipose tissue has many promising characteristics including easy accessibility and high yield [17].
MSCs can inhibit the proliferation of T lymphocytes, impair dendritic cell maturation and polarize macrophages toward the M2 type [18,19,20]. However, some studies have reported an immunostimulatory function [21]. These immunomodulatory properties could be due to direct cell-to-cell contacts, cytokines secretion or by a combination of both mechanisms [9].Previous studies have compared different MSCs and revealed different immunomodulatory capacity. They showed that adipose tissue-derived MSCs had a stronger inhibitory effect and that umbilical cord matrix-derived MSCs had little effect on B and NK cells [22]. MSCs from different tissues have been tested for the treatment of inflammatory and immunological disorders and showed a therapeutic effect in many autoimmune disease models, such as inflammatory bowel disease, multiple sclerosis and graft versus host reaction (GVHR) [23, 24].
Unfortunately, systematic evaluation of different kinds of MSCs in immunomodulation are lacking where different source, age and quality of cells could impact their immune properties [25]. Although the therapeutic effect is promising, it is unclear how the biological characteristics and immunomodulatory properties of MSCs will change with variation in cell origin and cell quality. Here, we compare the immunomodulatory properties of MSCs from two different origins, bone marrow (BM) and adipose tissue (AD), and two different qualities, early-passaged (p1) and late-passaged (p6) BM-MSCs.
Methods
Animals
Adult (10-12-week-old) male C57BL/6 mice (CLEA Japan, Inc.) were used for this study.
Isolation and culture of AD-MSCs
Mice were anaesthetized and epididymal fat was isolated through a midline incision. Isolated fat was digested at 37◦C in Hanks buffered salt solution (HBSS) containing 100 U/ml collagenase (collagenase I, Gibco) for 60 min. After adding DMEM (Dulbecco’s modified eagle’s medium) media and centrifugation, the pellet was suspended in RBC lysis buffer. After centrifugation and filtration through 100 µm and 40 µm nylon filter mesh, isolated cells were suspended in medium and plated in 10 cm tissue culture plates, and cultured at the presence of 10% foetal bovine serum (FBS)-containing DMEM. The dish was incubated at 37◦C in a 5% CO2 incubator for 4 days, and then non-adherent cells were removed. The cells were observed daily under an inverted phase-contrast microscope and were passaged after 80–90% confluence. The culture media was changed every 4 days.
Culture of BM-MSCs
Mouse BM-MSCs p1 were provided from the NIH-funded research at Scripps Research Institute (Scripps, Florida, USA) [26]. Cells were suspended in medium, plated in 10 cm tissue culture plates, and cultured in Minimum Essential Medium (MEM-α) in the presence of 10% foetal bovine serum (FBS), 100 units/ml penicillin G and 10 µg/ml streptomycin. Mouse BM-MSCs p6 were commercially available (#MUBMX-01001-Cyagen, Santa Clara, CA, USA). Cells were suspended in medium, plated in 10 cm tissue culture plates, and cultured in Dulbecco’s modified Eagle’s medium (DMEM) in the presence of 10% foetal bovine serum (FBS), 100 units/ml penicillin G and 10 µg/ml streptomycin. The dish was incubated at 37◦C in a 5% CO2 incubator. The cells were observed daily under an inverted phase-contrast microscope and were passaged after 80–90% confluence. The culture media was changed every 4 days.
Flow cytometric characterization of MSCs
MSCs were detached using Trypsin-EDTA (Gibco) and counted. The cells were pelleted by centrifugation for 5 min at 400×g and washed twice with FACS buffer. Then, the cells were stained with fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD34(RAM34), CD106 (429), CD90(53-2.1), and MHC-II class (I-A) (NIMR-4) and phycoerythrin (PE)-conjugated rat anti-mouse CD44(IM7), CD105 (MJ7/18) and CD45(30-F11) at 4 °C for 30 min. All antibodies were purchased from eBioscience. The cells stained with FITC rat anti-mouse IgG(eBR2a) or PE rat anti-mouse IgG (eBR2a) were considered as negative controls. The cells were pelleted and washed twice with PBS (phosphate buffer saline). Then, fluorescence-activated cell sorting (FACS) analysis was performed on a FACS Callibur cytometry (Becton Dickinson, San Jose, CA, USA) using Cell Quest software.
Mouse chemokine and cytokine RT2 Profiler™ PCR array
To analyse gene expression, equal amount of total RNA was isolated from cells of three independent experiment using ISOGEN II (#317-07361, NIPPON GENE) and mixed together. The concentration of RNA was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and 2 µg of RNA was used to generate cDNA using the RT2 First Strand Kit (#330401, SABiosciences, a Qiagen Company). The mouse chemokine and cytokine RT2 Profiler™ PCR array was used according to the manufacturer’s instructions (#PAMM-150Z, SABiosciences). This PCR array profiled the expression of 84 chemokine and cytokine genes. The fold change of expression was calculated using the SABiosciences web-based data analysis program.
MSCs and mononuclear cells co-culture
We collected splenocytes from the spleens of mice. Briefly, mice were sacrificed and spleens were removed in RPMI (Roswell Park Memorial Institute) medium supplemented with 10% FBS, 100 units/ml penicillin G and 10 µg/ml streptomycin. To collect the mononuclear cells, we pressed the minced spleen tissues through a 100 µm filter into a 50-ml tube. After the gradient, red blood cells were removed by re-suspending the cell pellet in erythrocyte lysis buffer (eBioscience) for 5 min.
For co-culture, MSCs were plated in complete DMEM media in a 24 well culture dish. When they reached 80% confluency, 2 × 106 mononuclear cells were added to complete RPMI media. MSCs and mononuclear cells were co-cultured for 3 days.
Flow cytometric analysis of mononuclear cells
Isolated cells were washed with FACS buffer. Next, cells were stained with primary rat anti-mouse CD4 (GK1.5) and CD8a (53-6.7) (eBioscience) for 20 min at 4 °C. Then, the cells were washed twice and stained with fluorescein isothiocyanate (FITC)-conjugated donkey anti-rat secondary antibody. Fluorescein isothiocyanate–conjugated secondary antibody (eBioscience) was used as the isotype control. After staining, cells were washed with FACS buffer and analysed using a FACS Calibur flow cytometer.
ELISA
Conditioned media and co-culture supernatant were collected after 3 days of co-culture. Mononuclear cells were stimulated with 1 µg/ml phytohaemagglutinin (eBioscience). The mouse TGFβ-1, IL6 and IL10 ELISA kits (R&D Systems) were used to measure the levels of TGFβ-1, IL6 and IL10 in the conditioned medium and co-culture supernatant.
Immunocytochemistry
To evaluate the cell senescence the cells were fixed in 100% acetone for 10 min. After blocking, the cells were incubated with rabbit polyclonal anti-mouse telomerase reverse transcriptase-C-terminal antibody (#ab183105-Abcam) and followed by anti-rabbit Alexa Fluor 546 secondary antibodies(#A-11035-ebioscience). To detect the macrophages in mononuclear cells after MSCs coculture, cells were fixed with 4% PFA (paraformaldehyde). After blocking, cells were incubated with primary antibodies against CD206 (#Q2HZ94-R&D System) and CD86 (GL1-eBioscience) and followed by FITC fluorescence-conjugated secondary antibodies(#A-21208-ebioscience). The cell nuclei were labelled with DAPI [(4′,6-diamidino-2-phenylindole)]. Positively stained cells were counted and viewed by confocal laser scanning microscopy (FV10i-LIV, Olympus), and digital images were acquired using FV10-ASW software (Olympus) with a 60-fold magnification lens. Ten fields per section were randomly selected for quantitative counting. The results represent data from three independent experiments.
Cell proliferation assay
Cells were seeded in 96-well culture plates at a density of 1 × 104 cells per well and cultured overnight. Proliferation assay was performed using the Cell Proliferation Kit I (MTT) (#11465007001, Roche Life Science). Briefly, MTT was added and incubated for 4 h then the formation of formazan from MTT was stopped by adding solubilisation solution, and the absorbance of formazan was measured at 570 nm using a microplate reader.
Statistical analysis
All the results are presented as the mean ± SEM. Statistical significance was determined by Student’s t-test (JMP®, Version 13.0 JMP®, Version 13.0. SAS Institute Inc., Cary, NC, USA). Differences were considered significant when P < 0.05.
Results
Morphologic and phenotypic characteristics of MSCs
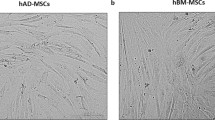
We isolated AD-MSCs p1 from the adipose tissues of 12-week-old mice and expanded BM-MSCs p1 (gift from Professor Donald G. Phinney, Scripps Institute, USA) and BM-MSCs p6 (commercially available). We found no gross morphological changes between AD-MSCs and BM-MSCs. Both cell types were plastic-adherent and had an elongated spindle shape. Characteristically, early-passaged cells MSCs (p1) acquired a flat shape and their proliferative capacity significantly decreased, as we measured it by MTT assay (Online Resource 1). On the other side, late-passaged cells (p6) retain its spindle shape and its proliferative capacity (Fig. 1a). We evaluated the cell senescence by immunostaining with telomerase reverse transcriptase (TERT). Although the proliferative capacity of early-passaged cells decreased, their expression of TERT was significantly higher than late-passaged cells (Online Resource 2).

Effect of cell origin and cell quality on cell morphology and phenotypic characteristics. a The growth of cells was observed under a microscope (× 40, scale bars 200 µm). b Flow cytometry analysis of CD45, HLA-DR, CD34, CD44, CD90, CD105, and CD106. AD-MSCs: adipose mesenchymal stem cells. BM-MSCs: bone marrow mesenchymal stem cells. p1: passage one. p6: passage six. Blue line represents isotype control
The investigation of the surface expression of seven molecules (CD105, CD90, CD44, CD106, HLA-DR, CD45, CD34) showed that MSCs were poor/negative for haemopoeitic cell markers (CD45 and HLA-DR), highly/moderately positive for CD44 and CD106 and moderately/poor positive for CD34, CD90. Although early passaged cells were moderately positive for CD105, late passaged ones were negative (Fig. 1b).
Expression of chemokine/cytokine genes in MSCs
We investigated the expression of 84 chemokine and cytokine genes by mouse RT2 Profiler TM PCR array. The dot-plot graphs showed that the expression of these genes in MSCs was different depending on cell origin (Fig. 2a) and cell quality (Fig. 2b).

Differential expression of chemokine and cytokine genes. Yellow circles represent upregulated genes with more than a twofold change, whereas the blue circles represent downregulated genes with more than a twofold change. a The dot-plot graph of AD-MSCs p1 versus BM-MSCs p1. b The dot-plot graph of BM-MSCs p6 versus BM-MSCs p1. c Fold changes in chemokine and cytokine genes expression of AD-MSCs P1 versus BM-MSCs p1. d Fold changes in chemokine and cytokine genes expression of BM-MSCs p6 and BM-MSCs p1. AD-MSCs: adipose mesenchymal stem cells. BM-MSCs: bone marrow mesenchymal stem cells, p1: passage one, p6: passage six
Among the genes differently expressed over twofold between different cell origins (AD-MSCs vs. BM-MSCs p1), there was a threefold increase in Il6 expression, 36-fold increase in colony stimulating factor 2 (Csf2) and 21-fold increase in tumour necrosis factor receptor superfamily member 13b (Tnfsf13b) in AD-MSCs versus BM-MSCs. AD-MSCs and BM-MSCs varied in the expression of many chemokine genes, including Ccl5, Ccl7, Cxcl9 and Cxcl12 (Fig. 2c).
Among the genes differently expressed over twofold between different cell qualities (BM-MSCs p6 versus BM-MSCs p1), Tgfβ2 was overexpressed 17-fold in early- versus late-passaged BM-MSCs. Furthermore, tumour necrosis factor receptor superfamily member 11B (Tnfrsf11b) was overexpressed 1200-fold in early- versus late-passaged cells. Other immunomodulatory cytokines and chemokine genes were overexpressed in late-passaged cells, such as Ccl5, Cxcl10, Lta and Mif. However, the expression of Il-6 was detected at a comparable level between early- and late- passaged BM-MSCs (Fig. 2d).
In vitro assessments of the immunomodulatory properties of MSCs
AD-MSCs and BM-MSCs were co-cultured with spleen mononuclear cells (MNC) for 3 days, and then CD4+ and CD8+ cells were measured by flow cytometry. BM-MSCs significantly decreased CD4+ and CD8+ cells (p < 0.005 for CD4+ and p < 0.05 for CD8+). However, AD-MSCs significantly decreased CD4+ cells and increased CD8+ cells (p < 0.05 for CD4+ and p < 0.005 for CD8+) (Fig. 3a, b, Online Resource 3).

Effect of cell quality and cell origin on CD4+ and CD8+ mononuclear cells. a Flow cytometry analysis of CD4+ after 3 days’ co-culture with MSCs. b Flow cytometry analysis CD8+ after 3 days’ co-culture with MSCs. *p < 0.05 versus MNC, †p < 0.005 versus BM-MSCs p6, ‡p < 0.0005 versus BM-MSCs p1, paired t test. AD-MSCs: adipose mesenchymal stem cells, BM-MSCs: bone marrow mesenchymal stem cells, p1: passage one, p6: passage six, MNC: mononuclear cell control. All data represent mean ± SEM
Both early- and late-passaged cells decreased the percentage of CD4+ and CD8+ cells significantly (p < 0.05), but early passaged cell were more potent suppressor for CD4+ and CD8+ cells than late passaged ones (p < 0.005) (Fig. 3a, b).
The immunohistochemistry assessment of macrophages after 3 days co-culture showed that early-passaged cells shifted the immune response toward the M2 phenotype, while late-passaged ones shifted the immune response toward the M1 phenotype (p < 0.05) (Fig. 4a, b).

Effect of cell quality and cell origin on macrophage polarization. a Representative images showing the CD206-positive M2 macrophages and CD86-positive M1 macrophages in each group (× 60, scale bars 10 µm). b Quantitative data of CD206-positive M2 macrophages. *p < 0.05 versus BM-MSCs p1, paired t test. AD-MSCs: adipose mesenchymal stem cells, BM-MSCs: bone marrow mesenchymal stem cells, p1: passage one, p6: passage six, M2: M2 macrophage. All data represent mean ± SEM
We performed ELISA assays for three immunosuppressive cytokines: TGFβ, IL10 and IL6. We first measured the levels of the three cytokines after mononuclear cell stimulation with phytohaemagglutinin (PHA); then we measured them in the conditioned media and co-culture supernatant after 3 days of co-culture and under stimulation with PHA. IL6 was in conditioned media and the co-culture supernatant of MSCs and increased significantly under co-culture conditions (p < 0.0005). The increased level of IL6 in the co-culture supernatant was more significant in AD-MSCs than BM-MSCs (p < 0.005) and in early-passaged cells (p < 0.0005 vs. p6) (Fig. 5a). TGFβ was in conditioned media and the co-culture supernatant of MSCs but it increased after co-culture with AD-MSCs (p < 0.05) and decreased after co-culture with late passaged cells (p < 0.05) (Fig. 5b). IL10 was only detected in conditioned media and the co-culture supernatant of AD-MSCs and it increased significantly in co-culture conditions (p < 0.05) (Fig. 5c).

Effect of cell quality and cell origin on immunomodulatory cytokines. ELISA assay of conditioned media and co-culture supernatant after PHA stimulation and 3 days of co-culture. a IL6, b TGFβ, c IL10. *p < 0.05 versus CM, †p < 0.0005 versus BM-MSCs p6, ‡p < 0.005 versus BM-MSCs p1, paired t test. AD-MSCs: adipose mesenchymal stem cells, BM-MSCs: bone marrow mesenchymal stem cells, p1: passage one, p6: passage six, CM: conditioned media, MNC: mononuclear cells. All data represent mean ± SEM
Discussion
Although MSCs have promising therapeutic effects, poor clinical outcomes [27] have been reported. The lack of standardization of cells leads to variable and inconsistent results. In this study, we investigated MSCs from two different sources BM and AD and two different passages early and late-passaged cells. Consistent with other studies [28,29,30], we showed that BM-MSCs and AD-MSCs varied in their surface phenotypes and that although the cell surface makers for human MSCs have been well established by international society of cellular therapy [31], there is no consensus on the cell surface makers for mouse MSCs. Characteristically, cell quality has a dramatic effect on cell morphology. Unlike late-passaged cells, early-passaged ones showed a decrease in the proliferation capacity with repeated subculture [32].
We were interested to know how much the cell origin and the cell quality could change the cellular profile of immunocytokines and to what degree this change could affect immunological cells. The gene expression array of AD-MSCs revealed a 32-fold upregulation of Csf2. We suggested that Csf2 released from AD-MSCs increased the proliferation of macrophages and monocytes, which in turn released IL10. Previous studies suggested that monocytes were induced to release IL10 by hepatocyte growth factor (HGF) secreted from MSCS [33,34,35,36]. AD-MSCs secreted more IL6 and less TGFβ than BM-MSCs, which was consistent with array results that showed a threefold increase in Il6 gene expression and a 23-fold decrease in Tfgb2 gene expression in AD-MSCs versus BM-MSCs. AD-MSCs showed the upregulation of Il23a, which was mainly immunostimulatory, and the downregulation of Tnfrsf11b, which is an important inhibitor of the transcription of many immune-related genes [37].
In this study, the gene expression array of AD-MSCs showed a 21-fold increase in Tnfsf13b, which is a B-cell activating factor, so we supposed that the increased percentage of CD8+ cells after co-culture with AD-MSCs could be attributed to B lymphocyte activation. Although the effect of MSCs on B lymphocytes is controversial [38, 39], some studies reported the expression of CD8 markers on B lymphocytes in some diseased conditions and after in vitro activation [40, 41]. It was reported that both AD and BM-MSCs induce macrophage polarization toward the M2 phenotype [42], and in another study, it was found that aged MSCs were less effective inducers of M2 macrophages than young MSCs [43], but no study has investigated the effect of the prolonged passage of MSCs on macrophage polarization. Here, we found that late-passaged cells were less effective inducers of M2 macrophages as well as less effective suppressors of CD4+ and CD8+ cells.
Although the proliferative capacity of early-passaged cells significantly decreased, they were more potent immunosuppressors than late-passaged cells. Late-passaged cells downregulated many immunosuppressive genes: Tnfrsf11b was downregulated 1200-fold in p6 versus p1, Tgfb2 was downregulated 17-fold, and leukocyte inhibitory factor (Lif) was downregulated 31-fold.
This study is limited by variable sources of MSCs; unfortunately, BM-MSCs isolated in our lab were highly contaminated with haemopoietic cells, which could affect downstream analysis (Online Resource 4). Therefore, we used immunodepleted BM-MSCs p1 obtained from NIH-funded research at The Scripps Research Institute. We tried to expand BM-MSCs and AD-MSCs, but passaged cells were low in quantity and quality so we used commercially available BM-MSCs p6 which indeed had decreased telomerase reverse transcriptase activity (TERT) in comparison with p1 cells (Online Resource 2). Although it will be interesting to know the inhibitory effect of MSCs derived from other tissues, we only used BM and AD for our study because It was more feasible. In vivo replication of the results was difficult due to low cell yield and low proliferative capacity.
In conclusion, we demonstrated that not only the cell phenotype but also the immunomodulatory properties of MSCs are largely affected by the cell origin and cell quality, which could be reflected in clinical outcomes. In vivo and detailed immunological studies of MSCs from different origins and passages will help to unify the cell sources and passages. Efforts to isolate homogenous MSC populations should focus not only on phenotypic characteristics but also on functional characteristics. These functional variations must be taken into consideration in trials to optimize MSC-based therapy.
References
Friedenstein AJ, Chailakhjan RK, Lalykina KS (1970) The development of fibroblast colonies in monolayer cultures of guina-pig bone marrow and spleen cells. Cell Prolif 3:393–403
Pittenger MF (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Gao F, Chiu SM, Motan DA et al (2016) Mesenchymal stem cells and immunomodulation: status and prospects. Cell Death Dis. https://doi.org/10.1038/cddis.2015.327
Abreu SC, Antunes MA, Xisto DG et al (2017) Bone marrow, adipose, and lung tissue-derived murine mesenchymal stromal cells release different mediators and differentially affect airway and lung parenchyma in experimental asthma. Stem Cells Transl Med. https://doi.org/10.1002/sctm.16-0398
Wen L, Zhu M, Madigan MC et al (2014) Immunomodulatory effects of bone marrow-derived mesenchymal stem cells on pro-inflammatory cytokine-stimulated human corneal epithelial cells. PLoS ONE. https://doi.org/10.1371/journal.pone.0101841
Tomic S, Djokic J, Vasilijic S, Vucevic D et al (2011) Immunomodulatory properties of mesenchymal stem cells derived from dental pulp and dental follicle are susceptible to activation by toll-like receptor agonists. Stem Cells Dev. https://doi.org/10.1089/scd.2010.0145
Amable PR, Teixeira MV, Carias RB et al (2014) Gene expression and protein secretion during human mesenchymal cell differentiation into adipogenic cells. BMC Cell Biol 15:46
Engela AU, Baan CC, Dor FJ et al (2012) On the interactions between mesenchymal stem cells and regulatory T cells for immunomodulation in transplantation. Front Immunol. https://doi.org/10.1186/s12860-014-0046-0
Kyurkchiev D, Bochev I, Ivanova-Todorova E, Mourdjeva M et al (2014) Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells. https://doi.org/10.4252/wjsc.v6.i5.552
Anjos-Afonso F, Bonnet D (2011) Prospective identification and isolation of murine bone marrow derived multipotent mesenchymal progenitor cells. Best Pract Res Clin Haematol. https://doi.org/10.1016/j.beha.2010.11.003
Zhang L, Chan C (2010) Isolation and enrichment of rat mesenchymal stem cells (MSCs) and separation of single-colony derived MSCs. J Vis Exp. https://doi.org/10.3791/1852
Huang S, Xu L, Sun Y, Wu T et al (2015) An improved protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. J Orthop Transl. https://doi.org/10.3791/1852
Yu G, Wu X, Kilroy G, Halvorsen YD et al (2011) Isolation of murine adipose-derived stem cells. Methods Mol Biol. https://doi.org/10.1007/978-1-61737-960-4_3
Ellis KM, O’Carroll DC, Lewis MD et al (2014) Neurogenic potential of dental pulp stem cells isolated from murine incisors. Stem Cell Res Ther. https://doi.org/10.1186/scrt419
Lushaj EB, Anstadt E, Haworth R et al (2011) Mesenchymal stromal cells are present in the heart and promote growth of adult stem cells in vitro. Cytotherapy. https://doi.org/10.3109/14653249.2010.529890
Kang SG, Shinojima N, Hossain A et al (2010) Isolation and perivascular localization of mesenchymal stem cells from mouse brain. Neurosurgery. https://doi.org/10.1227/01.NEU.0000377859.06219.78
Strioga M, Viswanathan S, Darinskas A et al (2012) Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev. https://doi.org/10.1089/scd.2011.0722
Glennie S, Soeiro I, Dyson PJ et al (2005) Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. https://doi.org/10.1182/blood-2004-09-3696
Bach M, Schimmelpfennig C, Stolzing A (2014) Influence of murine mesenchymal stem cells on proliferation, phenotype, vitality, and cytotoxicity of murine cytokine-induced killer cells in coculture. PLoS ONE. https://doi.org/10.1371/journal.pone.0088115
Kudlik G, Hegyi B, Czibula A et al (2016) Mesenchymal stem cells promote macrophage polarization toward M2b-like cells. Exp Cell Res. https://doi.org/10.1016/j.yexcr.2016.08.022
Zhou Y, Day A, Haykal S et al (2013) Mesenchymal stromal cells augment CD4+ and CD8+ T-cell proliferation through a CCL2 pathway. Cytotherapy. https://doi.org/10.1016/j.jcyt.2013.05.009
Ribeiro A, Laranjeira P, Mendes S et al (2013) Mesenchymal stem cells from umbilical cord matrix, adipose tissue and bone marrow exhibit different capability to suppress peripheral blood B, natural killer and T cells. Stem Cell Res Ther. https://doi.org/10.1186/scrt336
Le Blanc K, Rasmusson I, Sundberg B et al (2004) Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. https://doi.org/10.1016/S0140-6736(04)16104-7
Duijvestein M, Vos AC, Roelofs H et al (2010) Autologous bone marrow-derived mesenchymal stromal cell treatment for refractory luminal Crohn’s disease: results of a phase I study. Gut. https://doi.org/10.1136/gut.2010.215152
Gao F, Chiu SM, Motan DA et al (2016) Mesenchymal stem cells and immunomodulation: current status and future prospects. Cell Death Dis. https://doi.org/10.1038/cddis.2015.327
Boregowda SV, Krishnappa V, Phinney DG (2016) Isolation of mouse bone marrow mesenchymal stem cells. Methods Mol Biol. https://doi.org/10.1007/978-1-4939-3584-0_11
Baldari S, Di Rocco G, Piccoli M et al (2017) Challenges and strategies for improving the regenerative effects of mesenchymal stromal cell-based therapies. Int J Mol Sci. https://doi.org/10.3390/ijms18102087
Trounson A, Thakar RG, Lomax G, Gibbons D (2011) Clinical trials for stem cell therapies. BMC Med. https://doi.org/10.1186/1741-7015-9-52
Da Silva Meirelles L, Nardi NB (2003) Murine marrow-derived mesenchymal stem cell: ISolation, in vitro expansion, and characterization. Br J Haematol. https://doi.org/10.1046/j.1365-2141.2003.04669.x
Peister A, Mellad JA, Larson BL et al (2004) Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood. https://doi.org/10.1182/blood-2003-09-3070
Dominici M, Le Blanc K, Mueller I et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. https://doi.org/10.1080/14653240600855905
Hsu PJ, Liu KJ, Chao YY et al (2015) Assessment of the immunomodulatory properties of human mesenchymal stem cells (MSCs). J Vis Exp. https://doi.org/10.3791/53265
Ivanova-Todorova E, Bochev I, Dimitrov R et al (2012) Conditioned medium from adipose tissue-derived mesenchymal stem cells induces CD4 + FOXP3 + cells and increases IL-10 secretion. J Biomed Biotechnol. https://doi.org/10.1155/2012/295167
Deng Y, Zhang Y, Ye L et al (2016) Umbilical cord-derived mesenchymal stem cells instruct monocytes towards an IL10-producing phenotype by secreting IL6 and HGF. Sci Rep. https://doi.org/10.1038/srep37566
Puissant B, Barreau C, Bourin P et al (2016) Immunomodulatory effect of human adipose tissue-derived adult stem cells: comparison with bone marrow mesenchymal stem cells. Br J Haematol. https://doi.org/10.1111/j.1365-2141.2005.05409.x
Chen PM, Liu KL, Hsu PJ et al (2014) Induction of immunomodulatory monocytes by human mesenchymal stem cell-derived hepatocyte growth factor through ERK1/2. J Leukoc Biol. https://doi.org/10.1189/jlb.3A0513-242R
Ryan EJ, Bengtsson AK (2002) Immune function of the decoy receptor osteoprotegerin. Crit Rev Immunol 22:15
Fan L, Hu C. Chen J, Cen P et al (2016) Interaction between mesenchymal stem cells and B-cells. Int J Mol Sci. https://doi.org/10.3390/ijms17050650
Rosado MM, Bernardo ME, Scarsella M et al (2015) Inhibition of B-cell proliferation and antibody production by mesenchymal stromal cells is mediated by T cells. Stem Cells Dev. https://doi.org/10.1089/scd.2014.0155
Schlesinger M, Rabinowitz R, Levy P, Maayan S (1996) The expression of CD8 on B lymphocytes in HIV-infected individuals. Immunol Lett 50:23–27
Rabinowitz R, Massiah E, Hadar R, Schlesinger M (1995) In vitro activation leads to the binding of T-cell markers to the surface of B-lymphocytes. Clin Immunol Immunopathol 76:148–154
Hu Y, Qin C, Zheng G et al (2016) Mesenchymal stem cell-educated macrophages ameliorate LPS-induced systemic response. Mediators Inflamm. https://doi.org/10.1155/2016/3735452
Yin Y, Wu RX, He XT et al (2017) Influences of age-related changes in mesenchymal stem cells on macrophages during in-vitro culture. Stem Cell Res Ther. https://doi.org/10.1186/s13287-017-0608-0
Funding
This study was supported by the Ministry of Higher Education, Egypt and a Grant-in-Aid from the Ministry of Education, Science, Sports, Culture and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethics approval
All experiments were approved by the Institutional Animal Care and Use Committee of Nagasaki University and performed in accordance with the institutional and national guidelines.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
El-Sayed, M., El-Feky, M.A., El-Amir, M.I. et al. Immunomodulatory effect of mesenchymal stem cells: Cell origin and cell quality variations. Mol Biol Rep 46, 1157–1165 (2019). https://doi.org/10.1007/s11033-018-04582-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-04582-w