
Abstract
The interleukin 23 receptor (IL-23R) polymorphisms have been already discussed in rheumatoid arthritis (RA) repeatedly, but the results are conflict. The purpose of this meta-analysis was to assess whether IL-23R gene polymorphisms are associated with RA. We retrieved the available data from Pubmed, Medline, CNKI and CBM. Our study evaluated the effects of two polymorphisms (rs10489629, rs7517847) in European population. Pooling all the subjects, we found significant associations between the two polymorphisms and RA. For rs10489629, the pooled ORs (95 % CI) of C versus T, C/C+C/T versus T/T and C/C versus C/T+T/T were 1.092 (1.038–1.149), 1.146 (1.059–1.240) and 1.099 (1.008–1.199), respectively. For rs7517847, the combined ORs (95 % CI) of G versus T, G/G+G/T versus T/T and G/G versus G/T+T/T were 1.121 (1.063–1.183), 1.184 (1.092–1.283) and 1.133 (1.030–1.246), respectively. In conclusion, this meta-analysis demonstrates that the polymorphisms rs10489629 and rs7517847 of the IL-23R gene may be considered as risk factors for developing RA in European population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease of progressive joint destruction and impacts substantially 1 % of the world’s population [1]. Although the pathogenesis of RA is still unknown, the genetic factors seem to play an important role [2]. Human leukocyte antigen (HLA) region is the best-characterized gene susceptible to RA [3]. Nonetheless, the HLA locus only accounts for about one third of the total genetic factors of RA susceptibility [4]. It is indicated that non-HLA genes may take a prevalent part in RA.
Interleukin-23 (IL-23) has been classified as a necessary factor for the development of T cell-dependent inflammation and regulates the differentiation of naive CD4+ T cells into T helper cells [5, 6]. IL-23 plays a crucial role in the development of pathogenic Th17 cells which produce the cytokine IL-17. IL-17 can stimulate the production of several inflammatory cytokines, such as IL-6, and tumor necrosis factor (TNF)-α [7, 8]. IL-23 also leads to activation of janus kinases (Jak2) and tyrosine kinase (Tyk2) when combining with a complex, including IL-23 receptor (IL-23R) and IL-12 receptor beta-1 (IL-12Rβ1). Jak kinases phosphorylate IL-23R at discrete positions and emerge docking sites for the signal transducers and activators of transcription (STATs). Then the STATs are phosphorylated, dimerized and translocated to the nucleus where they effect the transcription of crucial pro-inflammatory genes such as IL-17 [9, 10]. IL-23/IL-17 axis is believed to be a new pathway which is involved in inflammatory responses [11]. Moreover, the serum and synovial fluid levels of IL-23 correlate with the IL-17 concentration in RA [12].
IL-23R, encoding on chromosome 1 (1p31.3), is a type I transmembrane protein and consists of 629 amino acids [9]. Recent studies suggest that the IL-23R variants might be affected with serum cytokine concentrations and subsequently impact on dependent cytokines [13]. There are four single nucleotide polymorphisms (SNPs) which have been studied most in RA (rs10489629, rs7517847, rs11209026, rs1343151). Although meta-analysis of the four SNPs had been reported before [14–16], new researches on the role of rs10489629 and rs7517847 in RA have been published and provided new proofs which were not involved in the previous meta-analysis. Due to small sample size of the former studies, the results of the new articles were contradictory with the previous ones. To better understand the relation of IL-23R gene polymorphisms (rs10489629, rs7517847) and RA risk, we performed the meta-analysis of these relevant studies to explore more accurate results.
Materials and methods
Identification of eligible studies
Available articles about the relation between IL-23R polymorphisms and RA were cautiously collected. A literature search was made using the key words “Interleukin 23 receptor’’ (IL-23R), “rheumatoid arthritis” (RA) and “polymorphism”, and only fully published studies until February 2012 were selected. All data were gathered from the following electronic databases: Pubmed, Medline, China National Knowledge Infrastructure database (CNKI), Chinese Biomedical database (CBM). When the studies contained overlapping data, we just chose the most recent subjects with largest sample. Eligible studies should approach to the following inclusion criteria: (a) all patients with RA were diagnosed by the 1987 American College of Rheumatology criteria, (b) it was a case–control design, (c) the study demonstrated the association between the IL-23R gene polymorphisms and RA, (d) the frequencies of alleles were available.
Data extraction
All information was extracted cautiously by two authors. Discrepancy was addressed by discussion. From each study, we extracted the following information: first author’s name, year of publication, country, ethnicity, the numbers of cases and controls, genotype method and allele frequencies of the IL-23R SNPs.
Quality assessment
Two authors evaluated the quality of the selected studies with the Newcastle–Ottawa Scale (NOS) [17]. In the case–control study, this scale describes three aspects of quality: selection of cases and controls, comparability of cases and controls, and ascertainment of exposure. Maximums of four stars, two stars and three stars are distributed to selection, comparability and exposure respectively. So, nine stars are the highest. The score higher than six is defined as high quality. Disagreement was settled by discussion.
Statistical analyses
We assessed the heterogeneity between studies with Q statistic as well as I2 statistic which measures the degree of heterogeneity (I2 = 100 % × (Q − df)/Q). I2 value varies from 0 to 100 %. Below 25 % is representative of no heterogeneity. From 25 % to 50 % stands for moderate heterogeneity. There is a large heterogeneity over 50 % [18]. p ≤ 0.10 was suggested to a statistically significance. We obtained the pooled odds ratio (OR) and 95 % confidence interval (CI) of all studies from fixed or random effect model. When there was no heterogeneity, we used the fixed effect model for meta-analysis [19]. Otherwise, a random effect model was used [20].
Evaluation of publication bias
The funnel plot was conducted on publication bias, and Egger’s linear regression test was to estimate funnel plot asymmetry [21]. The statistical analyses were completed by Stata 10.0 (Stata Corporation, College Station, TX, USA). All the p values were two-sided. p values less than 0.05 were regarded as significant.
Result
Selection and characteristics of studies
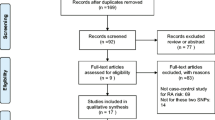
The process of study selection was showed in Fig. 1. After an initial search, we got 17 reports. According to the inclusion criteria, six reports were identified (excluded seven other autoimmune diseases, two reviews, and two other design reports). Three were excluded because two were not related to the SNPs (rs10489629, rs7517847) and one was not in Hardy–Weinberg equilibrium (HWE) [22]. In the remaining three reports, one comprised four different populations of each SNP, another contained two different populations of each SNP, and the other included one population of rs7517847. At last, six studies of rs10489629 and seven studies of rs7517847 were available in our meta-analysis [14–16]. All cases and controls in the studies were from European. Characteristics of all studies were illustrated in Table 1. The quality assessments of the studies were shown in Table 2. All studies were high quality for NOS score above six.

Process of study selection
Associations between polymorphisms and RA
Eligible studies were all pooled in this meta-analysis. A summary of the result was presented in Table 3:
-
1.
rs10489629. Six studies were involved with 5,786 patients and 7,326 controls from European. Compared with T allele, C allele was risk for RA (OR = 1.092, 95 %CI = 1.038–1.149, p = 0.001). Distribution of the ORs was presented in the forest plot (Fig. 2). We also discovered significant associations in the dominant and recessive modeling (C/C+C/T vs. T/T, OR = 1.146, 95 % CI = 1.059–1.240, p = 0.001, Fig. 3; C/C vs. C/T+T/T, OR = 1.099, 95 % CI = 1.008–1.199, p = 0.032, Fig. 4). In addition, a fixed effect model was applied to the studies for no significant heterogeneity between the comparisons (C vs. T, p = 0.122, I2 = 42.5 %, Fig. 2; C/C+C/T vs. T/T, p = 0.183, I2 = 33.8 %, Fig. 3; C/C vs. C/T+T/T, p = 0.303, I2 = 17.1 %, Fig. 4) (Table 3).
Fig. 2 
Forest plot for meta-analysis of C versus T (fixed effect model) with rs10489629
Fig. 3 
Forest plot for meta-analysis of C/C+C/T versus T/T (fixed effect model) with rs10489629
Fig. 4 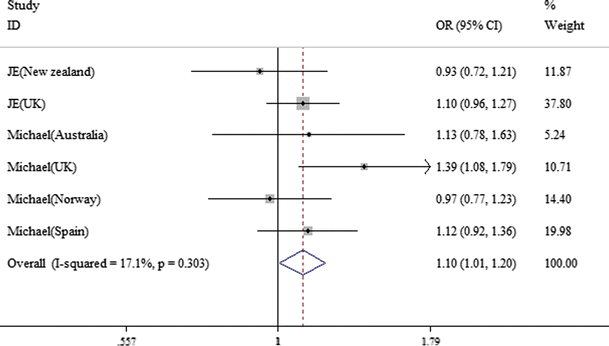
Forest plot for meta-analysis of C/C versus C/T+T/T (fixed effect model) with rs10489629
-
2.
rs7517847. Pooling the seven studies, a significant heterogeneity was found (p = 0.078, I2 = 47.2 %), so a random effect model was used (OR = 1.087, 95 % CI = 1.009–1.170, p = 0.027). Owing to a significant heterogeneity in overall studies, we calculated the combined ORs repeatedly after removing one study at a time. Excluding the Norway research, the result showed no significant heterogeneity (p = 0.483, I2 = 0.0 %, Fig. 5). The remaining six studies comprised of 5,089 cases and 6,711 controls from Europe. The G allele was risk for RA (G vs. T, OR = 1.121, 95 % CI = 1.063–1.183, p < 0.001, Fig. 5). The dominant and recessive modeling were also susceptible to RA (G/G+G/T vs. T/T, OR = 1.184, 95 % CI = 1.092–1.283, p < 0.001, Fig. 6; G/G vs. G/T+T/T, OR = 1.133, 95 % CI = 1.030–1.246, p = 0.010, Fig. 7). For no significant heterogeneity between comparisons after omitting the Norway population, we chose a fixed effect model (G/G+G/T vs. T/T, p = 0.506, I2 = 0.0 %, Fig. 6; G/G vs. G/T+T/T, p = 0.509, I2 = 0.0 %, Fig. 7) (Table 3).
Fig. 5 
Forest plot for meta-analysis of G versus T (fixed effect model) with rs7517847
Fig. 6 
Forest plot for meta-analysis of G/G+G/T versus T/T (fixed effect model) with rs7517847
Fig. 7 
Forest plot for meta-analysis of G/G versus G/T+T/T (fixed effect model) with rs7517847






Publication bias
The method of Egger’s linear regression test was used for accessing the funnel plot asymmetry. When publication bias does not exist, the regression line will pass through the origin. The results of Egger’s linear regression test shown in Table 3 indicated that there was no publication bias.
Discussion
The standard form of IL-23R is encoded by about 12 exons. It is suggested that at least six spliced isoforms could be generated (IL-23R1 to 6) via alternative splicing [23]. The SNPs rs10489629 located in intron seven and rs7517847 was in intron six [24]. The IL-23R gene expressed at least six alternatively spliced mRNAs, which lead to various isoforms of the receptor protein. The intronic polymorphisms (rs10489629, rs7517847) might exert their influence through regulating the specific splicing [25]. A genome-wide association study suggested a strong association with Crohn’s disease (CD) and IL-23R gene polymorphisms in 2006. Theoretical considerations implied potential relation of these SNPs with other autoimmune diseases such as RA [26]. A series of researches have been undertaken to replicate this association in RA. It has been confirmed that rs1343151 is risk for RA but rs11209026 is not associated with RA susceptibility [14, 16]. However, the associations of rs10489629 and rs7517847 with RA are still conflict. In this present study, we updated previous studies of the association between the IL23R polymorphisms (rs10489629, rs7517847) and RA in European population.
In this meta-analysis, we performed three genetic models for each SNP (rs10489629: C vs. T, C/C+C/T vs. T/T, C/C vs. C/T+T/T; rs7517847: G vs. T, G/G+G/T vs. T/T, G/G vs. G/T+T/T). Our research indicated C allele of rs10489629 was risk for RA which was consistent with the previous meta-analysis, and G allele of rs7517847 was susceptible to RA, while there was no significant association between rs7517847 and RA in the previous one [16]. Compared with the previous researches, we eliminated the study which exhibited a small deviation from HWE in the controls. Perhaps it contributed to the contradiction of rs7517847 between previous and present meta-analysis. In addition, the quality assessments of the studies were evaluated by the NOS. Although the number of the combined studies is rather small for a meta-analysis, these studies are all high quality.
However, the two SNPs are protective factors in ankylosing spondylitis (AS) [27–30]. All noninfectious inflammatory diseases depend on a spectrum from autoinflammatory (inducing by the innate immune system) to autoimmune (mediating by the adaptive immune system). Relative actions of each can define the type of diseases [31]. AS is considered to be a typical autoinflammatory disease, while RA is proposed to be autoimmune in nature [32, 33]. It is similar to PTPN22 C1858T which is associated with systemic autoimmune diseases such as RA, but not associated with organ-specific autoimmune diseases like AS [34, 35]. The same genetic variant may not share a common mechanism in different autoimmune diseases. Further researches are needed to clarify the exact mechanisms of IL-23R polymorphisms in RA.
By the way, some limitations of our study should be paid attention to. Primarily, the number of studies is small, and the sample was limited to European ancestry. Perhaps the allelic frequencies are different from European to others. Consequently, the relation in other ethnic groups can not be conducted. Secondly, our literature searching was based on English and Chinese, and language bias might be occurred. Next, significant heterogeneity was detected among several comparisons. The differences of experimental methods may cause the heterogeneity. Ultimately, owing to only extracting published studies, publication bias might occur, even if statistical test implied no significance.
In a word, this meta-analysis suggests the IL-23R polymorphisms rs10489629 and rs7517847 are susceptible to RA in European populations. Further gene–gene and gene–environment interactions researches on large case–control design in all races are still required.
References
Gabriel SE, Crowson CS, O’Fallon WM (1999) The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955–1985. Arthritis Rheum 42:415–420
Firestein GS (2003) Evolving concepts of rheumatoid arthritis. Nature 423:356–361
Weyand CM, Goronzy JJ (2000) Association of MHC and rheumatoid arthritis. HLA polymorphisms in phenotypic variants of rheumatoid arthritis. Arthritis Res 2:212–216
Cornélis F, Fauré S, Martinez M et al (1998) New susceptibility locus for rheumatoid arthritis suggested by a genome-wide linkage study. Proc Natl Acad Sci USA 95:10746–10750
Izcue A, Hue S, Buonocore S et al (2008) Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 28:559–570
Aggarwal S, Ghilardi N, Xie MH et al (2003) Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 278:1910–1914
Cornelissen F, van Hamburg JP, Lubberts E (2009) The IL-12/IL-23 axis and its role in TH17cell development, pathology and plasticity in arthritis. Curr Opin Investig Drugs 10:452–462
Bettelli E, Korn T, Oukka M, Kuchroo VK (2008) Induction and effector functions of T(H)17 cells. Nature 453:1051–1057
Parham C, Chirica M, Timans J et al (2002) A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol 168:5699–5708
Lankford CS, Frucht DM (2003) A unique role for IL-23 in promoting cellular immunity. J Leukoc Biol 73:49–56
Iwakura Y, Ishigame H (2006) The IL-23/IL-17 axis in inflammation. J Clin Invest 116:1218–1222
Kageyama Y, Kobayashi H, Kato N (2009) Infliximab treatment reduces the serum levels of interleukin-23 in patients with rheumatoid arthritis. Mod Rheumatol 19:657–662
Sarin R, Wu X, Abraham C (2011) Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci USA 108:9560–9565
Varade J, Ramón Lamas J, Rodríguez L et al (2009) IL23R and IL12B genes: susceptibility analysis in rheumatoid arthritis. Ann Rheum Dis 68:1230–1232
Hollis-Moffatt JE, Merriman ME, Rodger RA et al (2009) Evidence for association of an interleukin 23 receptor variant independent of the R381Q variant with rheumatoid arthritis. Ann Rheum Dis 68:1340–1344
Chen-Xu M, Topless R, McKinney C et al (2012) Replication of association of the interleukin 23 receptor rs1343151 variant with rheumatoid arthritis in Caucasian sample sets. Ann Rheum Dis 71:155–157
Wells GA, Shea B, O’Connell D et al. (2012) The Newcastle–Ottawa Scale (NOS) for assessing the quality of nonrandomized studies in meta-analyses. http://www.ohri.ca/programs/clinical_epidemiology/oxford. Accessed 2 March 2012
Higgins JP, Thompson SG (2002) Quantifying heterogeneity in a meta-analysis. Stat Med 21:1539–1558
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22:719–748
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7:177–188
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. Br Med J 315:629–634
Orozco G, Rueda B, Robledo G, Garcia A, Martin J (2007) Investigation of the IL23R gene in a Spanish rheumatoid arthritis cohort. Hum Immunol 68:681–684
Zhang XY, Zhang HJ, Zhang Y et al (2006) Identification and expression analysis of alternatively spliced isoforms of human interleukin-23 receptor gene in normal lymphoid cells and selected tumor cells. Immunogenetics 57:934–943
Kim HS, Kim I, Kim JO, Bae JS, Shin HD, Bae SC (2009) No association between interleukin 23 receptor gene polymorphisms and systemic lupus erythematosus. Rheumatol Int 30:33–38
Safrany E, Melegh B (2009) Functional variants of the interleukin-23 receptor gene in non-gastrointestinal autoimmune diseases. Curr Med Chem 16:3766–3774
Duerr RH, Taylor KD, Brant SR et al (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314:1461–1463
Burton PR, Clayton DG, Cardon LR et al (2007) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 39:1329–1337
Lee YH, Choi SJ, Ji JD, Song GG (2012) Associations between interleukin-23R polymorphisms and ankylosing spondylitis susceptibility: a meta-analysis. Inflamm Res 61:143–149
Karaderi T, Harvey D, Farrar C et al (2009) Association between the interleukin 23 receptor and ankylosing spondylitis is confirmed by a new UK case–control study and meta-analysis of published series. Rheumatology (Oxford) 48:386–389
Rahman P, Inman RD, Gladman DD, Reeve JP, Peddle L, Maksymowych WP (2008) Association of interleukin-23 receptor variants with ankylosing spondylitis. Arthritis Rheum 58:1020–1025
McGonagle D, McDermott MF (2006) A proposed classification of the immunological diseases. PLoS Med 3(8):e297. doi:10.1371/journal.pmed.0030297
Hull KM, Shoham N, Chae JJ, Aksentijevich I, Kastner DL (2003) The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol 15:61–69
Tan AL, Tanner SF, Conaghan PG et al (2003) Role of metacarpophalangeal joint anatomic factors in the distribution of synovitis and bone erosion in early rheumatoid arthritis. Arthritis Rheum 48:1214–1222
Lee YH, Rho YH, Choi SJ et al (2007) The PTPN22 C1858T functional polymorphism and autoimmune diseases – a meta-analysis. Rheumatology (Oxford) 46:49–56
Pearce SH, Merriman TR (2006) Genetic progress towards the molecular basis of autoimmunity. Trends Mol Med 12:90–98
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (30830089, 81102192) and the Anhui Provincial Natural Science Foundation (11040606M183).
Conflict of interest
The authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhai, Y., Xu, K., Huang, F. et al. Association of interleukin 23 receptor gene polymorphisms (rs10489629, rs7517847) with rheumatoid arthritis in European population: a meta-analysis. Mol Biol Rep 39, 8987–8994 (2012). https://doi.org/10.1007/s11033-012-1768-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-012-1768-8