
Abstract
Laccases are strong oxidizing enzymes that oxidize chlorinated phenols, synthetic dyes, pesticides, polycyclic aromatic hydrocarbons as well as a very wide range of other compounds with high redox potential. Based on the bias of genetic codons between fungus and yeast, we synthesized a laccase gene GlLCCI, originated from Ganoderma lucidum using optimized codons and a PCR-based two-step DNA synthesis method. The recombinant laccase, GlLCCI was successfully over-expressed in yeast, Pichia pastoris, with an alcohol oxidase1 promoter. The recombinant GlLCCI has a molecular mass of approximately 58 kDa. The K m values of GlLCCI for 2-2′-azino-bis-(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) and guaiacol were 0.9665, and 1.1122 mM, respectively. The V max of GlLCCI for both substrates was 3,024 and 82.13 μM mg−1 min−1. When ABTS was used as a substrate, the enzyme had an optimal temperature of approximately 55°C. The enzyme was detected over pH values from 2 to 8. The enzyme was strongly activated by K+, Na+, Cu2+ and mannitol. Six amino acids (alanine, histidine, glycine, arginine, aspartate and phenylalanine) increased the catalytic ability of the enzyme. The activity of laccase was obviously inhibited by Fe2+, Fe3+, sodium hydrosulphite, and sodium azide. Additionally, under optimal conditions, GlLCCI decolorized 37.62 mg l−1 of azo dye methyl orange (MO) in cultural medium. With a high MO degradation ability, GlLCCI may have potential in the treatment of industrial effluent containing azo dye MO.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Laccases (EC1.10.3.2), of the blue copper oxidase enzyme family, catalyze reactions for a broad range of substrates. Amino acid-sequence analysis of different laccases indicates that the copper-binding sites are highly conserved [1]. Substrates for laccases vary from organic to inorganic compounds, including aromatic amines, diphenols, and polyphenols catalyzed by a one-electron transfer mechanism [2]. Guaiacol, 2,6-dimethoxyphenol [3], or the electron-rich, non-phenolic substrate 2-2′-azino-bis-(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) [4] are the most commonly used substrates for enzyme activity measurement. Laccase also possesses functions wherein it decolorizes a range of synthetic dyes, such as, acidic, basic, disperse, azo, diazo, anthraquinone-based and metallic complex dyes [5]. So far, many studies have been reported on several species such as Trametes versicolor [6], Pleurotus eryngii [7], and Phanerochaete chrysosporium [8], but very limited research has been reported for the laccase of the Ganoderma lucidum strain 7071-9.
Ganoderma lucidum, a member of the white-rot basidiomycete family, enjoys special veneration in Asia and has been found to produce chemicals such as hypoglycemic polysaccharides, immunomodulatory [9] and antitumor proteins [10]. In pharmaceutical and medicinal studies and commercially cultivated isolates, it is generally named G. lucidum. This name, however, contains several laccate Ganoderma species that might differ in their bioactive compound composition. In 1996, different strains of G. lucidum were first grouped by internally transcribed spacer sequences [11]. A G. lucidum gene (GlLCC) was first cloned by Joo et al. [12]. However, studies on laccases from the G. lucidum species are limited to the analysis of laccase isoenzymes [13]. In 1999, Joan Lin Cereghino and James M. Cregg elaborated the process of heterologous protein expression in the methylotrophic yeast Pichia pastoris and described the methanol pathway in P. pastoris [14].
In this research, levels of codon usage in G. lucidum were determined, and G. lucidum 7071-9 gene (GlLcc) was synthesized through PTDS method, which was a newly developed PCR-based two-step DNA synthesis method for long gene sequences, the first step needed to synthesize individual fragments of the DNA of interest: ten to twelve 60mer oligonucleotides with 20 bp overlap were mixed and a PCR reaction was carried out to produce DNA fragments that were 500 bp in length; the second step needed to synthesize the entire sequence: five to ten PCR products from the first step were combined and used as the template for a second PCR reaction, with the two outermost oligonucleotides as primers. After linked with the secretory expression vector, the gene GlLCCI was heterogeneously expressed in P. pastoris and then characterized.
Materials and methods
Codon-usage and sequence-data analysis
The original DNA sequence of GlLCC gene (GenBank accession No. AF185275) from G. lucidum was downloaded from GenBank (ftp://ftp.ncbi.nih.gov) and were deposited in the software DNAStar v 8.02 earlier. Codon-usage processing was performed with an NCBI-related database at http://www.kazusa.or.jp/codon. The entire 1,563-bp sequence of the G. lucidum strain 7071-9 laccase gene was used. The DNAman v 6.0 software was used for relative adaptiveness analysis.
Strains, culture and chemicals
The Escherichia coli strain DH5α (Invitrogen, USA) was used in all plasmid manipulations. All enzymes of DNA or RNA manipulation were purchased from TaKaRa (Dalian, China). P. pastoris strain GS115 (his4) and plasmid pPIC9K were purchased from Invitrogen (San Diego, CA, USA). Yeast extract–peptone–dextrose (YPD), buffered minimal methanol (BMM), buffered minimal glycerol (BMG), minimal dextrose (MD), and other media were prepared following the instructions in the manufacturer’s manual in the Pichia Expression Kit (Invitrogen, USA). Oligonucleotides of Gl. 60 bp each were ordered from the Shanghai Sangon Biological Engineering Technology and Service Co., Ltd (Supplementary Table 1).
Synthesis, cloning and vector construction of the laccase gene
Based on the sequence of the GlLcc gene (GenBank accession No. AF185275), we modified the gene by deleting a 5′-end fragment coding for the N-terminal 22 amino acids signal sequence and added a BamHI and a SacI site at the 5′- and 3′-end, respectively (Supplementary Fig. 1). After codon optimization, we re-synthesized the recombinant gene using the PTDS technique [15] (Fig. 1). The synthesis included two subsequent PCR steps: (1) Thirty-six overlapped PCR primers were added in three PCR tubes, consisting of 50 μl PCR reaction, 200 nM of each outer primer and 20 nM of each inner primer. The thermal cycling conditions were: one cycle of 1 min at 94°C, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 54°C for 30 s and extension at 72°C for 1.5 min. Followed by an additional 10 min at 72°C to ensured a complete extension of the PCR products; (2) Aliquots of the three PCR products were used as templates for the second-step PCR, to which only the first forward primer (P1) and the last reverse primer (P36) were added to the reaction. All thermal cycling conditions were the same as the first-step PCR. Thereafter, the PCR product was cloned and sequenced. The synthetic gene of G. lucidum strain 7071-9 laccase was named as GlLCCI. After digestion by the restriction enzymes, GlLCCI was cloned into the secretory expression vector pYM 7909 using a modified pPIC9K (Invitrogen) in which the SacI site in the AOX 1 promoter and XhoI site in the G418 cassette were removed by site-directed mutagenesis. Meanwhile, a 6× His-tag was added after KEX2 protease cleavage at the site of the MF α-signal sequence (Supplementary Fig. 2).

The PTDS strategy for the synthesis of the GlLCCI gene. Oligonucleotides of Gl. 60 bp were assembled as the graph described. The gray oligonucleotides represent sense oligonucleotides while the black oligonucleotides represent anti-sense oligonucleotides. There was a 20 bp overlap for each of the oligonucleotides used
Transformation and screening
The plasmid pYM 7909 was cloned by E. coli DH5α. Three microgram of the purified plasmid pYM 7909 was linearized by digestion with enzyme SalI and the transformed into competent cells of P. pastoris through electroporation (Bio-Rad Genepulser, Hercules, USA) at 1,750 V with a 0.2-cm cuvette. After electroporation, P. pastoris cells were plated on selective plates (SD, 0.8 M sorbitol, 5% glucose, and 2% agar) and incubated at 28°C for 72 h. The surviving cells were picked up and streaked on MD (1.34% YNB, 0.000004% biotin, 0.5% methanol, and 2% agar) plates. After incubation at 28°C for 48 h, clones that appeared on the MD plates were amplified through liquid BMGY and expression was induced by liquid BMM (1% methanol added), and the activity of the enzyme in supernatants was detected by 10 mM ABTS.
Enzyme expression and purification
Laccase from one positive clone exhibiting high enzymatic activity was extracted by incubating the colony in 250 ml of BMGY liquid (with 1% glucose) at 28°C overnight with continuous shaking (220 rpm). The cells were harvested by centrifugation at 4,000×g, 4°C for 3 min when the OD600 of the culture reached 0.8–1. The cell pellets were re-suspended within BMM liquid by rotary shaking (120 rpm), methanol was added (1% final concentration) every 24 h for 3 days; CuSO4 was also added (1 mM final concentration) to induce the expression of the enzyme. The supernatant was harvested by centrifugation (4,000×g, 4°C, and 3 min).
Ammonium sulphate was added to the clean supernatant. After a centrifugation at 12,000 rpm at 4°C for 15 min, the precipitation was filtered through Microza (filter membrane) and dialyzed supernatant was loaded on to a nickel-charged iminodiacetic acid column pre-equilibrated with 50 mM PBS buffer, containing 0.5 M NaCl, 10% glycerol, 1% Tween, 1% Triston-X100, 0.5% β-ME, DD H2O at 4°C. The column was first washed with the same buffer containing 10 mM imidazole, and then 150 mM imidazole, finally the 6× His-tagged enzyme was eluted with the buffer containing 250 mM imidazole. Fractions were collected and dialyzed at 4°C overnight against SD, and fractions containing the laccase activity were pooled subsequently.
Enzyme characterization
The specific activity of the enzyme was measured with ABTS and guaiacol, respectively. The reagents 10 mM ABTS and 0.2 M citrate–phosphate buffer were incubated at 45°C for 10 min prior to starting the reaction with 50 μl of purified enzyme; reactions were terminated with 50 μl of 1 M sodium fluoride after 15 min and OD420 was immediately measured (TECAN Tecan i-control spectrophotometer). Similarly, 1.37 mM guaiacol and 0.2 M citrate–phosphate buffer were incubated at 45°C for 10 min prior to starting the reaction with 100 μl of purified enzyme and reactions were terminated after 12 h with 50 μl of 1 M sodium fluoride and immediately measured the OD at 480 nm.
Specific activity was defined as units per milligram of protein. One unit of laccase activity was defined as the enzyme amount required to catalyze 1 μmol of substrate per min. Suitable amounts of enzyme were used to obtain absorbance of 0.1–1 after approximation of time was used in all photometric measurements applying common test conditions. Protein concentrations were determined using the Bradford method (Bio-Rad protein assay dye reagent protocol). The enzyme molecular mass was estimated by 12% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) electrophoresis compared with the protein markers (Watson Bio-tech, Shanghai, China). Gel staining was performed with Coomassie Brilliant R-250. All assays were performed at least in triplicates. The graphs represented the averages of the values obtained and the error bars show the ranges of values.
The thermo dependence of enzymatic activity was measured between 25 and 45°C with ABTS. Thermostability assays were performed by incubating aliquots of the enzyme at 25, 35, 45 and 55°C for 10 min. Aliquots were removed in a time course and enzyme activity measured with ABTS as described above at 55°C for 15 min. The effect of pH on enzyme activity was determined with ABTS by assaying in the 0.2 M citrate–phosphate buffer ranging from pH 2 to 7. The pH stability was examined by assaying with 1.5 mM ABTS after incubating the enzyme for 24 h in the 0.2 M citrate–phosphate buffer at 4°C. Relative activity was measured as described above.
Effects of metallic ions were examined at two different concentrations (1 and 10 mM). Aliquots of enzyme 0.2 M citrate–phosphate buffer (pH 2.6) containing FeSO4, FeCl3, NaCl, CaCl2, KCl, MgCl2, ZnSO4, AlCl3, MnSO4, CuSO4, SDS, or EDTA were cultured for 1.5 h at 4°C. Activities of the treated enzyme was measured at 55°C for 15 min with 10 mM ABTS. Effects of organic and inorganic compounds were measured in 0.1 and 1 mM two different concentrations after incubation at 4°C for 1.5 h with the following four compounds: sodium thiosulfate, sodium azide, mannitol, and sodium hydrosulphite, or six l-amino acids: alanine, histidine, glycine, arginine, aspartate and phenylalanine. The relative activity was measured by methods described above.
MO decolorization was carried out in citrate–phosphate buffer (pH 4.0, 0.2 M) containing MO at 65 mg−1 l and 0.025 mM ABTS as a redox mediator. After adding 140 μl purified enzyme, enzyme activity of the reaction mixture (400 μl with 5 mM Cu2+) was measured at 25, 35, 45 and 55°C from 3 to 48 h. To study the metal ions effects on MO decolorization, different concentrations of K+, Mg2+, Cu2+ and Zn2+ were used in a total volume of 400 μl in a test tube. The reaction mixtures were incubated at 35°C for 3 h and the dye decolorization was measured at 470 nm.
Kinetic studies
Kinetic studies were performed using ABTS concentrations from 0.5 to 4 mM. The absorbance at 420 nm was monitored for 15 min at 55°C and guaiacol concentrations from 0.0343 to 0.343 mM, the absorbance at 480 nm was monitored for 12 h at 55°C. The K m and V max were calculated using the Enzyme Kinetics computer program GraphPad Prism (version 4.0, GraphPad Software Inc., San Diego, California).
Results
Parameterization of relative adaptiveness and codon usage in Ganoderma lucidum
Codons from G. lucidum were analyzed to determine the codon usage. The total coding GC content in the final modified gene is estimated at 53.79%. The relative adaptiveness of codon usage between G. lucidum and P. pastoris were calculated (mean difference = 24.76%). After gene modification, a new gene of GlLCCI that was more appropriate for expression of P. pastoris was designed.
Reconstruction, transformation and screening
Following two-step overlapped PCR amplification of the entire ORF of GlLCCI, a unique band of 1,512 bp was obtained (not shown) and sequenced to be correct. The reconstructed pYM 7909 was assessed by BamHI and SacI, and two right bands of 1,512 and 9,258 bp were shown on an agarose gel. The positively transformed P. pastoris cells, which contained correct reconstructed plasmids, were successfully selected on MD screen plate. Following activity tests of the crude laccase, 12 out of 1,500 P. pastoris colonies were shown to be active with ABTS and one of the vigorous colonies was chosen for further studies.
GlLCCI purification and kinetic properties
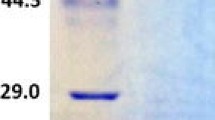
We purified the enzyme and examined its biochemical properties. The enzyme was stable at 4°C. SDS-PAGE analysis of the recombinant GlLCCI enzyme preparation showed that it was at least 85% pure and had an apparent molecular mass of 58 kDa (Fig. 2), which is smaller than that of three laccase isozymes derived from a different strain of G. lucidum [13]. The K m values of GlLCCI for ABTS and guaiacol were 0.9665 and 1.1122 mM, respectively, and the V max values of the GlLCCI at 55°C with a pH 2.6 for both substrates is 3,024 and 82.13 μM mg−1 min−1, respectively (Table 1).

SDS-PAGE of recombinant GlLCCI expressed via P. pastoris. Lane Marker protein molecular mass marker, Lane 1 unpurified GlLCCI, Lane 2, GlLCCI purified with nickel-charged iminodiacetic acid column at 4°C
Effects of pH and temperature on laccase activity and stability
The optimal pH value of the purified GlLCCI enzyme was 2.6 (Fig. 3b), reaching an undetectable level of laccase activity at pH greater than 8.0. When a pH of 9.0 was reached, GlLCCI activity was completely depleted. However, the activity of GlLCCI was still stable after incubation for 24 h at 4°C in 0.2 M citrate–phosphate buffer at pH ranging from 2 to 7; and 65% of activity persisted at pH 7 (Fig. 3d). The thermodependence of activity results showed that a maximum activity with ABTS was observed at 55°C and the enzyme still showed activity at 70°C (Fig. 3a). Slight loss of the activity was shown during the preincubation stage of the enzyme for 1 h at with temperature at 25°C. However, the activity decreased rapidly when pre-incubating at 55°C for 20 min (Fig. 3c).

Optimal temperature and pH, thermostability and pH stability of GlLCCI. a Thermodependence of purified GlLCCI was determined at various temperatures from 35°C to 70°C in increments of 5°C. b Effects of pH dependence were measured with 0.2 M Na2HPO4–citric acid buffer from pH 2 to 8. c Effect of pH on the stability of the recombinant GlLCCI, assayed after incubating the enzyme for 24 h at 4°C. The gray bar indicates that the enzyme was more stable at the optimum pH 2.6. The activity of the enzyme remained at 65% when incubated at pH 7. d The thermostability of purified GlLCCI was assayed via incubation of the same volume of purified enzyme at 25, 35, 45 or 55°C with different durations (10-min increment) and determined the remaining activity with ABTS as described above. The specific activity corresponding to 100% activity level was 141.28 U/mg. Values are means ± SD of three individual experiments
Effects of metallic ions on enzymatic activity
The effect of metallic ions on laccase activity was assessed with ABTS. Two metal ions concentrations of 1 and 10 mM were chosen for experiments. We noted that laccase activity remained stable in the presence of Mg2+, Mn2+ and EDTA at a low concentration of 1 mM. However, 10 mM Mg2+ could also increase the enzymatic activity. It is noteworthy that 10 mM K+ or Na+ enhanced laccase activity to 159.71 and 171.79%, which were higher than with Cu2+. Compared with the control, when the concentration of Cu2+ increased from 1 to 10 mM, the laccase activity increased by 24.14 and 49.14%, respectively (Table 2). This may be attributed to the sufficient occupation of copper binding sites with copper ions [16]. Both concentrations of Fe2+, Fe3+, Al3+, Zn2+ and SDS have been noted to decrease the activity of laccase. With increasing concentration of EDTA, the activity of laccase decreased to 45.85% at 10 mM, and had little effect at 1 mM.
Effects of organic and inorganic compounds on the enzyme activity
The effect of different chemical compounds on enzymatic activity was shown in Fig. 4. The most potent inhibitor for the enzyme was sodium hydrosulphite; when concentration increased from 0.1 to 1 mM, enzymatic activity decreased from 39.75 to 2.21%. Sodium thiosulfate and sodium azide also inhibited enzymatic activity at 1 mM concentration, but had minimal effect at 0.1 mM. Mannitol increased enzymatic activity to 115.62% at a concentration of 1 mM, but had no effect at 0.1 mM. All of the 1 mM six l-amino acids (alanine, histidine, glycine, arginine, aspartate and phenylalanine) stimulated the activity of the laccase to variable extents, but at the low concentration of 0.1 mM, the effect was not significant.

Effects of organic and inorganic compounds effects were measured in gradient concentrations. 1 sodium thiosulfate, 2 sodium azide, 3 mannitol, 4 sodium hydrosulphite, 5 l-alanine, 6 l-histidine, 7 l-glycine, 8 l-arginine, 9 l-aspartate, and 10 l-phenylalanine. The relative activity was measured as described above. Values are means ± SD of three individual experiments. The significant (P < 0.05) and extremely significant (P < 0.01) data were annotated as * and **
Decolorization of methyl orange
The effect of metal ions on laccase mediated decolorization of azo dye MO is shown in Fig. 5. In the presence of metal ions, 5 mM Cu2+ caused 8.8% degradation of MO in 3 h, which appeared to be the most effective. Five multimolar Mg2+, 15 mM Zn2+ and 20 mM K+ could also enhanced decolorization to a various extent (Fig. 5a). During 48 h incubation at four different temperatures, maximum of 57.48% decolorization of MO was observed at 35°C (Fig. 5b). Without ABTS as redox mediator, no decolorization was detected.

Metal ions, temperature and time effects on decolorization of MO. a Among the four metal ions used in the reactive system, Cu2+ activated the decolorization of MO most effectively at 5 mM, 8.8% of MO was degradable in 3 h. Five multimolar Mg2+, 15 mM Zn2+ and 20 mM K+ decolorized MO by 6.07, 2.77 and 2.41% respectively. b During 48 h incubation at four different temperatures, maximum of 57.48% decolorization of MO was observed at 35°C. All values represent the mean of triplicate measurements with a sample mean deviation of less than 3%
Discussion
During the stage of codon-usage adaptive modification, various biological parameters, such as expression level, mutation frequency, protein length and protein translation signals, are affected by codon bias [17]. The bias in the usage of the Ala, Leu, Arg and Gly codons is closer between G. lucidum and P. pastoris, and this suggest that host codon bias should be correlated to the expression for maximal heterologous gene expression [18]. After analysis of gene adaptation, we modified the original sequence and synthesized the 1512-bp GlLCCI gene rapidly through PTDS [15], which offered a simple, rapid, high-fidelity and low-cost alternative method of synthesizing and assembling long DNA fragments. P. pastoris was used to secrete foreign protein as the low levels of native proteins [19]. An inducible expression system is particularly important because the strongly regulated promoter (AOX1) is induced by this promoter [20]. Given the secretive signal for “MVKFQSLLSCVTLLFAASAHAG” in pYM 7909, the GlLCCI gene successfully expresses in the heterologous host P. pastoris. Compared with the reported molecular weights of most of fungal laccases, ranged from 55 to 90 kDa including carbohydrates [21], a 58 kDa of GlLCCI are quite similar to those of other fungal laccases.
Although GlLCCI is stable over a wide acidic pH range, the optimal pH value of the purified enzyme was 2.6, which was quite similar to other laccase enzymes from Agaricus blazei (pH 2) [22], Albatrellus dispansus (pH 4) [23], Lentinula edodes (pH 4) [16], and Hericium erinaceum (pH 5) [24], the optimal pH value of the purified enzyme was 2.6 and GlLCCI is stable over a wide acidic pH range, maintaining over 65% activity following incubation in buffers ranging from pH 2 to 7 for 24 h, which indicates that the pH-stable GlLCCI may be a good candidate for the development of a biotechnological tool. The specific activity of GlLCCI on ABTS was 1413-fold greater than that of guaiacol, suggested that the GlLCCI had better affinity to ABTS compared with the laccase of Pleurotus sajor-caju [1]. Unfortunately, the V max value of GlLCCI on guaiacol was relatively low [25].
Results of ion studies with the enzyme were similar to those reported for other laccases that were highly sensitive to Fe2+and Fe3+ [26]; this may be attributed to the ability of the ions of iron to interact with the electron transportation system of laccase. When a concentration of EDTA was increased, enzyme activity had decreased. This failure to restore activity may be due to an inability to maintain the proper conformation necessary for the reincorporation of metal ions that may be required to ensure structural stability, but not catalytic activity. Surprisingly, high concentrations of K+ and Na+ did not inhibit GlLCCI activity but promoted laccase activity distinctly, which digressed with the research data of a laccase from the Melanocarpus albomyces [27]. This can be attributed to the ions of K+ and Na+ may be required for laccase catalytic activity, but not for structural stability. A 10 mM high concentration of Mn2+, Al3+, and Zn2+ suppressed enzymatic activity obviously; this may because these ions had been incorporated with the enzyme wherein the active sites were blocked at the stage of pre-incubation.
After an independent-samples t test, we found that all the 1 mM six l-amino acids tested increased enzyme activity of laccase apparently. GlLCCI was more sensitive to 1 mM histidine, which demonstrated an ability to increase activity by 26%. Alanine, at 1 mM, enhanced activity by 13% but had no significant effect at a concentration of 0.1 mM. In the research reported by O’Callaghan [28], the appropriate addition of alanine into culture has increased the unitary output of the laccase. The mechanism of l-amino acids increased laccase activity level may correlate with their chemical structure of l-amino acids residue. These chemical structures increased the dielectric constant of water, diminished polarity effect and the Coulomb force which made electriferous substrate and enzyme easier to attract each other.
In conclusion, the results from this study clearly showed that laccase GlLCCI from the G. lucidum strain 7071-9 is a new member of the growing family of laccase enzymes. We report the purification and characterization of the recombinant laccase, GlLCCI from P. pastoris, which will be a new expression system. The strong ability of its decolorization of MO suggested that GlLCCI can be a potential enzyme for the removal of color from reactive textile dye effluent.
References
Soden DM, O’Callaghan J, Dobson AD (2002) Molecular cloning of a laccase isozyme gene from Pleurotus sajor-caju and expression in the heterologous Pichia pastoris host. Microbiology 148(Pt 12):4003–4014
Thurston CF (1994) The structure and function of fungal laccase. Microbiology 140:19–26
Prillinger H, Esser K (1975) The phenoloxidases of the ascomycete Podospora anserina XIII. Action and interaction of genes controlling the formation of laccase. Mol Gen Genet 156(3):333–345
Childs RE, Bardsley WG (1975) The steady-state kinetics of peroxidase with 2,2′-azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem J 145(1):93–103
Murugesan K (2006) Decolorization of reactive dyes by a thermostable laccase produced by Ganoderma lucidum in solid state culture. Enzyme Microb Technol 40(2007):1662–1672
Jonsson LJ, Saloheimo M, Penttila M (1997) Laccase from the white-rot fungus Trametes versicolor: cDNA cloning of lcc1 and expression in Pichia pastoris. Curr Genet 32(6):425–430
Wang HX, Ng TB (2006) Purification of a laccase from fruiting bodies of the mushroom Pleurotus eryngii. Appl Microbiol Biotechnol 69(5):521–525. doi:10.1007/s00253-005-0086-7
Baldrian P (2006) Fungal laccases—occurrence and properties. FEMS Microbiol Rev 30(2):215–242. doi:10.1111/j.1574-4976.2005.00010.x
Paterson RR (2006) Ganode—a therapeutic fungal biofactory. Phytochemistry 67(18):1985–2001. doi:10.1016/j.phytochem.2006.07.004
Gao Y, Gao H, Chan E, Tang W, Xu A, Yang H, Huang M, Lan J, Li X, Duan W, Xu C, Zhou S (2005) Antitumor activity and underlying mechanisms of ganopoly, the refined polysaccharides extracted from Ganoderma lucidum, in mice. Immunol Invest 34(2):171–198
Hseu RS, Wang HH, Wang HF, Moncalvo JM (1996) Differentiation and grouping of isolates of the Ganoderma lucidum complex by random amplified polymorphic DNA-PCR compared with grouping on the basis of internal transcribed spacer sequences. Appl Environ Microbiol 62(4):1354–1363
Joo SS, Ryu IW, Park JK, Yoo YM, Lee DH, Hwang KW, Choi HT, Lim CJ, Lee do I, Kim K (2008) Molecular cloning and expression of a laccase from Ganoderma lucidum, and its antioxidative properties. Mol Cells 25(1):112–118
Ko EM, Leem YE, Choi HT (2001) Purification and characterization of laccase isozymes from the white-rot basidiomycete Ganoderma lucidum. Appl Microbiol Biotechnol 57(1–2):98–102
Cereghino JL, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24(1):45–66
Xiong AS, Yao QH, Peng RH, Li X, Fan HQ, Cheng ZM, Li Y (2004) A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res 32(12):e98. doi:10.1093/nar/gnh09432/12/e98
Nagai M, Sato T, Watanabe H, Saito K, Kawata M, Enei H (2002) Purification and characterization of an extracellular laccase from the edible mushroom Lentinula edodes, and decolorization of chemically different dyes. Appl Microbiol Biotechnol 60(3):327–335. doi:10.1007/s00253-002-1109-2
Biro JC (2008) Does codon bias have an evolutionary origin? Theor Biol Med Model 5:1–15
Kliman RM, Irving N, Santiago M (2003) Selection conflicts, gene expression, and codon usage trends in yeast. J Mol Evol 57(1):98–109. doi:10.1007/s00239-003-2459-9
Fu XY, Zhao W, Xiong AS, Tian YS, Peng RH (2010) High expression of recombinant Streptomyces sp. S38 xylanase in Pichia pastoris by codon optimization and analysis of its biochemical properties. Mol Biol Rep. doi:10.1007/s11033-010-0644-7
Cereghino GP, Cregg JM (1999) Applications of yeast in biotechnology: protein production and genetic analysis. Curr Opin Biotechnol 10(5):422–427. doi:S0958-1669(99)00004-X
Peter MG, Wollenberger U (1997) Phenol-oxidizing enzymes: mechanisms and applications in biosensors. EXS 80:63–82
Ullrich R, Huong le M, Dung NL, Hofrichter M (2005) Laccase from the medicinal mushroom Agaricus blazei: production, purification and characterization. Appl Microbiol Biotechnol 67(3):357–363. doi:10.1007/s00253-004-1861-6
Wang HX, Ng TB (2004) A novel laccase with fair thermostability from the edible wild mushroom (Albatrella dispansus). Biochem Biophys Res Commun 319(2):381–385. doi:10.1016/j.bbrc.2004.05.011
Wang HX, Ng TB (2004) A new laccase from dried fruiting bodies of the monkey head mushroom Hericium erinaceum. Biochem Biophys Res Commun 322(1):17–21. doi:10.1016/j.bbrc.2004.07.075
Eggert C, Temp U, Eriksson KE (1996) The ligninolytic system of the white rot fungus Pycnoporus cinnabarinus: purification and characterization of the laccase. Appl Environ Microbiol 62(4):1151–1158
Murugesan K, Kim YM, Jeon JR, Chang YS (2009) Effect of metal ions on reactive dye decolorization by laccase from Ganoderma lucidum. J Hazard Mater 168(1):523–529. doi:10.1016/j.jhazmat.2009.02.075
Kiiskinen LL, Viikari L, Kruus K (2002) Purification and characterisation of a novel laccase from the ascomycete Melanocarpus albomyces. Appl Microbiol Biotechnol 59(2–3):198–204. doi:10.1007/s00253-002-1012-x
O’Callaghan J, O’Brien MM, McClean K, Dobson AD (2002) Optimisation of the expression of a Trametes versicolor laccase gene in Pichia pastoris. J Ind Microbiol Biotechnol 29(2):55–59. doi:10.1038/sj.jim.7000268
Acknowledgments
This research study was supported with funding from the Science and Technology Commission of Shanghai Municipality (09dz2200800). Another financial support was the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sun, J., Peng, RH., Xiong, AS. et al. Secretory expression and characterization of a soluble laccase from the Ganoderma lucidum strain 7071-9 in Pichia pastoris . Mol Biol Rep 39, 3807–3814 (2012). https://doi.org/10.1007/s11033-011-1158-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-1158-7