
Abstract
Lactobacillus plantarum (LP) has previously been used for the treatment and prevention of intestinal disorders and disease. However, the role of the LP surface layer adhesive protein (SLAP) in inhibition of epithelial cell disruption is not fully understood. The aim of the present study was to investigate the protective effects of purified SLAP on Caco-2 cells infected with enteropathogenic Escherichia coli (EPEC). The role of ERK in LP-mediated inhibition of tight junction (TJ) injury was also evaluated in order to determine the molecular mechanisms underlying the protective effects of LP in epithelial cells. SLAP was extracted and purified from LP cells using a porcine stomach mucin-Sepharose 4B column. SLAP-mediated inhibition of bacterial adhesion was measured using a competition-based adhesion assay. Expression of TJ-associated proteins, maintenance of TJ structure, and levels of extracellular signal regulated kinase (ERK) and ERK phosphorylation were assessed in SLAP-treated cells by a combination of real-time PCR, western blotting, and immunofluorescence microscopy. Cell permeability was analyzed by measurement of trans-epithelial electrical resistance (TER) and dextran permeability. The effect of SLAP on levels of apoptosis in epithelial cells was assessed by flow cytometry. Results from these experiments revealed that treatment with SLAP decreased the level of adhesion of EPEC to Caco-2 cells. SLAP treatment also enhanced expression of TJ proteins at both the mRNA and protein levels and affected F-actin distribution. Although ERK levels remained unchanged, ERK phosphorylation was increased by SLAP treatment. Caco-2 cells treated with SLAP exhibited increased TER and decreased macromolecular permeability, which was accompanied by a decrease in the level of apoptosis. Together, these results suggest that LP-produced SLAP protects intestinal epithelial cells from EPEC-induced injury, likely through a mechanism involving ERK activation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lactobacillus bacteria appear to benefit the human body through several mechanisms, including maintaining the balance of the intestinal microflora, modulating the intestinal immune system, detoxifying colonic toxins, lowering serum cholesterol levels, promoting lactose tolerance, and producing metabolites essential to the function of intestinal epithelial cells (IECs) [1, 2]. Furthermore, studies have demonstrated that Lactobacillus can improve pathological disorders in the intestine through modulation of intestinal functions. The proposed mechanisms for these improvements were shown to be multi-factorial and include increased mucin secretion by host cells [3], immuno-modulatory effects [4], and competitive exclusion of pathogens from the surface of IECs [5]. Additionally, Lactobacillus is also one of the best-characterized probiotic bacteria studied in clinical trials assessing the treatment and prevention of several intestinal disorders and diseases, including inflammatory bowel disease (IBD) and diarrhea [6].
Recent studies have shown that interactions between Lactobacillus and IECs are critical for adhesion of the bacteria to the epithelium. These interactions are thought to be initially mediated by non-specific physical interactions, but are rapidly followed by a second level of specific interactions between bacterial ligands and corresponding cell surface receptors on IECs, which may help to induce the protective effects of Lactobacillus on the intestinal barrier and intercellular tight junctions (TJ) [7, 8]. The formation of these interactions may inhibit subsequent adhesion of pathogenic bacteria, such as enteropathogenic Escherichia coli (EPEC).
The identity of the precise components required for interactions between Lactobacillus and IECs remain unclear. Recently, the bacterial surface layer adhesive protein (SLAP) has been explored as a potential mediator of Lactobacillus adhesion to IECs [9–11]. SLAP forms ubiquitous cell envelope structures that are composed of numerous identical subunits held together through interactions with the underlying cell surface [12]. SLAP subunits exhibit an intrinsic ability to spontaneously form regular layers, either in solution or on solid supports under specific conditions [13]. However, due to the complex hydrophilic and hydrophobic properties of this protein, few studies have explored the role of SLAP in adhesion of Lactobacillus to IECs. Granato, et al. found that the LiCl-extracted, 50-kDa SLAP protein could adhere to cells and to mucin [14]. Furthermore, SLAP isolated from Lactobacillus crispatus inhibited adherence of enterotoxigenic E. coli to a synthetic basement membrane [15]. A proteinaceous substance present in the culture supernatant from several probiotic strains was also observed to reduce colonization of the enterotoxigenic E. coli strain O157:H7 on HT-29 human colon carcinoma cells [16]. Pretreatment of polarized IECs with SLAP extract prior to EPEC infection reduced both dextran flux and trans-epithelial electrical resistance (TER) [17]. Johnson-Henry, et al. demonstrated that treatment with SLAP reduced the number and rearrangement of α-actin foci, and attenuated bacterial colonization on IECs and pathogen-induced changes in cellular permeability [18]. The p75 and p40 proteins, two soluble factors extracted from Lactobacillus rhamnosus GG culture broth supernatant, have been shown to mediate Akt activation and regulate anti-apoptotic responses through the phosphatidylinositol-3-kinase pathway [19, 20].
TJ proteins are associated with numerous intracellular signaling molecules [21]. For example, extracellular signal-regulated kinase (ERK) directly interacts with the C-terminal region of occludin to protect TJs from epithelial growth factor-induced oxidative stress [22]. Furthermore, interactions with ERK may regulate phosphorylation of specific TJ proteins and signaling molecules associated with the TJ to maintain the integrity of TJs and the epithelial barrier [23, 24].
Our previous studies have shown that Lactobacillus plantarum (LP) can prevent injury of Caco-2 cells induced by enteroinvasive E. coli through maintaining the expression level of TJ proteins and the characteristic structure of TJs [4–6]. However, the mechanistic roles of SLAP in the adhesive process are complex and generally unknown. Therefore, the aim of the present study was to investigate the protective effects of purified SLAP from LP on EPEC-infected Caco-2 cells and to evaluate the role of ERK in this process.
Materials and methods
Bacterial strains, cell line, and reagents
The LP strain (CGMCC 1258) was provided by the Institute of Bio-medicine, Shanghai Jiao Da Onlly Company Ltd. The EPEC strain (O111:NM, ATCC 43887) was obtained from the Shanghai Municipal Center for Disease Control and Prevention. The human colon adenocarcinoma cell line Caco-2 was obtained from the Shanghai Institute of Cell Biology, Chinese Academy of Science. Polyvinylidene fluoride (PVDF) membrane was purchased from Millipore (Billerica, MA, USA). Primer synthesis and sequencing was performed by Sangon (Shanghai, China).
LP and Caco-2 cell culture
LP was cultured in MRS broth (Difco, Sparks, MD, USA) at 37°C. Caco-2 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 20 mM Hepes, 50 U/ml penicillin, 50 mg/ml streptomycin, and 10% fetal bovine serum (Gibco, California, USA), referred to as complete DMEM. The cells were cultured in 25 cm2 plastic culture dishes (Corning, Inc., Corning, NY, USA) or on filters (culture plate inserts; 0.4 μm pore size; 0.6 cm2 area, Millipore) and were maintained in a humidified atmosphere containing 5% CO2 at 37°C. The cells were passaged at pre-confluent densities using 0.05% trypsin and 0.5 mm EDTA (Invitrogen).
Isolation and purification of SLAP
Fresh LP cultures were incubated at 37°C for 24 h. The cells were collected by centrifugation at 3500×g at 4°C for 20 min, followed by incubation with 2 M guanidine hydrochloride for 3 h. The resulting solution was then subjected to centrifugation at 6000×g at 4°C for 20 min. The supernatant was dialyzed in 0.01 M phosphate-buffered saline (PBS) overnight, followed by ultra-centrifugation at 40000×g at 4°C for 60 min. The resulting pellet was dissolved in 100 μl 0.1 M PBS, incubated at 70°C for 30 min, and subjected to centrifugation at 16000×g at 4°C for 20 min. The supernatant was then pre-incubated with Sepharose 4B (75 μl Sepharose/0.5 ml extract) for 30 min to remove proteins that non-specifically bound the solid support. The supernatant was next loaded onto a porcine stomach mucin (PSM)-Sepharose 4B column equilibrated with 10 mM Tris–HCl, pH 8, containing 1 mM MgCl2, 1 mM CaCl2, and 0.02% (w/v) NaN3. Following washing, bound proteins were eluted with 20 mM 1,3-diaminopropane (DAP). The final fractions containing purified SLAP were then diluted to 1 ng/ml for further studies.
Competition-based adhesion assay
Two bacterial strains were used for the adhesion assay, LP and EPEC. LP was cultured in MRS broth under anaerobic conditions in anaerobic jars (Oxoid, Cambridge, UK) at 37°C for 18–20 h. EPEC was cultured in blood-agar plates at 37°C for 18–20 h under aerobic conditions. Based on optical density values used to measure turbidity, LP and EPEC were diluted to a concentration of approximately 1 × 108 cells/ml and were incubated with Caco-2 cells for 3 h at 37°C in 5% CO2 and 95% humidity. After trypsinization, the numbers of bacteria adhering to the Caco-2 cells were quantified by plating diluted bacterial suspensions on MRS or blood-agar plates and were expressed as colony forming units (CFU). The level of adhesion was calculated by dividing the number of bacteria by the number of Caco-2 cells. A total of four treatment groups were compared in this assay: (1) control group, Caco-2 cells with no pre-treatment; (2) EPEC group, Caco-2 cells incubated with 0.1 mL EPEC (1 × 108 cells/ml) for 3 h; (3) LP group, Caco-2 cells pre-incubated with 0.1 ml LP (1 × 108 cells/ml) for 2 h, followed by incubation with 0.1 ml EPEC (1 × 108 cells/ml) for 1 h; and (4) SLAP group, Caco-2 cells pre-incubated with 0.1 ml 1 ng/ml SLAP for 2 h, followed by incubation with 0.1 ml EPEC (1 × 108 cells/ml) for 1 h.
Infection of Caco-2 cells with EPEC
Caco-2 cells were washed three times in Hank’s solution (Life Technologies, Carlsbad, CA, USA) to remove media containing antibiotics. An average of 1 × 106 Caco-2 cells were present in each monolayer. For rapid infection of the monolayer, 100 μl EPEC (1.0 × 109 cells/ml) was added to the apical side of the cell culture insert, and the insert was placed in a 50-ml tube and centrifuged at 200×g for 4 min in preparation for further analyses described below. The inoculation ratio of EPEC to Caco-2 cells was approximately 100:1. Prior to EPEC infection, two of the groups of Caco-2 cell monolayers were pre-incubated with 100 μl LP (1.0 × 1010 cells/ml) or 100 μl SLAP (1 ng/ml solution) for 24 h. The ratio of LP to EPEC was 10:1. The concentration of SLAP was chosen based on preliminary studies of five concentrations of SLAP, ranging from 0.01 ng/ml to 10 ng/ml, and represents the median concentration from these studies (data not shown). Untreated Caco-2 monolayers served as the control group; Caco-2 cells infected with EPEC served as the EPEC group; and Caco-2 cells infected with EPEC and incubated with LP or SLAP served as the LP and SLAP groups, respectively.
Quantitative real-time PCR
Total RNA was extracted from Caco-2 cells, treated as described above, using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and was quantified spectrophotometrically (Gene Quant II; Amersham Pharmacia Biotech, Piscataway, NJ, USA). Primer sequences are shown in Table S1. First-strand cDNA was synthesized from 1 μg total RNA by reverse transcription using oligo-dT primers and reverse transcriptase (superscript II; Invitrogen) according to the manufacturer’s recommended instructions. Real-time PCR reactions were performed in a 20 μl total volume and contained 1 μl cDNA, 1× PCR mix (iQ SYBR Green Supermix; Bio-Rad, Hercules, CA), and 500 nmol of each primer. Reactions were incubated in a thermocycler (iCycler iQ system; Bio-Rad) under the following conditions: one cycle of 95°C for 5 min, followed by 50 cycles of 95°C for 15 s, 65°C for 15 s, and 72°C for 15 s. The fluorescence threshold (Ct) was calculated using corresponding software (Bio-Rad). Absence of nonspecific products was confirmed by analyzing the melting-point curves. GAPDH was also amplified as an internal control for normalization purposes.
SDS-polyacrylamide gel electrophoresis and western blotting
Control and treated Caco-2 cells were homogenized in chilled RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 0.5% sodium deoxycholate, 1% Triton X-100, 1 mM EDTA) containing protease and phosphatase inhibitors (1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, and 5 g/ml each of aprotinin, leupeptin, and pepstatin) [25–27]. After centrifugation at 10,000×g for 10 min at 4°C, the supernatant was recovered, and total protein concentration was quantified using the DC protein assay (Bio-Rad, Hercules, CA, USA). Equivalent amounts of total protein were resolved on 10% SDS-polyacrylamide gels, followed by transfer to a PVDF membrane. After blocking overnight in Tris-buffered saline containing 0.05% Tween (TBS-T) and 5% dry powdered milk, the membranes were washed three times for 5 min each with TBS-T. The membranes were then incubated for 2 h at room temperature with the indicated primary antibody diluted 1:50 in TBT-T (rabbit anti-Claudin-1, rabbit anti-occludin, rabbit anti-junctional adhesion molecule 1 [JAM-1], rabbit anti-zona occludens 1 [ZO-1], rabbit anti-ERK, or rabbit anti-phosphorylated ERK [p-ERK]) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After three washes with TBS-T, the membranes were incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies (1:50). Following two washes with TBS-T and one wash with tris-buffered saline, bound antibodies were visualized using enhanced chemiluminescence reagent (Amersham, Princeton, NJ, USA). The relative density was defined as the density of each protein band (obtained by densitometry) normalized to the density of the constitutively-expressed β-actin band and was used to control for loading and transfer artifacts introduced in western blots.
Fluorescence staining and microscopy
Treated and untreated Caco-2 cell monolayers were fixed and permeabilized with methanol at −20°C, followed by incubation overnight at 4°C with the following polyclonal rabbit primary antibodies or reagents: anti-claudin-1 (1:100, Zymed, Carlsbad, CA, USA), anti-occludin (1:100, Zymed), anti-JAM-1 1:50, Zymed), anti-ZO-1 (1:50, Zymed), or FITC-conjugated phalloidin (1:50, Zymed). The cells were then incubated for 2 h with corresponding FITC-conjugated secondary antibodies (Sigma-Aldrich, St. Louis, MO, USA) at room temperature (RT) in the absence of light. Subsequently, the monolayers were washed several times with PBS, detached from the Anocell inserts (Millipore), and mounted with Vectashield mounting medium (Vector Laboratories, Inc., Burlingame, CA). Stained cells were imaged by confocal laser scanning microscopy (CLSM, Bio-Rad MRC 1024, Bio-Rad). Negative controls included cells that were treated similarly, but with the primary antibodies omitted.
Assessment of TER and dextran permeability
Control and treated Caco-2 cells were cultured on filters (Millicell culture plate inserts; 0.4 μm pore size; 0.6 cm2). When the cells reached complete confluence (15–18 days), measurements using a voltmeter (Millicell-ERS; Millipore) revealed that the monolayers achieved a TER value >450 Ω cm2. The integrity of the confluent polarized monolayers was assessed by measuring TER at different time intervals. TER (Ω cm2) = (Total resistance (Ω) − Blank resistance (Ω)) × Area (cm2). Because TER values often vary among individual Caco-2 cultures, the electrical resistance values were recorded for each membrane before and after experimental treatment, and the percentage decrease from the baseline measurement (% TER) was calculated for each membrane.
Integrity of Caco-2 monolayers was also assayed using the macromolecular conjugate probe, Alexa Fluor 647 dextran (10 kDa; Molecular Probes, Eugene, OR, USA). Briefly, 0.2 ml conjugated dextran suspended in DMEM (Invitrogen) was added to the apical compartment of each transwell in the plate, (Corning Costar Corp., Cambridge, MA, USA), and 0.4 ml DMEM alone was added to the basolateral compartment. After incubation for 5 h at 37°C, samples (200 μl) from the basolateral compartment were placed in a 96-well plate (Corning), and fluorescence intensity was measured using the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA) set to a wavelength of 700 nm. Integrated intensities were normalized to the integrated intensity of medium samples from untreated controls.
Assessment of apoptosis by flow cytometry
Control and treated Caco-2 cells were harvested by trypsinization, washed in PBS, and fixed in cold 70% ethanol overnight at −20°C. The cells were then washed twice in pre-chilled PBS, collected by centrifugation at 300×g for 5 min, and incubated in 300 μl propidium iodide (PI) solution (50 μg/ml PI and 50–100 μg/ml RNAse in PBS) in the absence of light for 20 min at 37°C. Cellular fluorescence was then analyzed using a flow cytometer (Beckman-Coulter, Miami, FL, USA) after filtering through 200 mesh nylon net.
Statistical analysis
For comparisons, one-way analysis of variance (ANOVA) was performed with the Tukey–Kramer post-hoc comparison. Significant differences were assessed using the Student’s t-test. All data are presented as means ± standard error (SE). A minimum of three independent experiments were carried out for each assay. Statistical analyses were performed using GraphPad Prism 5 software (San Diego, CA, USA). P-values less than 0.05 were considered to be statistically significant.
Results
SLAP decreases adhesion of EPEC to Caco-2 cells
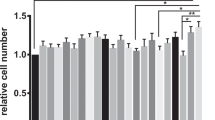
Pre-incubation of Caco-2 cells with LP for 2 h significantly decreased the level of EPEC adhesion to Caco-2 cells in comparison with incubation with EPEC alone (P < 0.05). A significant difference in the level of adhesion was also observed between cells pre-incubated with SLAP and cells incubated with EPEC alone (P < 0.05, Fig. 1).

Adhesion of EPEC to Caco-2 cells in the presence and absence of LP or SLAP. Pre-incubation of Caco-2 cells with LP or SLAP for 2 h significantly decreased EPEC adhesion to Caco-2 cells in comparison to cells incubated with EPEC alone. *P < 0.05 vs. EPEC group
SLAP treatment leads to increased mRNA levels of TJ proteins, but not ERK, in EPEC-treated Caco-2 cells
Caco-2 cells were incubated with EPEC alone, or were pretreated with LP or SLAP. Quantitative real-time PCR was then performed to analyze the occludin, claudin-1, JAM-1, ZO-1, and ERK levels in treated and untreated control cells. The expression of TJ-associated mRNAs was significantly decreased after treatment with EPEC in comparison to untreated controls. This decrease could be inhibited by pretreatment with LP or SLAP (P < 0.05, Fig. 2a–d). However, no significant changes were observed in levels of the ERK mRNA among the control, EPEC, LP, and SLAP groups (P > 0.05, Fig. 2e).

SLAP inhibits EPEC-induced decreases in the mRNA expression of TJ-associated proteins. The mRNA levels of TJ proteins were significantly decreased in Caco-2 cells after treatment with EPEC in comparison to control untreated cells. This decrease could be reversed by pre-treatment with LP or SLAP. In contrast, ERK mRNA expression levels did not change significantly among the control, EPEC, LP, and SLAP groups. *P < 0.05 vs. EPEC group
SLAP treatment increases the expression of TJ proteins and ERK phosphorylation in EPEC-treated Caco-2 cells
Western blot analysis was used to determine the relative levels of the occludin, claudin-1, JAM-1, ZO-1, ERK, and p-ERK proteins in control and EPEC-treated Caco-2 incubated with or without LP or SLAP. Levels of TJ proteins were markedly decreased after treatment with EPEC compared to untreated control cells (P < 0.05). However, pre-incubation of EPEC-treated cells with LP or SLAP significantly inhibited this decrease (P < 0.05, Fig. 3a, b).

Decreases in expression of TJ proteins and increases in ERK phosphorylation induced by EPEC infection were ameliorated by SLAP treatment. a A representative experiment showing western blot analysis of claudin, occludin, JAM-1, ZO-1, ERK, and p-ERK expression. Expression of TJ proteins was markedly decreased after treatment with EPEC compared to the control group. However, treatment with LP or SLAP restored expression of TJ proteins in comparison to the EPEC group. Increased p-ERK expression was observed in the EPEC group compared with the control group, but the addition of LP or SLAP inhibited this increase. ERK expression did not change significantly among the control, EPEC, LP, and SLAP groups. b Quantification of experimental results described in (a). Protein expression was quantified by densitometry for three independent experiments. The results were normalized to β-actin to control for loading and transfer artifacts introduced in western blotting. *P < 0.05 vs. EPEC group
Treatment of Caco-2 cells with EPEC resulted in significantly increased ERK phosphorylation in comparison to the control group, which could be inhibited by pretreatment with LP or SLAP (P < 0.05). However, expression of the ERK did not change significantly among the control, EPEC, LP, and SLAP groups (P > 0.05, Fig. 3a, b).
Fluorescence microscopy confirms that SLAP treatment reverses EPEC-induced disruption of TJs and cytoskeletal elements
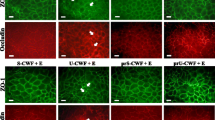
Fluorescence microscopy revealed that claudin-1, occludin, and JAM-1 localized to the cell membrane, while ZO-1 was observed in the cytoplasm in untreated Caco-2 cells. Additionally, in control Caco-2 cells, both ZO-1 and occludin were present at the apical intercellular borders in a belt-like pattern, encircling the cell surface. However, in the EPEC group, the green fluorescence associated with the TJ proteins was more dispersed, with some loss from the membrane. In EPEC-treated cells pretreated with LP or SLAP, the membrane localization of TJ proteins was decreased in comparison to the control group, but was substantially more normal than that of the EPEC group (Fig. 4).

Fluorescence microscopy confirmed that SLAP reverses EPEC-induced alterations in TJ and cytoskeletal architecture. In control Caco-2 monolayers, both ZO-1 and occludin were present at the apical inter-cellular borders in a belt-like pattern, encircling the cellular borders. However, in the EPEC group, the green fluorescence was dispersed, with some loss from the membrane. In the LP and SLAP groups, the distribution of ZO-1 and occludin fluorescence was more continuous and intact than that of the EPEC group. Similar results were observed for F-actin. Disrupted TJ and F-actin are indicated by arrows. Original magnification, ×600
Phalloidin staining in the control group revealed a continuous distribution of F-actin at the cell borders. A high density of F-actin filaments was present at the apical peri-junctional regions, encircling the cells in a belt-like manner. In contrast, treatment with EPEC resulted in disorganization and disruption of the F-actin architecture, including centripetal retraction of the peri-junctional F-actin filaments and separation of F-actin from the apical cellular borders. However, EPEC-induced alterations in peri-junctional F-actin filaments were reversed by pre-treatment with LP or SLAP (Fig. 4).
SLAP treatment reverses EPEC-induced decreases in TER and increases in the macromolecular permeability of Caco-2 cells
TER of untreated Caco-2 monolayers remained at approximately 450 Ω cm2 after 24 h of culture. In contrast, infection with EPEC resulted in an approximate 48.00% decrease in TER (from 450 to 234 Ω cm2). However, pre-incubation of Caco-2 cells with LP or SLAP, resulted in a reduction in the EPEC-induced decrease in TER to 36.44% (286 Ω cm2) and 35.56% (290 Ω cm2), respectively (Fig. 5a).

SLAP treatment increased the TER and decreased the macromolecular permeability of EPEC-treated Caco-2 cells. a SLAP treatment increased the TER of EPEC-treated Caco-2 cells. After EPEC infection for 6 h, TER was significantly lower than that of the control group. However, treatment of Caco-2 cells with LP or SLAP significantly decreased the effects of EPEC on TER. Each point represents the mean value obtained from 10 to 12 individual Caco-2 monolayers. Error bars represent the standard deviation. b SLAP treatment decreased the macromolecular permeability of EPEC-treated Caco-2 cells. EPEC treatment increased dextran permeability in comparison with the control group after 60 min of culture, which was reversed by the addition of LP or SLAP. Three independent experiments were performed. (open diamond) control group, (filled square) EPEC group, (filled triangle) LP group, (asterisk) SLAP group. P < 0.05 for the control group, LP group, and SLAP group vs. the EPEC group
An infrared-sensitive dextran (10-kDa) probe was used to assess the macromolecular permeability of Caco-2 cell monolayers. Results from these assays demonstrated that EPEC-infected monolayers exhibited a significant increase in permeability to the dextran probe in comparison to the control group (P < 0.05). EPEC-induced increases in dextran permeability of Caco-2 cell monolayers were reduced when Caco-2 cells were pretreated with LP or SLAP (P < 0.05, Fig. 5b).
SLAP treatment decreased the levels of apoptosis of Caco-2 cells induced by EPEC
The percentage of apoptotic Caco-2 cells was increased significantly after treatment with EPEC for 48 h in comparison with the control group (P < 0.05), while pre-treatment with LP or SLAP significantly decreased the level of apoptosis in comparison with the EPEC group (P < 0.05). No significant difference in levels of apoptosis was observed between the LP and SLAP groups (P > 0.05, Fig. 6).

SLAP decreases the levels of apoptosis induced by EPEC treatment of Caco-2 cells. The level of apoptosis in Caco-2 cells was significantly increased after treatment with EPEC for 48 h in comparison to the control group. In the LP and SLAP groups, the levels of apoptosis were decreased significantly in comparison to the EPEC group. *P < 0.05 vs. the control group; # P < 0.05 vs. the EPEC group
Discussion
Although probiotics have been shown to induce remission and prevent recurrence of IBD in patients [28, 29] and in animal models of colitis [30, 31], a clinical trial designed to test the efficacy of LP as an adjunct to standard therapy in children with IBD showed no beneficial effects of LP in maintaining remission [32]. These results underscore a current problem regarding the use of probiotics, namely the difficulty of determining the bioavailability of bacteria in the gastrointestinal tract. In addition, use of live probiotics also raised concerns due to several reported cases of bacteremia associated with probiotic therapy in young [33] and immunocompromised patients [34, 35]. One approach to circumvent these issues may be the use of probiotic-derived proteins as novel therapeutic agents for treatment of IBD and other inflammation-related intestinal disorders. Surface molecules are likely to play an important role in establishment of colonization and may be involved in the exclusion of intestinal pathogens [14]. Given that SLAP forms the outermost layer of gram-positive bacteria, this protein may potentially play a role in competitive exclusion of pathogens [11, 36]. Therefore, the aim of the present study was to characterize a SLAP extract from LP and to determine the role of SLAP in attenuating EPEC-induced injury on the human epithelial Caco-2 cell line.
In the present study, SLAP was extracted from LP and was further purified using a mucin (PSM)-Sepharose 4B column. The protective effects of this purified SLAP on the functional integrity of intercellular TJs and their ability to modulate intercellular permeability of polarized Caco-2 monolayers were then investigated. Intimate adherence of EPEC to host epithelial cells appears to be an important virulence factor required for induction of disease pathogenesis, causing disruption of the microvilli and resulting in the formation of attaching-effacing lesions [17, 37]. In this study, we demonstrated that treatment with SLAP decreased adhesion of EPEC to Caco-2 cells and rescued EPEC-induced alterations in TJ structure and permeability of Caco-2 cell monolayers.
Interestingly, ERK activation appeared to be central to the effects of deformation of IEC. Intestinal epithelial ERK has been reported to be activated by deformation of both collagen and fibronectin substrates [38]. However, inhibition of ERK activity blocked both intestinal epithelial cell proliferation on collagen and intestinal epithelial cell restitution on fibronectin [39]. Many stimulatory signals can lead to ERK activation, including growth factors [40], mechanical forces, oxidative stress, and integrin-mediated signaling [41]. In different cell types, these signals can mediate their effects on ERK through different signaling mediators, including Src, focal adhesion kinase (FAK), and protein kinase C (PKC) [41]. Although basal ERK 1/2 activity is present in young adult mouse colon (YAMC) cells, pretreatment with conditioned media from the probiotic bacterium Lactobacillus rhamnosus GG activates ERK 1/2, as well as p38 and c-Jun N-terminal kinase (JNK), as efficiently as the phorbol ester PMA. Seth, et al. found that the probiotic proteins p40 and p75 prevented H2O2-induced disruption of TJ and increased para-cellular permeability by activating ERK 1/2 via MEK activity [42]. The present study confirmed that EPEC treatment increased levels of ERK phosphorylation in Caco-2 cells, which could be reversed by SLAP treatment. SLAP also improved TJ and cytoskeletal structure and increased expression of TJ-associated proteins, including claudin-1, occludin, JAM-1, ZO-1, in EPEC-treated Caco-2 cells. The mechanism underlying LP-mediated protection of TJ from EPEC-induced injury in this model may be similar to that described above. Interestingly, ERK expression was not changed by EPEC treatment. Therefore, our results suggested EPEC activates ERK, leading to disruption of the F-actin cytoskeleton and TJs and increased levels of apoptosis in Caco-2 cells. Furthermore, SLAP-induced decreases in the levels of p-ERK may play a crucial role in regulation of TJ-protein complexes, thereby preventing TJ injury induced by EPEC.
Certainly, other factors secreted by LP may also counteract pathogenic bacteria adhesion, including adhesins, such as lipoteichoic acid [43], exopolysaccharide [44], lactic acid, and other antimicrobial compounds [45]. Moreover, in addition to the mucin-related adhesive protein, other types of adhesive proteins are also present on the surface of human IECs. A randomized controlled trial is needed to further investigate the protective effects of SLAP on the human intestinal tract.
Conclusion
In summary, our study showed that purified SLAP from LP protected IECs from EPEC-induced injury, likely through a mechanism involving attenuation of ERK phosphorylation. Isolation of probiotic bioactive factors can ultimately culminate in the development of novel therapeutic agents, which could in turn be administered in a consistently and pharmacologically-sound manner for treatment of intestinal disorders.
References
Whelan K, Myers CE (2010) Safety of probiotics in patients receiving nutritional support: a systematic review of case reports, randomized controlled trials, and nonrandomized trials. Am J Clin Nutr 91(3):687–703
Zhang M, Wang XQ, Zhou YK et al (2010) Effects of oral Lactobacillus plantarum on hepatocyte tight junction structure and function in rats with obstructive jaundice. Mol Biol Rep 37(6):2989–2999
Mack DR, Ahrne S, Hyde L, Wei S, Hollingsworth MA (2003) Extracellular MUC3 mucin secretion follows adherence of Lactobacillus strains to intestinal epithelial cells in vitro. Gut 52(6):827–833
Liu ZH, Ma YL, Shen TY, et al (2010) Identification of DC-SIGN as the receptor during the interaction of lactobacillus plantarum CGMCC 1258 and dendritic cells. World J Microbiol Biotechnol. doi:10.1007/s11274-010-0495-3. [Epub ahead of print]
Qin H, Zhang Z, Hang X, Jiang Y (2009) L. plantarum prevents enteroinvasive Escherichia coli-induced tight junction proteins changes in intestinal epithelial cells. BMC Microbiol 9:63
Liu Z, Zhang P, Ma Y, et al (2010) Lactobacillus plantarum prevents the development of colitis in IL-10-deficient mouse by reducing the intestinal permeability. Mol Biol Rep. doi:10.1007/s11033-010-0237-5. [Epub ahead of print]
Garcia-Lafuente A, Antolin M, Guarner F, Crespo E, Malagelada JR (2001) Modulation of colonic barrier function by the composition of the commensal flora in the rat. Gut 48(4):503–507
Gareau MG, Jury J, MacQueen G, Sherman PM, Perdue MH (2007) Probiotic treatment of rat pups normalises corticosterone release and ameliorates colonic dysfunction induced by maternal separation. Gut 56(11):1522–1528
Buck BL, Altermann E, Svingerud T, Klaenhammer TR (2005) Functional analysis of putative adhesion factors in Lactobacillus acidophilus NCFM. Appl Environ Microbiol 71(12):8344–8351
Francius G, Alsteens D, Dupres V et al (2009) Stretching polysaccharides on live cells using single molecule force spectroscopy. Nat Protoc 4(6):939–946
Johnson-Henry KC, Hagen KE, Gordonpour M, Tompkins TA, Sherman PM (2007) Surface-layer protein extracts from Lactobacillus helveticus inhibit enterohaemorrhagic Escherichia coli O157:H7 adhesion to epithelial cells. Cell Microbiol 9(2):356–367
Avall-Jaaskelainen S, Hynonen U, Ilk N et al (2008) Identification, characterization of domains responsible for self-assembly, cell wall binding of the surface layer protein of Lactobacillus brevis ATCC 8287. BMC Microbiol 8:165
Sara M, Sleytr UB (2000) S-Layer proteins. J Bacteriol 182(4):859–868
Granato D, Bergonzelli GE, Pridmore RD et al (2004) Cell surface-associated elongation factor Tu mediates the attachment of Lactobacillus johnsonii NCC533 (La1) to human intestinal cells and mucins. Infect Immun 72(4):2160–2169
Horie M, Ishiyama A, Fujihira-Ueki Y et al (2002) Inhibition of the adherence of Escherichia coli strains to basement membrane by Lactobacillus crispatus expressing an S-layer. J Appl Microbiol 92(3):396–403
Gopal PK, Prasad J, Smart J, Gill HS (2001) In vitro adherence properties of Lactobacillus rhamnosus DR20 and Bifidobacterium lactis DR10 strains and their antagonistic activity against an enterotoxigenic Escherichia coli. Int J Food Microbiol 67(3):207–216
Johnson-Henry K, Wallace JL, Basappa NS et al (2001) Inhibition of attaching and effacing lesion formation following enteropathogenic Escherichia coli and Shiga toxin-producing E. coli infection. Infect Immun 69(11):7152–7158
Johnson-Henry KC, Donato KA, Shen-Tu G, Gordanpour M, Sherman PM (2008) Lactobacillus rhamnosus strain GG prevents enterohemorrhagic Escherichia coli O157:H7-induced changes in epithelial barrier function. Infect Immun 76(4):1340–1348
Yan F, Polk DB (2002) Probiotic bacterium prevents cytokine-induced apoptosis in intestinal epithelial cells. J Biol Chem 277(52):50959–50965
Yan F, Cao H, Cover TL et al (2007) Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology 132(2):562–575
Anderson JM, Van Itallie CM (1995) Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol 269(4 Pt 1):G467–G475
Basuroy S, Seth A, Elias B, Naren AP, Rao R (2006) MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J 393(Pt 1):69–77
Ballif BA, Blenis J (2001) Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ 12(8):397–408
Rosseland CM, Wierod L, Oksvold MP et al (2005) Cytoplasmic retention of peroxide-activated ERK provides survival in primary cultures of rat hepatocytes. Hepatology 42(1):200–207
Liu ZH, He YP, Qin H (2010) The growth-inhibition effect of tamoxifen in the combination chemotherapeutics on the human cholangiocarcinoma cell line QBC939. Mol Biol Rep 37(6):2693–2701
Liu ZH, He YP, Zhou Y, Zhang P, Qin H (2010) Establishment and identification of the human multi-drug-resistant cholangiocarcinoma cell line QBC939/ADM. Mol Biol Rep. doi:10.1007/s11033-010-9975-7. [Epub ahead of print]
Liu ZH, Ma YL, He YP et al. (2010) Tamoxifen reverses the multi-drug-resistance of an established human cholangiocarcinoma cell line in combined chemotherapeutics. Mol Biol Rep. doi:10.1007/s11033-010-0291-z. [Epub ahead of print]
Prisciandaro L, Geier M, Butler R, Cummins A, Howarth G (2009) Probiotics and their derivatives as treatments for inflammatory bowel disease. Inflamm Bowel Dis 15(12):1906–1914
Haller D, Antoine JM, Bengmark S et al (2010) Guidance for substantiating the evidence for beneficial effects of probiotics: probiotics in chronic inflammatory bowel disease and the functional disorder irritable bowel syndrome. J Nutr 140(3):690S–697S
Rioux KP, Madsen KL, Fedorak RN (2005) The role of enteric microflora in inflammatory bowel disease: human and animal studies with probiotics and prebiotics. Gastroenterol Clin North Am 34(3):465–482 ix
Dieleman LA, Goerres MS, Arends A et al (2003) Lactobacillus GG prevents recurrence of colitis in HLA-B27 transgenic rats after antibiotic treatment. Gut 52(3):370–376
Bousvaros A, Guandalini S, Baldassano RN et al (2005) A randomized, double-blind trial of Lactobacillus GG versus placebo in addition to standard maintenance therapy for children with Crohn’s disease. Inflamm Bowel Dis 11(9):833–839
Land MH, Rouster-Stevens K, Woods CR et al (2005) Lactobacillus sepsis associated with probiotic therapy. Pediatrics 115(1):178–181
Apostolou E, Kirjavainen PV, Saxelin M et al (2001) Good adhesion properties of probiotics: a potential risk for bacteremia? FEMS Immunol Med Microbiol 31(1):35–39
Liong MT (2008) Safety of probiotics: translocation and infection. Nutr Rev 66(4):192–202
Chen X, Xu J, Shuai J et al (2007) The S-layer proteins of Lactobacillus crispatus strain ZJ001 is responsible for competitive exclusion against Escherichia coli O157:H7 and Salmonella typhimurium. Int J Food Microbiol 115(3):307–312
Kaper JB, Nataro JP, Mobley HL (2004) Pathogenic Escherichia coli. Nat Rev Microbiol 2(2):123–140
Chaturvedi LS, Marsh HM, Shang X, Zheng Y, Basson MD (2007) Repetitive deformation activates focal adhesion kinase and ERK mitogenic signals in human Caco-2 intestinal epithelial cells through Src and Rac1. J Biol Chem 282(1):14–28
Zhang J, Owen CR, Sanders MA, Turner JR, Basson MD (2006) The motogenic effects of cyclic mechanical strain on intestinal epithelial monolayer wound closure are matrix dependent. Gastroenterology 131(4):1179–1189
Howe KL, Reardon C, Wang A, Nazli A, McKay DM (2005) Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol 167(6):1587–1597
Chaturvedi LS, Gayer CP, Marsh HM, Basson MD (2008) Repetitive deformation activates Src-independent FAK-dependent ERK motogenic signals in human Caco-2 intestinal epithelial cells. Am J Physiol Cell Physiol 294(6):C1350–C1361
Seth A, Yan F, Polk DB, Rao RK (2008) Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a PKC- and MAP kinase-dependent mechanism. Am J Physiol Gastrointest Liver Physiol 294(4):G1060–G1069
Walter J, Loach DM, Alqumber M et al (2007) D-alanyl ester depletion of teichoic acids in Lactobacillus reuteri 100-23 results in impaired colonization of the mouse gastrointestinal tract. Environ Microbiol 9(7):1750–1760
Sun J, Le GW, Shi YH, Su GW (2007) Factors involved in binding of Lactobacillus plantarum Lp6 to rat small intestinal mucus. Lett Appl Microbiol 44(1):79–85
Hayes M, Ross RP, Fitzgerald GF, Hill C, Stanton C (2006) Casein-derived antimicrobial peptides generated by Lactobacillus acidophilus DPC6026. Appl Environ Microbiol 72(3):2260–2264
Acknowledgments
The authors thank Shanghai Jiao Tong University Affiliated Sixth People’s Hospital for technical assistance with this study. This study was supported by the National Natural Science Foundation of China (No. 81070293) and the National Basic Research Program of China, Ministry of Science and Technology of the People’s Republic of China (No. 2008CB517403).
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhihua Liu and Tongyi Shen are co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Liu, Z., Shen, T., Zhang, P. et al. Lactobacillus plantarum surface layer adhesive protein protects intestinal epithelial cells against tight junction injury induced by enteropathogenic Escherichia coli. Mol Biol Rep 38, 3471–3480 (2011). https://doi.org/10.1007/s11033-010-0457-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-010-0457-8