
Abstract
Using long-polymerase chain reaction (Long-PCR) method, we determined the complete nucleotide sequence of the mitochondrial genome (mitogenome) of Phthonandria atrilineata. The complete mtDNA from P. atrilineata was 15,499 base pairs in length and contained 13 protein-coding genes (PCGs), 2 rRNA genes, 22 tRNA genes, and a control region. The P. atrilineata genes were in the same order and orientation as the completely sequenced mitogenomes of other lepidopteran species. The nucleotide composition of P. atrilineata mitogenome was biased toward A + T nucleotides (81.02%), and the 13 PCGs show different A + T contents that range from 73.25% (cox1) to 92.12% (atp8). Phthonandria had the canonical set of 22 tRNA genes, that fold in the typical cloverleaf structure described for metazoan mt tRNAs, with the unique exception of trnS(AGN). The phylogenetic relationships were reconstructed with the concatenated sequences of the 13 PCGs of the mitochondrial genome, which confirmed that P. atrilineata is most closely related to the superfamily Bombycoidea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insect mitochondrial DNA (mtDNA) is a closed-circular molecule ranging in size from 14 to 20 kb, which encodes 37 genes, including 13 protein-coding genes, two ribosomal RNA genes, and 22 tRNA genes [1]. Additionally, it contains at least one sequence of variable length known in insect mtDNA as the adenine (A) + thymine (T)-rich region, which contains initiation sites for transcription and replication [2]. The mtDNA of metazoan present the strand specific bias in nucleotide composition, where one strand is G rich, whereas the other strand is G poor, and because they show different buoyant densities in a cesium chloride gradient, they are respectively called heavy (H) and light (L) strands [3, 4]. To date, the complete or near-complete mitogenome have been sequenced from 73 insect species [5], including eight species from the order Lepidoptera: four species of Bombyx mori, Bombyx mandarina [6], Maduca sexta [5] and Antheraea pernyi (GenBank AY242996) belonging to the superfamily Bombycoidea, two species of Ostrinia furnacalis and Ostrinia nubilalis belonging to the superfamily Pyraloidea [7], two species of Coreana raphaelis [8] and Artogeia melete (GenBank EU597124) belonging to the superfamily Papilionoidea, and one specie belonging to the superfamily Tortricoidea, Adoxophyes honmai [9]. This study extends the description of the complete mitogenome to Phthonandria atrilineata, a new member of another superfamily of Lepidoptera, the Geometroidea. According to the most recent consensus view of lepidopteran relationships, the expected relationships of these species would be ((((Bombycoidea) + Papillionoidea + Geometroidea) + Pyraloidea) + Tortricoidea) [10].
The P. atrilineata is widely distributed in China, Korea, and Japan, and is a major pest to mulberry leaves. In the present study, we sequenced the complete sequence of mitochondrial genome of P. atrilineata. The gene order, the nucleotide composition of PCGs and the secondary structures of tRNA genes of mitochondrial genome of P. atrilineata were analyzed and compared with other lepidopteran sequences. In addition, a phylogenetic analysis of the lepidopteran superfamilies was performed.
Materials and methods
DNA samples extraction
Larvae of P. atrilineata were collected from mulberry leaves in Hefei, China. According to the manufacturer’s instructions, total DNA was isolated from single specimens using the Takara Genomic DNA Extraction Kit (Takara Co., Dalian, China), and the DNA of the larvae of P. atrilineata was used for amplification of the fragments of the complete genome.
Primers design, PCR amplification, and sequencing
In order to amplify the whole mitochondrial genome of P. atrilineata, 24 primers were designed and synthesized (Shanghai Sangon Biotechnology Co., Ltd., Shanghai, China). According to the One-Step PCR Amplification of Complete Arthropod Mitochondrial Genomes method [11], two degenerate primers 16SAA and 16SBB (Table 1) were used for amplification of the long fragment of the mitochondrial genome. The other degenerate and specific primers were designed based on the conserved nucleotide sequences of the known mitochondrial sequences in Lepidoptera [5–9] or the known sequences of fragments of mitochondrial genome of P. atrilineata that we have previously sequenced (GenBank: DQ235691 and DQ132998).
The fragment of PF1 (about 15 kb) was obtained by long-polymerase chain reaction (Long-PCR) using Takara LA Taq (Takara Co., Dalian, China): an initial denaturation for 2 min at 96°C, followed by 30 cycles of 10 s at 98°C and 10 min at 58°C, and a subsequent 10 min final extension at 72°C. The long PCR product was used as a template to amplify the overlapping fragments PF2 to PF12 (Table 1).
The fragments that range from 1.3 to 2.3 kb (Table 1) were amplified using Takara LA Taq (Takara Co., Dalian, China) by PCR: an initial denaturation for 2 min at 96°C, followed by 30 cycles of 10 s at 98°C and 2 min at 58°C, and a subsequent 10 min final extension at 72°C. The above PCR products were separated by electrophoresis in a 1% agarose gel and purified using a DNA gel extraction kit (Takara Co., Dalian, China). The purified PCR product was ligated into T-vector (Takara Co., Dalian, China) and then transformed into XL-1 blue competent bacteria [12]. The positive recombinant clone with an insert was sequenced using the dideoxynucleotide chain termination method (Takara Co., Dalian, China) at least 3 times.
Gene identification and sequence analysis
Protein-encoding genes of the P. atrilineata mitochondrial genome were identified by sequence similarity of other lepidopteran species [5–9]. Identification of tRNA genes was verified using the program tRNAscan-SE. The potential stem–loop secondary structures within these tRNA gene sequences were calculated using the tRNAscan-SE Search Server available online (http://lowelab.ucsc.edu/tRNAscan-SE/) [13]. Alignments of the protein-coding genes for each of the available lepidopteran mitogenomes were made in MEGA ver4.0 [14]. All regions involving ambiguity for the position of the gaps were excluded from the analyses to avoid erroneous hypotheses of primary homology [4]. The reduced alignment of mt sequences consists of 11,450 nucleotides and 3,802 amino acids. Based on the concatenated amino acids and nucleotide data sets from the 13 PCGs, the phylogenetic analyses were performed using maximum likelihood (ML), maximum parsimony (MP) and neighbor-joining (NJ) methods. MP and NJ analyses were carried out using MEGA ver4.0 [14]. ML analyses and bootstrap resampling were performed using the set of programs in the PHYLIP package [15]. The mitochondrial genomes of Drosophila melanogaster [16] and Locusta migratoria [17] were used as outgroups.
For ML analyses (PHYLIP package), the concatenated amino acids /nucleotide sequence of P. atrilineata mitochondrial genome was aligned with the sequences of other species, using CLUSTALX 1.83 [18]. These aligned sequences were entered into the subprogram SEQBOOT (bootstrap sequence data sets) of the PHYLIP package to create 100 datasets by bootstrap resampling. These 100 datasets were used as input to generate the 100 most parsimonious trees using the subprogram PROML/DNAML. During the creation of the 100 most parsimonous trees, all the sequences were randomly entered into each dataset 10 times, based on the random number 3. Thus, the resulting output was based on 100 × 10 runs of all the sequences, with D. melanogaster [16] and L. migratoria [17] used as the outgroups. The output of these 100 × 10 runs of PROML/DNAML was entered into the program CONSENSE to calculate a majority-rules strict consensus tree with confidence intervals [13]. For NJ and MP analyses, a distance matrix was constructed from the aligned sequences using Kimura two-parameter formula [19]. The bootstrap test (1,000 repeats) was used to determine the reliability of the different branches in the NJ dendrogram [14].
Results and discussion
Genome organization and base composition
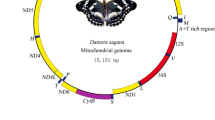
The mitogenome of P. atrilineata is a closed-circular molecule of 15,499 bp (GenBank EU569764) (Fig. 1), the size of which is comparable to other lepidopteran mitogenomes, which range from 15,314 bp in Coreana raphaelis [8] to 15,928 bp in Bombyx mandarina [6]. The gene content is typical of other metazoan mitochondrial genomes: 13 PCGs (cox1-3, nad1-6, nad4L, cob, atp6 and atp8), 22 tRNA genes (one for each amino acid, two for Leucine and Serine), and two for mitochondrial ribosomal RNAs (rrnS and rrnL). There is a 457 bp A + T-rich region between rrnS and trnI, and it was deemed homologous to the control region (CR) by positional homology, general structure, and base content. Gene order follows the other Lepidoptera arrangement. When searching tRNA genes, tRNA-Scan-SE failed to find a copy of trnS(UCN) and trnL(UUR), which was determined by sequence comparison of the region in which this gene occurs in other insect species.

Circular map of the mitochondrial genome of Phthonandria atrilineata. The abbreviations for the genes are as follows: cox1, cox2, and cox3 refer to the cytochrome oxidase subunits, cob refers to cytochrome b, and nad1-6 refers to NADH dehydrogenase components. tRNAs are denoted as one-letter symbol according to the IUPAC-IUB single-letter amino acid codes. Direction of gene transcription is indicated with an arrow
Likewise to other insect mtDNA sequences, the nucleotide composition of the P. atrilineata mitogenome is also biased toward A + T nucleotides (81.02%), which is lower than M. sexta (81.79%), B. mandarina (81.68%), B. mori (81.2%) and C. raphaelis (82.66%), and higher than the other five lepidopterans. Within 13 protein-coding genes (PCGs), the A + T composition in the atp8 gene is the highest (92.12%), and 73.25% for the cox1 gene, with this value the lowest (Table 2).
Protein-coding genes
The mitochondrial genome of P. atrilineata encodes the regular set of 13 PCGs (Fig. 1) found, with few exceptions, in all insect mitochondrial genomes. Reading frames for protein-coding genes were determined by comparison with alignments of other insect mitochondrial genes and by looking at start and stop codons.
The start and stop codons of the 13 protein-coding genes in the P. atrilineata mitochondrial genome are shown in Table 3. Six protein-coding genes share the start codon ATG (cox2, atp6, cox3, nad4, nad4L and cob), four genes start with ATA (nad1, nad2, nad6, nad8 and atp8), and nad3 and nad5 start with ATT, while cox1 starts with CGA (Table 3). For cox1 none of the lepidopterans start with a typical ATN codon, rather all start with CGA and D. melanogaster starts with TCG. The atypical start codon of mitochondrial cox1 gene had also been observed and extensively discussed in other insect and arthropod species; suggestions include one hexanucleotide (ATTTAA) [20] and some tetranucleotides (ATAA, TTAA, and ATTA) [16, 17]. Eight of the protein-coding genes stop with TAA; nad4L uses TAG instead of TAA as a stop codon. Four of the PCGs had incomplete stop codons consisting of just a T-nucleotide, including cox1, cox2, nad4 and nad5 (Table 3). There is also a high degree of conservation of incomplete stop codons across the order; cox1 and cox2 have incomplete stop codons in all lepidopteran species and nad5 has one in all species with the exception of Bombyx [5–9].
Transfer RNA and ribosomal RNA genes
Phthonandria had the typical set of 22 tRNA genes, which are interspersed between ribosomal RNAs and protein-encoding regions, range in size from 64 to 71 nucleotides. About 14 tRNAs are codified by the H-strand and eight are codified by the L-strand, which similar to the organization in other lepidopterans. All P. atrilineata tRNAs have the typical cloverleaf structure of mt tRNAs (Fig. 2), except for the trnS(AGN), wherein the dihydrouridine (DHU) arm forms a simple loop (Fig. 2) as seen in many metazoan mtDNAs [21, 22]. The anticodons of P. atrilineata tRNAs were all identical to their counterpart tRNAs of the sequenced lepidopterans [5–9]. As reported previously, the trnM is quite well conserved, showing high identity with the sequence found in M. sexta [5], B. mandarina, B. mori [6] and C. raphaelis [8].


Predicted secondary cloverleaf structure for the tRNA genes of Phthonandria atrilineata
The rrnL and rrnS genes in P. atrilineata are 1,400 and 803 nucleotides long, respectively. Both ribosomal RNA genes show great sequence similarity to other lepidopterans. The base composition of the rrnL is 42.92% A, 9.51% C, 4.72% G, and 42.85% T. The rrnS composition is 42.82% A, 9.36% C, 4.37% G, and 43.45% T. The A + T content of rrnL and rrnS gene are 85.77% and 86.27%, respectively. These values are high but within the range of those found in the insect species sequenced to date.
Phylogenetic analysis
In the present study, the sequences of the 13 PCGs of the mitochondrial genome were concatenated, rather than analyzed separately, to reconstruct the phylogenetic relationships, which may result in a more complete analysis [23]. All the ML, MP and NJ analyses clustered P. atrilineata, B. mandarina, B. mori, A. pernyi, and M. sexta in one branch (Fig. 3a–f). The phylogenetic relationships confirmed that P. atrilineata is most closely related to the superfamily Bombycoidea, which was in accordance with the phylogeny of lepidopteran superfamilies proposed by Kristensen and Skalski [10].

Phylogeny of the lepidopteran species. Phylogenetic analysis were performed by maximum likelihood (a, b), maximum parsimony (c, d) and neighbor-joining methods (e, f), using the nucleotide (b, d and f) and amino acid sequences (a, c and e) of the concatenated 13 PCGs. The sequences of Locusta migratoria (NC_001712) and Drosophila melanogaster (NC_001709) were used as outgroups. Phthonandria atrilineata (EU569764); Coreana raphaelis (NC_007976); Adoxophyes honmai (NC_008141); Maduca sexta (EU286785); Antheraea pernyi (AY242996); Bombyx mandarina (NC_003395); Bombyx mori (AB070264); Ostrinia furnacalis (NC_003368); Ostrinia nubilalis (NC_003367); Artogeia melete (EU597124)
Exception of the ML tree based on the concatenated amino acid sequences, the other five trees showed similar topology (Fig. 3b–f). The 10 species of Lepidoptera was divided into three clades: two species of the superfamily Papilionoidea (I); two species of the superfamily Pyraloidea and A. honmai of Tortricoidea (II); four species of the superfamily Bombycoidea and P. atrilineata of Geometroidea (III). The ML tree based on amino acid sequences (Fig. 3a) showed that A. honmai was not clustered to the two species of the superfamily Pyraloidea firstly. Thus we divided A. honmai as a separate clade (IV). In addition, the bootstrap score of the clades were different when reconstructed based on the two type sequences, or using different methods.
In our results, the two butterflies (A. melete and C. raphaelis) are the sister to the remaining lepidopteran superfamilies (Fig. 3a–f); A. honmai is the sister group to the crambids (Ostrinia) (Fig. 3b–f), which was different to the lepidopteran relationships in Kristensen and Skalski [10]. Regier et al. [24] demonstrated that Tortricoidea was the sister to the other 5 superfamilies included in the present study when their phylogenetic relationships were reconstructed using the period gene [24]. In contrast to our findings, butterflies (Papillionoidea) are typically considered to be very highly derived lepidopterans, not the sister of the remaining macrolepidopteran families; Adoxophyes would be expected to be the sister to all other families based on previous work, not the sister group to the crambids (Ostrinia) [10].
References
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780. doi:10.1093/nar/27.8.1767
Taanman JW (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410:103–123. doi:10.1016/S0005-2728(98)00161-3
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465. doi:10.1038/290457a0
Hassanin A, Léger N, Deutsch J (2005) Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst Biol 54:277–298. doi:10.1080/10635150590947843
Cameron SL, Whiting MF (2008) The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408:112–123. doi:10.1016/j.gene.2007.10.023
Yukuhiro K, Sezutsu H, Itoh M, Shimizu K, Banno Y (2002) Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori. Mol Biol Evol 19:1385–1389
Coates BS, Sumerford DV, Hellmich RL, Lewis LC (2005) Partial mitochondrial genome sequence of Ostrinia nubilalis and Ostrinia furnicalis. Int J Biol Sci 1:13–18
Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS et al (2006) The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol Biol 15:217–225. doi:10.1111/j.1365-2583.2006.00630.x
Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB (2006) The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene 373:52–57. doi:10.1016/j.gene.2006.01.003
Kristensen NP, Skalski AW (1999) Phylogeny and paleontology. In: Kristensen NP (ed) Lepidoptera: moths and butterflies. 1. Evolution, systematics, and biogeography. Handbook of zoology, vol IV, Part 35. De Gruyter, Berlin and New York, pp 7–25
Hwang UW, Park CJ, Yong TS, Kim W (2001) One-step PCR amplification of complete arthropod mitochondrial genomes. Mol Phylogenet Evol 19:345–352. doi:10.1006/mpev.2001.0940
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi:10.1093/nar/25.5.955
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599. doi:10.1093/molbev/msm092
Felsenstein J (2004) PHYLIP (Phylogeny inference package) version 3.6. Department of Genome Sciences, University of Washington, Seattle. Distributed by the author
Lewis DL, Farr CL, Kaguni LS (1995) Drosophila melanogaster mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol Biol 4:263–278. doi:10.1111/j.1365-2583.1995.tb00032.x
Flook PK, Rowell CH, Gellissen G (1995) The sequence, organization, and evolution of the Locusta migratoria mitochondrial genome. J Mol Evol 41:928–941. doi:10.1007/BF00173173
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTER-X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. doi:10.1093/nar/25.24.4876
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245. doi:10.1093/bioinformatics/17.12.1244
Junqueira AC, Lessinger AC, Torres TT, da Silva FR, Vettore AL, Arruda P et al (2004) The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene 15(339):7–15. doi:10.1016/j.gene.2004.06.031
Ji X, Wu X, Yan P, Amato G (2007) Complete sequence and gene organization of the mitochondrial genome of Siamensis Crocodile (Crocodylus siamensis). Mol Biol Rep. doi:10.1007/s11033-007-9062-x
Zhou Z, Huang Y, Shi F, Ye H (2007) The complete mitochondrial genome of Deracantha onos (Orthoptera: Bradyporidae). Mol Biol Rep. doi:10.1007/s11033-007-9145-8
Hassanin A (2006) Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol Phylogenet Evol 38:100–116. doi:10.1016/j.ympev.2005.09.012
Regier JC, Fang QQ, Mitter C, Peigler RS, Friedlander TP, Solis MA (1998) Evolution and phylogenetic utility of the period gene in Lepidoptera. Mol Biol Evol 15:1172–1182
Acknowledgments
We appreciate helpful comments provided by the anonymous reviewers. This study is supported by the grant from the National Natural Science Foundation of China (30500374), the Program for New Century Excellent Talents in University (NCET-07-0251), and the grant from Science and Technological Fund of Anhui Province for Outstanding Youth (08040106803).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Yang, L., Wei, ZJ., Hong, GY. et al. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol Biol Rep 36, 1441–1449 (2009). https://doi.org/10.1007/s11033-008-9334-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-008-9334-0