
Abstract
Aflatoxin is a mycotoxin produced by the fungus Aspergillus flavus (Link:Fr), an opportunistic ear-rot pathogen of maize (Zea mays L. ssp. mays). Pre-harvest contamination of maize grain with aflatoxin is a chronic problem worldwide and particularly in the Southeastern US. Quantitative trait loci (QTL) were mapped by multiple interval mapping (MIM) in a population consisting of 250 F2:3 lines derived from the cross Mp715 × Va35. Mp715 is resistant to the accumulation of aflatoxin and Va35 is susceptible. The population was genotyped with 1200 single-nucleotide polymorphism (SNP) and simple sequence repeat (SSR) molecular markers and phenotyped for the accumulation of total aflatoxins under artificial inoculation in four environments. Both parents contributed resistance alleles. Two QTL in bins 6.06 and 7.03 were the most promising for the marker-assisted introgression of the resistance present in Mp715. They were the most consistent across individual environments and together were responsible for nearly 30% of the phenotypic variance when data was combined across all four environments. In addition to those two QTL, Mp715 was also the source of the beneficial aflatoxin-reducing allele for several smaller effect QTL. Once their effect is validated in further experiments, the identification of these relatively large effect QTL should facilitate the utilization of this aflatoxin accumulation-resistant germplasm in applied maize breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mycotoxins are toxic secondary metabolites produced by filamentous fungi (Bennett and Klich 2003). They are important contaminants of foods and feeds and cause diseases (mycotoxicoses), including carcinomas, in humans and in animals (CAST 2003; Reddy et al. 2010; Wu et al. 2014). Over 100 countries have regulations in place to limit mycotoxin contamination with the majority of those regulations focused on aflatoxins (FAO 2004; van Egmond and Jonker 2004; van Egmond et al. 2007). Aflatoxins are the major class of mycotoxins produced by Aspergillus spp. and are the most toxic of the known mycotoxins (Payne 2016). The US Food and Drug Administration sets “action levels” for aflatoxin in maize grain: 20 ng g−1 for grain used in human food and varying levels, ranging from 20 to 300 ng g−1, for different classes of animal feed (Park and Liang 1993; USFDA 2010). In the USA, aflatoxin-contaminated maize is estimated to cost growers, on average, $163 million (Wu 2006) to $225 million (CAST 2003) every year while the annual cost of aflatoxin testing and mitigation is $20–50 million (Robens and Cardwell 2003; Schmale and Munkvold 2009). Due to the enforcement of regulations, the aflatoxin-contaminated grain is largely an economic problem in most developed countries, but it remains a human health problem in many developing countries (Payne and Yu 2010). Aflatoxin is associated with both acute and chronic toxicities (aflatoxicoses) in humans and in animals, with the liver, the primary target organ in both cases (Bennett et al. 2007). Acute aflatoxicosis causes death while chronic exposure to aflatoxin causes liver cancer and immune suppression (Williams et al. 2004; Bennett et al. 2007; Kew 2013). Aflatoxins are classified as group 1 human carcinogens (IARC 2012).
Several species of Aspergillus are capable of producing aflatoxins (Frisvad et al. 2005), but the economically important species are A. flavus and A. parasiticus (CAST 2003; Klich 2007). They are capable of colonizing most food products during storage but are also pre-harvest pathogens of oilseed crops including maize, peanut, cottonseed, and tree nuts (Scheidegger and Payne 2003; Klich 2007). In maize, A. flavus is much more common than A. parasiticus (Payne 2016). Aspergillus flavus is a ubiquitous, saprophytic, soil-borne fungus capable of acting as a weak, opportunistic, ear-rot pathogen of maize during periods of heat and drought stress (Payne and Yu 2010; White 2016). Aspergillus ear rot rarely causing economically significant reductions in yield or losses in grain quality, but A. flavus remains an important pathogen of maize due to pre-harvest aflatoxin contamination (White 2016).
Pre-harvest aflatoxin contamination of maize is a chronic problem in the Southeastern US and can reach epidemic proportions in some years (Payne 1992; Widstrom 1996). Over 90% of the Southeastern corn harvest was contaminated by aflatoxin in 1977 after a severe drought and increased insect pressure (Zuber and Zuber and Lillehoj 1979). In 1988, after a drought in the Midwest, one-third of the test samples in the Corn Belt exceeded FDA limits (Payne 1992; Clements and White 2004). A severe outbreak occurred across the Southeast in 1998 due to drought and unusually high temperatures (Windham and Williams 1999). That year in Mississippi, 20% of corn was sold at reduced prices and 4% abandoned completely due to aflatoxin contamination (Robens and Cardwell 2003).
Strategies to reduce pre-harvest aflatoxin contamination of maize grain have centered on both agronomic practices and the development of host plant resistance. Pre-harvest contamination is associated with biotic and abiotic stresses, such as drought and insect damage, and agronomic management of aflatoxin is aimed at mitigating those sources of stress (Bruns 2003). There are also biological control strategies that utilize non-toxin producing strains of A. flavus (Cotty 2006; Dorner 2010). Host plant resistance is widely considered a promising approach to reducing aflatoxin contamination (Moreno and Kang 1999; Williams et al. 2003, 2015; Brown et al. 2013), but current commercial hybrids lack adequate resistance (Abbas et al. 2002, 2006; Daves et al. 2010). Sources of resistance have been identified, and publicly developed germplasm and breeding lines have been registered and released (Scott and Zummo 1990, 1992; McMillian et al. 1993; Williams and Windham 2001, 2006, 2012; Llorente et al. 2004; Guo et al. 2007, 2011; Menkir et al. 2008; Mayfield et al. 2012; Scully et al. 2016).
The resistant breeding lines that have been released were bred from tropical sources or from older Southern US germplasm (Mayfield et al. 2012; Williams et al. 2003, 2015). Originating outside the Corn Belt dent maize populations, these breeding lines are considered exotic. They are un-adapted because, despite contributing stable resistance to aflatoxin accumulation in testcrosses, they also contribute undesirable agronomic characteristics: late flowering, high ear and plant height, increased lodging, late maturity, high grain moisture at harvest, and poor combining ability for yield (Brooks et al. 2005; Mayfield et al. 2012). These traits are typical of exotic maize when used in temperate US maize breeding (Hallauer 1978; Holland and Goodman 1996). Germplasm lines are intended to contribute favorable alleles to breeding populations, and host plant resistance is the most common use of exotic germplasm in US maize breeding (Goodman 1999; Betran et al. 2006). Unfortunately, transferring resistance to aflatoxin accumulation has proven difficult due to the highly quantitative nature of the trait: polygenic control, low heritability, and large genotype by environment interaction effects (Warburton et al. 2013). Mapping quantitative trait loci (QTL) and identifying closely linked molecular markers for use in marker-assisted selection (MAS) should allow for the “targeted introgression” of QTL from exotic germplasm to elite, adapted lines (Edwards 1992).
In order to identify molecular markers for use in marker-assisted selection, several bi-parental QTL mapping studies have been conducted on maize host plant resistance to pre-harvest aflatoxin accumulation (Paul et al. 2003; Widstrom et al. 2003; Busboom and White 2004; Brooks et al. 2005; Alwala et al. 2008; Warburton et al. 2009, 2011; Mayfield et al. 2011; Willcox et al. 2013; Yin et al. 2014; Dhakal et al. 2016). A meta-QTL analysis of some of those studies has been conducted (Mideros et al. 2014) and there have also been three genome-wide association (GWAS) mapping studies published (Warburton et al. 2013, 2015; Farfan et al. 2015; Zhang et al. 2016). Mp715, the resistant parent included in the present study, was the subject of two previous bi-parental QTL mapping studies (Warburton et al. 2011; Dhakal et al. 2016), and Va35, the susceptible parent, was included in another study in which Mp313E was the resistant parent (Willcox et al. 2013). By mapping the resistance alleles in Mp715 using a different genetic background, we can find which QTL are consistent across backgrounds as well as find novel QTL not detected in the previous studies. The present study also aims to map QTL with more precision than the previous studies of Mp715 through a combination of larger population size and greater marker density. The present study includes 250 families, genotyped with 1200 markers and phenotyped in four environments, as opposed to 225 families genotyped with 103 markers and phenotyped in four environments (Warburton et al. 2013) and 210 families genotyped with 136 markers and phenotyped in two environments (Dhakal et al. 2016). Using Va35 as the susceptible parent should allow for a comparison of the alleles for resistance being contributed by Mp715 and Mp313E.
Materials and methods
Population formation
An F2:3 mapping population was derived from the cross Mp715 × Va35. Mp715 is an aflatoxin accumulation-resistant breeding line developed from open-pollinated Tuxpan through eight generations of selfing and selecting for reduced aflatoxin accumulation after field inoculation (Williams and Windham 2001). Va35 is a Southern US adapted, non-Stiff Stalk inbred, developed in Virginia by selfing out of the backcross [(C103 × T8) × T8] (Gerdes et al. 1993). T8 is an inbred from Tennessee derived from open-pollinated Jarvis Golden Prolific, and C103 is derived from open-pollinated Lancaster Sure Crop (Gerdes et al. 1993). Va35 is susceptible to aflatoxin accumulation and is routinely used as a tester and as a susceptible check in aflatoxin research (e.g., Williams et al. 2008) as well as serving as the susceptible parent in a previous bi-parental QTL mapping study (Willcox et al. 2013). Va35 has also been used in breeding crosses to improve the agronomics of resistant germplasm lines (Williams and Windham 2012). Seed from one ear of a single self-pollinated F1 plant was planted as an F2 population. F2 plants were self-pollinated and individually harvested, creating 250 ear-to-row F2:3 families. Families were sib-mated to increase seed while maintaining within family variation.
Field conditions and experimental design
The study was planted in four environments: 2015, 2016, and 2017 at the R.R. Foil Plant Science Research Center, Mississippi State, Mississippi (MS) and 2017 at the Quaker Research Farm, Texas A&M AgriLife Extension Center, Lubbock, Texas (TX). Test entries included the 250 F2:3 families and 5 checks: the inbred parents (Mp715 and Va35), their F1 hybrid, and two inbreds (Mp718 and Mp719) derived from the cross Mp715 × Va35 (Williams and Windham 2012). Entries were grown in a randomized complete block design with three replications. All 250 F2:3 families were planted in the 2015 test, but only 244 families were included in the 2016 test, 237 in the MS 2017 test, and 241 in the TX 2017 test due to a shortage of seed for some families. The experimental unit consisted of a plot. Each plot was a single 5.1-m row planted to a single entry with 0.96-m row spacing. Rows were overplanted and thinned to 20 plants row−1. Management of test plots followed standard agronomic practices.
Phenotyping
Inoculum preparation and in-field inoculation of developing ears were performed using the side-needle technique according to Zummo and Scott (1989) and Windham and Williams (2002). Aflatoxigenic A. flavus strain NRRL 3357 (ATCC 200026) was used. The fungal inoculum was increased on sterile corn-cob grits (size 2040, Grit-O-Cobs, The Anderson Co., Maumee, OH) in 500-mL flasks, each containing 50 g of grits and 100 mL of sterile, distilled water, and incubated at 28 °C for 3 weeks. Conidia from each flask were washed from the grits using 500-mL sterile distilled water containing 20 drops L−1 of Tween 20 (Atlas Chemical Industry, Inc., London, UK) and filtered through four layers of cheesecloth. Concentrations of conidia were determined with a hemacytometer and adjusted with sterile distilled water to 9 × 107 conidia mL−1. Inoculum not used immediately was stored at 4 °C. The top ear of each plant was inoculated with the A. flavus conidial suspension 10 days after mid-silk (50% of the plants in a plot had emerged silks). A 3.4-mL suspension containing 3 × 108 conidia was injected through the husk with an Idico tree marking gun (Idico Products Co., New York, NY) fitted with a 35-mm 14-gauge needle. Inoculated ears in each plot were harvested by hand at kernel maturity, bulked by plot, and dried at 53 °C for 7 days in order to reach a uniform 13–15% moisture concentration. Ears (bulked by plot) were machine shelled and the grain mixed by pouring through a sample splitter twice. The entire grain sample was ground using a Romer subsampling mill (Romer Industries Inc., Union, MO) and a 50-g sample of ground grain was used for analysis. Total aflatoxin concentration was determined using the VICAM AflaTest (VICAM, Watertown, MA) in compliance with the USDA test protocol (USDA 2002).
Statistical analysis of phenotypes
Aflatoxin concentrations were log transformed using ln(y + 1) to normalize the distribution, and the transformed aflatoxin concentrations were the response variables for each plot. The five check genotypes were subjected to analysis of variance (ANOVA) and means were separated by pairwise t tests (α = 0.05) using type III analysis in the Proc Mixed function of SAS 9.4 (SAS Institute, Cary, NC). The effect due to genotype, environment, and genotype × environment interaction were all treated as a fixed effect, while block nested in the environment was treated as a random effect. When presenting the results of the multiple comparisons, phenotypes were transformed back to the original scale using antilogs and presented as geometric means of the original data (Steel and Torrie 1980).
The F2:3 family means, within and across environments, were calculated as best linear unbiased predictors (BLUPs) using restricted maximum likelihood (REML) in the PROC Mixed function of SAS 9.4. When analyzing data within a single environment, genotypes and blocks were treated as random effects. When analyzing data combined across all four environments, genotypes, environments, genotype × environment interactions, and blocks nested in environments were all treated as random effects. The significance of model terms was determined by likelihood ratio tests (LRT, α = 0.05) (Isik et al. 2017). Each family’s BLUP was used as its phenotype during QTL analysis. Variance components were estimated by REML in PROC Mixed and family mean heritabilities, within and across environments, were calculated as the immediate response to selection (Holland et al. 2003).
Genotyping
Leaf tissue was bulk collected from all F2:3 plants in a plot from the first replication of the test in 2015. Tissue samples were frozen in liquid nitrogen, lyophilized (Freezone Benchtop System, Labconco, Kansas City, MO), and ground using a Tecator Cyclotec-1093 sample mill (FOSS, Inc., Eden Prairie, MN). DNA was extracted by the CTAB method (Murray and Thompson 1980). The DNA concentration was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE) and samples were diluted to a common stock concentration in TE buffer. Genotyping was conducted using simple sequence repeat (SSR) and single-nucleotide polymorphism (SNPs) markers. The SNP markers were run as KASP assays (LGC Genomics, Teddington, UK) and as whole-genome genotyping assays on a custom designed Infinium array (Illumina, San Diego, CA). The custom array was run by Corteva Agriscience (Agriculture Division of DowDuPont, Johnston, IA) and was created by combining assays from the Maize 768 plex (Jones et al. 2009) and MaizeLD BeadChip (Rousselle et al. 2015).
The SSR primer pairs were synthesized by Integrated DNA Technologies (IDT Inc., Coralville, IA). Markers were amplified via polymerase chain reaction (PCR) in thin-walled 96 well plates using RedTaq ReadyMix PCR reaction mix and its recommended protocols (Sigma-Aldrich Co., Saint Louis, MO). The PCR products were separated by electrophoresis in × 1 TBE agarose gels (4% w/v) and visualized with ethidium bromide using an AlphaImager Gel Imaging System (Alpha Innotech, San Leandro, CA). Marker genotypes were scored visually from imaged gels.
For the KASP SNP assays (Semagn et al. 2014), a polymerase chain reaction was performed in 384-well plates loaded with an epMotion 5073m automated liquid handling system (Eppendorf AG, Hamburg, Germany). Template DNA (25.0 ng) was loaded into the wells and allowed to dehydrate on the benchtop overnight. 2.5-μL molecular grade H20 (Sigma-Aldrich Co., Saint Louis, MO), 2.5-μL KASP master mix, and 0.7-μL KASP SNP assay (LGC Genomics Limited, Teddington, UK) were added to each well pre-loaded with dehydrated template DNA. Prior to PCR, the plates were sealed with an optically clear seal in a K-seal heat-based plate sealer (KBioScience, Beverly, MA). The PCR was performed in a 65–57 °C touchdown protocol according to LGC Genomics’ KASP thermal cycling conditions manual (Teddington, UK). The assay’s allele-specific fluorescent signal was read on a FLUOstar Omega microplate reader (BMG-Labtech, Ortenberg, Germany) and the resulting data was processed in MARS Data Analysis software (BMG-Labtech, Ortenberg, Germany). The MARS output was imported into KlusterCaller software (LGC Genomics, Teddington, UK) and interpreted as allele calls.
The Infinium whole-genome genotyping assay is designed to interrogate a large number of SNPs at many loci simultaneously. Genotyping was performed as per Illumina’s protocol (Steemers and Gunderson 2007). Template DNA (200 ng) was amplified using the reagents supplied by Illumina and incubated overnight at 37 °C. Amplified DNA was enzymatically fragmented to around 300 base pairs using end point fragmentation followed by precipitation using 2-propanol. The DNA was resuspended and hybridized to the BeadChips and incubated in a humidified chamber overnight at 48 °C. Unhybridized and non-specifically hybridized DNA samples were washed from the BeadChips. Labeled nucleotides were added to the hybridized DNA. Allele specificity is conferred by enzymatic base extension. Products were immunohistochemically stained in TeFlow chambers on Tecan liquid handlers (Illumina, San Diego, CA). BeadChips were coated for protection and imaged on the iScan system using a two-color confocal laser system (0.8-μm resolution). The iScan reader uses a laser to excite the fluor of the single-base extension product on the BeadChip. Light emissions from these fluors are then recorded in high-resolution images of the BeadChip sections. Data from these images were analyzed using Illumina’s Genome Studio genotyping module which is based on the cluster file created from a set of reference samples. The allele calls were made for each sample and marker combination.
Linkage map construction
Genotypic data were analyzed in JoinMap 4 mapping software (van Ooijen 2006) in order to calculate segregation distortion, determine linkage groups, and estimate linkage distances within groups. Linkage groups were determined using the LOD value of the test for independence. Recombination frequencies within linkage groups were calculated using JoinMap’s Monte Carlo maximum likelihood mapping algorithm (Jansen et al. 2001) and converted to centimorgans (cM) by the Haldane mapping function. Locus order was confirmed according to the markers’ physical position in version 3 of the maize reference genome in MaizeGDB (Andorf et al. 2015).
QTL analysis
Quantitative trait locus mapping was conducted using QTL Cartographer version 2.5 (Wang et al. 2012). Composite interval mapping (CIM) (Jansen 1993; Jansen and Stam 1994; Zeng 1993, 1994) was implemented to locate QTL that were then used as the initial model terms in multiple interval mapping (MIM) (Kao et al. 1999; Zeng et al. 1999). For the CIM procedure, LOD thresholds were empirically estimated for each trait by 1000 permutations (Churchill and Doerge 1994; Doerge and Churchill 1996) in order to maintain a genome-wide 0.05 level of significance. Forward and backward stepwise regression was used for cofactor selection with a 10-cM window size for the genome scans. Tests to add and to remove markers from the set of cofactors were conducted at α = 0.1 level of significance. The walk speed, the grid of positions to be tested for putative QTL, was set at 0.5 cM. The LOD score at each test position is the log10 of the ratio of the likelihood of the full model to the likelihood of the reduced model (Jansen 2007). The reduced model included only cofactor effects, while the full model included cofactor effects and the additive and dominance effects of a putative QTL (Silva et al. 2012). The QTL peaks with LOD scores clearing the permutation-based threshold, with a minimum of 5 cM between QTL, were declared significant, and the significant QTL from CIM were used as initial model terms in MIM. The MIM models were refined by stepwise testing according to the guidelines of Silva et al. (2012): (1) search for new main effect QTL, (2) search for epistatic interactions between identified QTL, (3) test for the significance of epistatic terms; (4) test for the significance of main effect QTL without interactions, and (5) optimize the positions of the final QTL. After each cycle of testing, new terms (main effect QTL or epistatic interactions) were added to the model only if they decreased the Bayesian information criterion (BIC) (Schwarz 1978). The BIC favors models with higher likelihoods but avoids overfitting by including a penalty for each additional parameter added. After the final model was chosen, all QTL effects (additive, dominance, and epistasis) were simultaneously estimated and the partitioning of the genotypic variance calculated (Kao and Zeng 2002). Overfitting was then further avoided by not allowing the proportion of the total variation due to genetic effects to exceed the heritability. When this occurred, QTL making the least contribution to the genetic variance were removed from the model, and the remaining terms were re-estimated (Robertson-Hoyt et al. 2006).
Results and discussion
Phenotypes
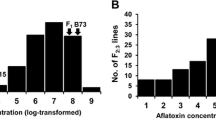
Five genotypes were planted as checks in each environment: the resistant parent (Mp715), the susceptible parent (Va35), their F1, and two inbreds derived from Mp715 × Va35 used as a breeding cross (Mp718 and Mp719). Although the five checks were planted in all four environments, too many inbred plots were missing in MS 2016 to include in the ANOVA. Consequently, data from the check genotypes were only analyzed in MS 2015, MS 2017, and TX 2017. Across those three environments, block nested in the environment was not a significant source of variance (p = 0.4886; Table 1). The effect of the interaction between genotype and environment was significant (p = 0.0030; Table 1). Due to the significant interaction, genotype means were contrasted as simple effects by the environment, not as main effects (Table 2). In all three environments analyzed, the resistant parent (Mp715) had significantly lower aflatoxin levels than the susceptible parent (Va35), but their performance relative to the F1, Mp719, and Mp718 varied by the environment (Table 2).
Prior to transforming the data, mean aflatoxin concentrations plot−1 for the F2:3 families were 40.6 ± 3.6 ppb (x̅ ± s.e.) in MS 2015, 397.8 ± 27.4 ppb in MS 2016, 131.1 ± 8.6 ppb in MS 2017, and 204.9 ± 9.1 ppb in TX 2017. The lower levels of aflatoxin observed in MS 2015 were not explained through obvious weather patterns. Daily temperatures and precipitation were not appreciably different in MS 2015 than in MS 2016 or MS 2017 (Supplemental Fig. 1). In addition to having the lowest mean aflatoxin concentration, MS 2015 was also the environment with the largest coefficient of variability (Table 3).
The F2:3 family means were calculated as BLUPs within and across all four environments (Supplemental Table 1), and these BLUPs were used as the phenotypes during QTL analysis. The models used to calculate the BLUPs treated all terms, other than the overall mean, as random effects and the significance of the model terms were tested in LRTs. When data was combined across all four environments, all terms were significant (Supplemental Table 2). When data was analyzed within environments, the variance due to genotype was always significant but the significance of blocking varied by the environment (Supplemental Table 3). Variance components were estimated and used to calculate family mean heritabilities within and across environments (Table 4). The heritability across all four environments was 0.69, while the within environment heritabilities ranged from 0.48 in MS 2015 to 0.70 in MS 2017 (Table 4). The family mean heritabilities were calculated as the immediate response to selection (Holland et al. 2003).
Genotyping and linkage map construction
Ten linkage groups corresponding to the ten chromosomes of maize were identified at a LOD of 4 and all ten groups remained unbranched at the most stringent threshold tested (LOD = 10). The final map (Supplemental Table 4) consisted of 1200 markers: 25 SSRs, 73 KASP SNPs, and 1102 modified MaizeLD Beadchip SNPs. The map spanned a total length of 1367 cM with an average distance of 1.15 cM between markers. Of the 139 markers mapped to chromosome 3, the first 124 showed significant segregation distortion (α = 0.05), with fewer than expected homozygous genotypes for the Va35 allele (Supplemental Table 4). This region of segregation distortion included bins 3.01–3.09. Bins 3.03–3.07 are known to include segregation distortion regions in other maize populations (Lu et al. 2002). Segregation distortion regions are no less likely to contain QTL, but the segregation distortion will generally decrease the power of QTL mapping (Xu 2008).
QTL analysis
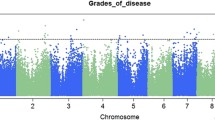
Composite interval mapping was conducted within and across all four environments and the significant QTL (Supplemental Table 5) were used as the initial model terms in MIM. The QTL included in the final MIM models along with their estimated additive, dominance, and epistatic effects and their contribution to the phenotypic variance are presented in Table 5. The MIM models explained between 35% (TX 2017) and 55% (MS 2017) of the phenotypic variance within environments and 61% of the phenotypic variance when all four environments were combined.
When MIM was conducted within individual environments, every chromosome except chromosome 9 contained at least one significant QTL in at least one environment. In every environment, both parents contributed beneficial (aflatoxin-reducing) alleles, but Mp715 (the resistant parent) always contributed more beneficial alleles than Va35 and the QTL for which the Mp715 alleles were beneficial always accounted for a larger cumulative share of the phenotypic variance than the QTL for which the Va35 alleles were beneficial. In all four environments, at least one QTL for which Va35 was the source of the beneficial allele was identified on the short arm of chromosome 1. These QTL were located in bins 1.01, 1.02, 1.03, and 1.05. This was consistent with a previous QTL mapping study in which Va35 served as the susceptible parent and contributed the beneficial allele for a QTL in bin 1.02 (Willcox et al. 2013). In that study, the QTL in bin 1.02 was the only QTL for which the Va35 allele was beneficial when the data was averaged across environments, although Va35 was the source of the resistance alleles for other QTL that were significant in individual environments. In the present study, Va35 also contributed beneficial alleles on chromosomes 2, 3, 7, and 10 but none of these QTL were significant in more than one environment.
In Mississippi 2016 and Texas 2017, no individual QTL accounted for more than 10% of the phenotypic variance (Table 5). In Mississippi 2015 and 2017, one QTL in bin 6.06 was responsible for more than 10% of the phenotypic variance (17.9% and 13.7%, respectively). A QTL in bin 7.03 was observed in all four environments and explained a range of 6.7% (MS 2016) to 8.6% (TX 2017) of the phenotypic variance. Mp715 was the source of the beneficial allele for the QTL in bins 6.06 and 7.03. A significant QTL for which Mp715 contributed the resistance allele was detected on chromosome 10 in all 3 years, the study was conducted in Mississippi, but no significant QTL was detected on chromosome 10 in Texas.
When the data was combined across all four environments, eight QTL and two epistatic interactions were identified (Table 5). The QTL were present on every chromosome except 2, 5, and 9. Va35 contributed the beneficial allele for two of the eight QTL: a QTL on the short arm of chromosome 1 (bin 1.03) that was responsible for nearly 10% of the phenotypic variance and a QTL in bin 7.05 of chromosome 7. The QTL in bin 7.05 had a very small effect but was important to the model due to its interaction with the QTL in bin 1.03. Both of the epistatic terms were composed of interactions between the two QTL contributed by Va35. The QTL, for which Va35 was the source of the beneficial allele, and the interactions between those QTL, accounted for 12.8% of the phenotypic variance and 20.8% of the genotypic variance as modeled in the MIM.
The QTL in bins 6.06 and 7.03, discussed above, had the largest effect size when the data was combined across environments (Table 5). They accounted for 14.3% and 15.4% of the phenotypic variance, respectively. Since the MIM model explained 61.4% of the phenotypic variance, the combined effect of these two QTL accounted for nearly half (23.3% and 25%, respectively) of the genotypic variance, as modeled, present in the population. They are therefore the most logical targets for marker-assisted introgression of the resistance to aflatoxin accumulation present in Mp715.
The QTL in bins 4.08 and 10.05 were responsible for 6.1% and 7.2% of the phenotypic variance, respectively (11.1% and 11.7% of the genotypic variance). These QTL could also be targeted in marker-assisted introgression. The QTL in bin 3.03 had a larger effect size but a smaller component of the phenotypic variance than the QTL in bins 4.08 and 10.05. This lack of agreement between relative effect size and relative impact on the variance is due to the segregation distortion present on chromosome 3. At position 63.9 cM on chromosome 3, there were 215 homozygotes for the Mp715 allele, eight heterozygotes and five homozygotes for the Va35 allele (Supplemental Table 4). This extreme segregation distortion makes the estimate of effect size unreliable for this locus, but also means that even if the locus has a large effect on the phenotype, we would expect very little phenotypic variance in the population to be due to the locus since most individuals have the same genotype at this locus. Based on its effect size, one could choose to target this QTL during marker-assisted introgression despite its small contribution to the phenotypic variance. However, this effect size is estimated from a skewed sample and is therefore unreliable.
Comparison with previous QTL analyses
Mp715 was previously mapped using T173 (Warburton et al. 2011) and B73 (Dhakal et al. 2016) as susceptible parents. In those two previous studies and in the present study, aflatoxin-reducing alleles were contributed by both Mp715 and the susceptible parent. In the present study and in Warburton et al. (2011), the majority of beneficial alleles was contributed by Mp715, while in Dhakal et al. (2016), the susceptible parent contributed as much of the resistance as Mp715. All three studies report QTL for aflatoxin accumulation on the short arm of chromosome 1. In the present study, Va35 was consistently the source of the beneficial alleles in that region. In the previous work, Mp715 was the source of the aflatoxin-reducing alleles in that region when mapped against T173 (Warburton et al. 2011) and it was the source of the beneficial allele for one of two QTL on the short arm of chromosome 1 when mapped against B73 (Dhakal et al. 2016). It is possible that different alleles are being contributed by the four parents so that the Mp715 alleles were beneficial relative to the T173 and B73 alleles but not the Va35 alleles. A QTL was detected in bin 10.05 in the present study and in Dhakal et al. (2016) and one in 10.04 in Warburton et al. (2011) but again, the three studies did not all agree on the Mp715 allele being the beneficial allele.
The QTL with the largest and most consistent effects in the present study were found in bins 6.06 and 7.03. No QTL was found on chromosomes 6 or 7 in any environment in Warburton et al. (2011) and only a small effect QTL was detected on chromosome 7, though not in bin 7.03, in Dhakal et al. (2016). Whether this inconsistency is due to differences in genetic background, marker density, or test environment is not known. Also, whether to view this as an inconsistency versus the “detection of novel” QTL between studies is open to interpretation. When Va35 served as the susceptible parent in across with Mp313E, the resistance alleles contributed by Mp313E were on chromosomes 2, 3, and 4 (Willcox et al. 2013). One of those QTL was at 98.8 cM on chromosome 4 and is possibly the same locus detected in bin 4.08 at position 92.2 cM in the present study. Otherwise, the resistance alleles contributed by Mp313E and by Mp715 do not co-locate. This suggests that the alleles for resistance in Mp313E and Mp715 could potentially be pyramided in a common background to further increase resistance to aflatoxin accumulation.
Potential for marker-assisted selection
The results of the present analysis, specifically the identified QTLs’ location and magnitude, need to be refined and validated before they can be utilized in practical marker-assisted breeding. A breeder must be confident that a QTL has a large enough effect to warrant marker-assisted introgression and must know how large and precisely which region of a chromosome to introgress. However, the effect size is often overestimated in QTL analysis (Beavis 1994; Schon et al. 2004) and a single term in a statistical QTL model may in fact be the result of multiple biological QTL (Jansen 2007). One method for testing the effect size is to create near-isogenic lines (NILs) through marker-assisted backcrossing using the susceptible parent (Va35) as the recurrent parent and Mp715 as the donor parent. While creating those same NILs, the region of chromosome possessing the QTL can be refined by selecting different sets of marker haplotypes. It is customary to report the location of QTL as the 2-LOD interval around the peak of a LOD profile, but it is possible that the detected effect is the result of multiple QTL, and there is no reason to assume they all fall into that 2-LOD region (Jansen 2007). The 2-LOD interval is more akin to a statistical confidence interval around the peak position (Jansen 2007) than a biological description of the region of chromosome possessing the causal polymorphisms. If one studies the LOD profiles for the QTL reported in this study (Supplemental Fig. 2), one finds that the regions of the chromosome with a high likelihood of containing QTL are generally wider than the 2-LOD support intervals (Table 5). Assuming the effect size is validated, the region of chromosome necessary for that effect is an empirical question. Using the susceptible parent as the recurrent parent is appealing since that is the genetic background in which the marker-trait associations were identified. However, if the analysis is to be useful in practical breeding, it must be possible to achieve similar effects when introgressing the QTL into genetic backgrounds not included in the QTL analysis. Additional NILs must also be created using additional recurrent parents. The present study is the first step in the genetic analysis necessary to support the marker-assisted introgression of the resistance to aflatoxin accumulation present in Mp715.
Conclusion
Beneficial, aflatoxin-reducing alleles were contributed by both parents in the cross Mp715 × Va35. Multiple QTL for which the alleles for resistance were contributed by Va35 were found on the short arm of chromosome 1 in every environment tested. A QTL for which the allele for resistance was contributed by Mp715 was found in bin 7.03 in every environment tested. A QTL in bin 6.06 was the only QTL to account for more than 10% of the phenotypic variance in any test environment, doing so in two of four. When the data was analyzed across environments, the QTL in bins 6.06 and 7.03 together caused nearly 30% of the phenotypic variance and nearly 50% of the genotypic variance. These two QTL could be targeted for marker-assisted introgression of the resistance to pre-harvest aflatoxin accumulation present in maize inbred Mp715. Other smaller effect QTL in bins 10.05, 4.08, and possibly 3.03 could also be targeted depending on the breeding objectives.
References
Abbas HK, Williams WP, Windham GL, Pringle HC, Xie W, Shier WT (2002) Aflatoxin and fumonisin contamination of commercial corn (Zea mays) hybrids in Mississippi. J Agric Food Chem 50:5246–5254
Abbas HK, Cartwright RD, Xie W, Shier WT (2006) Aflatoxin and fumonisin contamination of corn (maize, Zea mays) hybrids in Arkansas. Crop Prot 25:1–9
Alwala S, Kimbeng CA, Williams WP, Kang MS (2008) Molecular markers associated with resistance to Aspergillus flavus in maize grain: QTL and discriminant analyses. J New Seeds 9:1–18
Andorf CM, Cannon EK, Portwood JL, Gardiner JM, Harper LC, Schaeffer ML, Braun BL, Campbell DA, Vinnakota AG, Sribalusu VV, Huerta M, Cho KT, Wimalanathan K, Richter JD, Mauch ED, Rao BS, Birkett SM, Sen TZ, Lawrence-Dill CJ (2015) MaizeGDB update: new tools, data and interface for the maize model organism database. Nucleic Acids Res 44:1195–1201
Beavis WD (1994) The power and deceit of QTL experiments: lessons from comparative QTL studies. In: 49th Annual Corn and Sorghum Industry Research Conference. ASTA, Washington, DC, pp 250–266
Bennett JW, Klich M (2003) Mycotoxins. Clin Microbiol Rev 16:497–516
Bennett JW, Kale S, and Yu J. 2007. Aflatoxins: background, toxicology, and molecular biology. In: Simjee S (ed.) Foodborne diseases. Humana Press. p355–373
Betran FJ, Mayfield K, Isakeit T, Menz M (2006) Breeding maize exotic germplasm. In: Lamkey KR, Lee M (eds) Plant breeding: the Arnel R. Hallauer international symposium. Blackwell Publishing, Ames, IA, pp 352–367
Brooks TD, Williams WP, Windham GL, Willcox MC, Abbas HK (2005) Quantitative trait loci contributing resistance to aflatoxin accumulation in maize inbred Mp313E. Crop Sci 45:171–174
Brown RL, Bhatnagar D, Cleveland TE, Chen ZY, and Menkir A. 2013. Development of maize host resistance to aflatoxigenic fungi. In: M. Razzaghi-Abyane, editor, Aflatoxins: recent advances and future prospects. InTech. p. 3-23
Bruns HA (2003) Controlling aflatoxin and fumonisin in maize by crop management. J Toxicol 22:153–173
Busboom KN, White DG (2004) Inheritance of resistance to aflatoxin production and Aspergillus ear rot of corn from the cross of inbreds B73 and Oh516. Phytopathology 94:1107–1115
CAST. 2003. Mycotoxins: risks in plant, animal, and human systems. CAST Task Force Rep. 139. Counc. Agric. Sci. Technol., Ames, IA
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Clements MJ, White DG (2004) Identifying sources of resistance to aflatoxin and fumonisin contamination in corn grain. J Toxicol 23:381–396
Cotty PJ (2006) Biocompetitive exclusion of toxigenic fungi. In: Barug D et al (eds) The mycotoxin factbook. Wageningen Academic Publ., Wageningen, Netherlands, pp 179–197
Daves C, Windham GL, and Williams WP. 2010. Aflatoxin accumulation in commercial corn hybrids artificially inoculated with Aspergillus flavus in 2008 and 2009. Res. Rep. 24(9). MS Agric. For. Exp. Stn., Mississippi State, MS.
Dhakal R, Windham GL, Williams WP, Subudhi PS (2016) Quantitative trait loci for reducing aflatoxin accumulation in corn. Mol Breed 36:164
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Dorner JW (2010) Efficacy of a biopesticide for control of aflatoxins in corn. J Food Prot 73:495–499
Edwards MD (1992) Use of molecular markers in the evaluation and introgression of genetic diversity for quantitative traits. Field Crop Res 29:241–260
van Egmond HP, Jonker MA (2004) Worldwide regulations on aflatoxin: the situation in 2002. Toxin Rev 23:273–293
van Egmond HP, Schothorst RC, Jonker MA (2007) Regulations relating to mycotoxins in food – perspectives in a global and European context. Anal Bioannal Chem 389:147–157
FAO, 2004. Worldwide regulations for mycotoxins in food and feed in 2003. FAO Food and Nutrition Paper 81. Food and Agric. Organ. of the United Nations, Rome, Italy
Farfan IDB, de la Fuente GD, Murray SC, Isakeit T, Huang PC, Warburton M, Williams P, Windham GL, Kolometis M (2015) Genome wide association study for drought, aflatoxin resistance, and important agronomic traits of maize hybrids in the sub-tropics. PLoS One 10:e0117737. https://doi.org/10.1371/journal.pone.0117737
Frisvad JC, Skoube R, Samson RA (2005) Taxonomic comparison of three different groups of aflatoxin producers and a new efficient producer of aflatoxin B1, sterigmatocystin, and 3-O-methylsterigmatocystin Aspergillus rambelli sp nov. Syst Appl Microbiol 28:442–453
Gerdes JT, Behr CF, Coors JG, Tracy WF (1993) Compilation of North American maize breeding germplasm. CSSA, Madison, WI
Goodman MM (1999) Broadening the genetic diversity in maize breeding by use of exotic germplasm. In: Coors JG, Pandey S (eds) The genetics and exploitation of heterosis in crops. ASA, CSSA, and SSSA, Madison, WI, pp 139–148
Guo BZ, Widstrom NW, Lee RD, Coy AE, Lynch RE (2007) Registration of maize germplasm GT601 (AM-1) and GT602 (AM-2). J Plant Reg 1:153–154
Guo BZ, Krakowsky MD, Ni X, Scully BT, Lee RD, Coy AE, Widstrom NW (2011) Registration of maize inbred line GT603. J Plant Reg 5:211–214
Hallauer AR (1978) Potential of exotic germplasm for maize improvement. In: Walden DB (ed) Maize breeding and genetics. John Wiley & Sons, New York, NY, pp 229–247
Holland JB, Goodman MM (1996) Combining ability of tropical maize accessions with U.S. germplasm. Crop Sci 35:767–773
Holland JB, Nyquist WE, Cervantes-Martinez CT (2003) Estimating and interpreting heritability for plant breeding: an update. Plant Breed Rev 22:9–112
IARC, 2012. Aflatoxins. IARC monographs on the evaluation of carcinogenic risks to humans: a review of human carcinogens, Vol. 100F, Chemical agents and related occupations. Int. Agency for Res. on Cancer. Lyon, France. p. 225–248
Isik F, Holland JB, Maltecca C (2017) Genetic data analysis for plant and animal breeding. Springer, New York
Jansen RC (1993) Interval mapping of multiple quantitative trait loci. Genetics 135:205–211
Jansen RC 2007. Quantitative trait loci in inbred lines. In: D.J. Balding et al., editors, Handbook of statistical genetics, vol. 2. 3rd ed. John Wiley & Sons, West Sussex, England. p. 589–622
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Jansen RC, de Jong AG, van Ooijen JW (2001) Constructing dense genetic linkage maps. Theor Appl Genet 102:1113–1122
Jones E, Chu WC, Ayele M, Ho J, Bruggeman E, Yourstone K, Rafalski A, Smith OS, McMullen MD, Bezawada C, Warren J, Babayev J, Basu S, Smith S (2009) Development of single nucleotide polymorphism (SNP) markers for use in commercial maize (Zea mays L.) germplasm. Mol. Breeding 24:165–176
Kao CH, Zeng ZB (2002) Modeling epistasis of quantitative trait loci using Cockerham’s model. Genetics 160:1243–1261
Kao CH, Zeng ZB, Teasdale RD (1999) Multiple interval mapping for quantitative trait loci. Genetics 152:1203–1216
Kew MC (2013) Aflatoxin as a cause of hepatocellular carcinoma. J Gastrointestin Liver Dis 22:305–310
Klich M (2007) Aspergillus flavus: the major producer of aflatoxin. Mol Plant Pathol 8:713–722
Llorente CF, Betran FJ, Bockholt A, Fojt F (2004) Registration of Tx772 maize. Crop Sci 44:1036–1037
Lu H, Romero-Severson J, Bernardo R (2002) Chromosomal regions associated with segregation distortion in maize. Theor Appl Genet 105:622–628
Mayfield KL, Murray SC, Rooney WL, Isakeit T, Odvody GA (2011) Confirmation of QTL reducing aflatoxin in maize testcrosses. Crop Sci 51:2489–2498
Mayfield KL, Betran FJ, Isakeit T, Odvody G, Murray SC, Rooney WL, Landivar JC (2012) Registration of maize germplasm lines Tx736, Tx739, and Tx740 for reducing preharvest aflatoxin accumulation. J Plant Reg 6:88–94
McMillian WW, Widstrom NW, Wilson DM (1993) Registration of GT-MAS:gk maize germplasm. Crop Sci 33:882
Menkir A, Brown RL, Bandyopadhyay R, Cleveland TE (2008) Registration of six tropical maize germplasm lines with resistance to aflatoxin contamination. J Plant Reg 2:246–250
Mideros SX, Warburton ML, Jamann TM, Windham GL, Williams WP, Nelson RJ (2014) Quantitative trait loci influencing mycotoxin contamination of maize: analysis by linkage mapping, characterization of near-isogenic lines, and meta-analysis. Crop Sci 54:127–142
Moreno OJ, Kang MS (1999) Aflatoxins in maize: the problem and genetic solutions. Plant Breed 118:1–16
Murray MG, Thompson W (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4326
van Ooijen JW (2006) JoinMap 4, software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, Netherlands
Park DL, Liang B (1993) Perspectives on aflatoxin control for human food and animal feed. Trends Food Sci Technol 4:334–342
Paul C, Naidoo G, Forbes A, Mikkilineni V, White DG, Rocheford T (2003) Quantitative trait loci for low aflatoxin production in two related maize populations. Theor Appl Genet 107:263–270
Payne GA (1992) Aflatoxin in maize. Crit Rev Plant Sci 10:423–440
Payne GA (2016) Aflatoxins and aflatoxicosis. In: Munkvold GP, White DG (eds) Compendium of corn diseases, 4th edn. APS Press, St. Paul, MN, Am. Phytopathol. Soc., pp 97–98
Payne GA, Yu J (2010) Ecology, development, and gene regulation in Apsergillus flavus. In: Machida M, Gomi K (eds) Aspergillus: molecular biology and genomics. Caister Academic Press, Norfolk, UK, pp 151–171
Reddy KRN, Salleh B, Abbas HK, Abel CA, Shier WT (2010) An overview of mycotoxin contamination in foods and its implications for human health. Toxin Rev 29:3–26
Robens J, Cardwell K (2003) The costs of mycotoxin management to the USA: management of aflatoxins in the United States. Toxin Rev 22:139–152
Robertson-Hoyt LA, Jines MP, Balint-Kurti PJ, Kleinschmidt CE, White DG, Payne GA, Maragos CM, Molnar TL, Holland JB (2006) QTL mapping for fusarium ear rot and fumonisin contamination resistance in two maize populations. Crop Sci 46:1734–1743
Rousselle Y, Jones E, Charcosset A, Moreau P, Robbins K, Stich B, Knaak C, Flament P, Karaman Z, Martinant J-P, Fourneau M, Taillardat A, Romestant M, Tabel C, Bertran J, Ranc N, Lespinasse D, Blanchard P, Kahler A, Chen J, Kahler J, Dobrin S, Warner T, Ferris R, Smith S (2015) Study on essential derivation in maize: III. Selection and evaluation of a panel of single nucleotide polymorphism loci for use in European and North American germplasm. Crop Sci 55:1170–1180
Scheidegger KA, Payne GA (2003) Unlocking the secrets behind secondary metabolism: a review of Aspergillus flavus from pathogenicity to functional genomics. J Toxicol 22:423–459
Schmale DG and Munkvold GP. 2009. Mycotoxins in crops. The Plant Health Instructor. https://doi.org/10.1094/PHI-I-2009-0715-01
Schon CC, Utz HF, Groh S, Truberg B, Openshaw S, Melchinger AE (2004) Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics of complex traits. Genetics 167:485–498
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Scott GE, Zummo N (1990) Registration of Mp313E parental line of maize. Crop Sci 30:1378
Scott GE, Zummo N (1992) Registration of Mp420 germplasm line of maize. Crop Sci 32:1296
Scully BT, Krakowsky MD, Ni X, Tapp PJ, Knoll JE, Lee RD, Guo BZ (2016) Registration of maize inbred line GT888. J Plant Reg 10:87–92
Semagn K, Babu R, Hearne S, Olsen M (2014) Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): overview of the technology and its application in crop improvement. Mol Breed 33:1–14
Silva LDCE, Wang S, and Zeng Z. 2012. Composite interval mapping and multiple interval mapping: procedures and guidelines for using Windows QTL Cartographer. In: Rifkin SA, editor, Quantitative trait loci (QTL): methods and protocols. Methods in Molecular Biology, vol. 871. Springer Science Media, New York, NY. p. 75–118
Steel RGD, Torrie JH (1980) Principles and procedures of statistics: a biometrical approach, 2nd edn. McGraw Hill, New York
Steemers FJ, Gunderson KL (2007) Whole genome genotyping technologies on the BeadArrayTM platform. Biotechnol J 2:41–49
USDA (2002) Vicam test method. In: Aflatoxin handbook. Federal Grain Inspection Service, Grain Inspection, Packers, and Stockyards Administration, Washington, D.C, pp 8.1–8.17
USFDA. 2010. CPG sec. 683.100 Action levels of aflatoxins in animal feeds. http://www.fda.gov/ICECI/ComplianceManuals/CompliancePolicyGuidanceManual/ucm074703 (accessed 05/23/2018)
Wang S., C. J. Basten, and Z.B. Zeng. 2012. Windows QTL Cartographer 2.5. Dept. Statistics, North Carolina State University, Raleigh, NC.
Warburton ML, Brooks TD, Krakowsky MD, Shan X, Windham GL, Williams WP (2009) Identification and mapping of new sources of resistance to aflatoxin accumulation in maize. Crop Sci 49:1403–1408
Warburton ML, Brooks TD, Windham GL, Williams WP (2011) Identification of novel QTL contributing resistance to aflatoxin accumulation in maize. Mol Breed 27:491–499
Warburton ML, Williams WP, Windham GL, Murray SC, Xu W, Hawkins LK, Duran JF (2013) Phenotypic and genetic characterization of a maize association mapping panel developed for the identification of new sources of resistance to Aspergillus flavus aflatoxin accumulation. Crop Sci 53:2374–2383
Warburton ML, Tang JD, Windham GL, Hawkins LK, Murray SC, Xu W, Boykin D, Perkins A, Williams WP (2015) Genome-wide association mapping of Aspergillus flavus and aflatoxin accumulation resistance in maize. Crop Sci 55:1–11
White DG (2016) Aspergillus ear rot. In: Munkvold GP, White DG (eds) Compendium of corn diseases, 4th edn. APS Press, St. Paul, MN, Am. Phytopathol. Soc., pp 84–88
Widstrom NW (1996) The aflatoxin problem with corn grain. Adv Agron 56:220–280
Widstrom NW, Butron A, Guo BZ, Wilson DM, Snook ME, Cleveland TE, Lynch RE (2003) Control of preharvest aflatoxin contamination in maize by pyramiding QTL involved in resistance to ear-feeding insects and invasion by Aspergillus spp. Eur J Agron 19:563–572
Willcox MC, Davis GL, Warburton ML, Windham GL, Abbas HK, Betran J, Holland JB, Williams WP (2013) Confirming quantitative trait loci for aflatoxin resistance from Mp313E in different genetic backgrounds. Mol Breed 32:15–26
Williams WP, Windham GL (2001) Registration of maize germplasm line Mp715. Crop Sci 41:1374–1375
Williams WP, Windham GL (2006) Registration of maize germplasm line Mp717. Crop Sci 46:1407
Williams WP, Windham GL (2012) Registration of Mp718 and Mp719 germplasm lines of maize. J Plant Reg 6:1–3
Williams WP, Windham GL, Buckley PM (2003) Enhancing maize germplasm with resistance to aflatoxin contamination. J Toxicol 22:175–193
Williams JH, Phillips TD, Jolly PE, Stiles JK, Jolly CM, Aggarwal D (2004) Human aflatoxicosis in developing countries: a review of toxicology, exposure, potential health consequences, and interventions. Am J Clin Nutr 80:1106–1122
Williams WP, Krakowsky MD, Windham GL, Balint-Kurti P, Hawkins LK, Henry WB (2008) Identifying maize germplasm with resistance to aflatoxin accumulation. Toxin Rev 27:319–345
Williams WP, Krakowsky MD, Scully BT, Brown RL, Menkir A, Warburton ML, Windham GL (2015) Identifying and developing maize germplasm with resistance to accumulation of aflatoxins. World Mycotoxin J 8:193–209
Windham GL and Williams WP. 1999. Aflatoxin accumulation in commercial corn hybrids in 1998. Res. Rep. 22(8). MS Agric. For. Exp. Stn., Mississippi State, MS.
Windham GL, Williams WP (2002) Evaluation of corn inbreds and advanced breeding lines for resistance to aflatoxin contamination in the field. Plant Dis 86:232–234
Wu F (2006) Mycotoxin reduction in Bt corn: potential economic, health, and regulatory impacts. Transgenic Res 15:277–289
Wu F, Groopman JD, Pestka JJ (2014) Public health impacts of foodborne mycotoxins. Annu Rev Food Sci Technol 5:351–372
Xu S (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208
Yin Z, Wang Y, Wu F, Gu X, Bian Y, Wang T, Deng D (2014) Quantitative trait locus mapping of resistance to Aspergiullus flavus infection using a recombinant inbred line population in maize. Mol Breed 33:39–49
Zeng ZB (1993) Theoretical basis of separation of multiple linked gene effects on mapping quantitative trait loci. Proc Natl Acad Sci U S A 90:10972–10976
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1466
Zeng ZB, Kao CH, Basten CJ (1999) Estimating the genetic architecture of quantitative traits. Genet Res 74:279–289
Zhang Y, Cui M, Zhang J, Zhang L, Li C, Kan X, Sun Q, Deng D, Yin Z (2016) Confirmation and fine mapping of a major QTL for aflatoxin reduction in maize using a combination of linkage and association mapping. Toxins 8:258
Zuber MS, Lillehoj EB (1979) Status of the aflatoxin problem in corn. J Environ Qual 8:1–5
Zummo N, Scott GE (1989) Evaluation of field inoculation techniques for screening maize genotypes against kernel infection by Aspergillus flavus in Mississippi. Plant Dis 73:313–316
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Smith, J.S., Williams, W.P., Windham, G.L. et al. Identification of quantitative trait loci contributing resistance to aflatoxin accumulation in maize inbred Mp715. Mol Breeding 39, 91 (2019). https://doi.org/10.1007/s11032-019-0997-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-0997-0