
Abstract
Genomic selection (GS) is expected to increase the rate of genetic gain in oil palm. In a GS scheme, breeding cycles with progeny tests (phenotypic selection, PS) used to calibrate the GS predictive model and for selection alternate with GS cycles, making it possible to train the GS model with aggregated data from several cycles. To evaluate this possibility, we simulated four cycles of hybrid breeding for bunch production and compared two methods of calibrating the GS model, one using aggregated data from the two most recent cycles (Tr2Gen), the other using data from the last cycle (Tr1Gen). We also compared a GS scheme with two PS cycles and two GS cycles (2PT-2noPT), and a scheme with PS every other cycle and GS otherwise (PT-noPT). We showed that Tr2Gen had a 10.7% higher genetic gain per cycle than Tr1Gen, mostly due to increased selection accuracy, particularly in across-cycle selection, despite the decreased relationship between training individuals and selection candidates. After four cycles, Tr2Gen had a 5% higher cumulative genetic gain than Tr1Gen, with a lower coefficient of variation. PT-noPT benefited more from the advantages offered by Tr2Gen than 2PT-2noPT. Over four breeding cycles, combining PT-noPT and Tr2Gen largely outperformed conventional reciprocal recurrent selection (RRS), with an increase in annual genetic gain ranging from 37.6 to 57.5%, depending on the number of GS candidates. This study confirms the advantages of GS over RRS and indicated that oil palm breeders should train GS models using all data from past breeding cycles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Oil palm (Elaeis guineensis Jacq.) is the world’s number one oil crop, with current annual production at > 60 Mt (USDA 2017). It is a diploid, perennial, and naturally cross-pollinated species cultivated in humid tropical zones. Palm oil is extracted from the mesocarp of the fruits constituting the bunches. Bunch production is a major component of oil yield, and the hybrid oil palm cultivars display heterosis for this trait (Gascon and de Berchoux 1964). Bunch production is the mathematical product of bunch number (BN) and average bunch weight (BW), two quantitative traits with mostly additive inheritance and a strong negative genetic correlation (Gascon et al. 1966; Corley and Tinker 2016). Oil palm bunch production therefore illustrates the case of heterosis resulting from the multiplicative interaction between additive and negatively correlated components (Schnell and Cockerham 1992; Gallais 2009, pp. 68–71), like for crop yield, as the product of fruit weight and number, or plant height, as the product of internode number and length. In such cases, the heterosis in the multiplicative trait can appear even in the absence of dominance at the gene level. Oil palm populations can be organized in heterotic groups that show complementarity for these BN and BW. The cultivars are thus interpopulation hybrids, selected in a reciprocal recurrent selection (RRS) scheme. This approach has been applied since the 1950s (Gascon and de Berchoux 1964; Meunier and Gascon 1972), and many oil palm breeding programs now rely on it (Corley and Tinker 2016). Populations with a small number of big bunches constitute the group A. The major population in this group is the Deli population, which originated from four oil palms planted in Indonesia in 1848. The populations with a large number of small bunches constitute the group B. This comprised African populations from different countries. In particular, the La Mé population, which originated from a survey made in the 1920s in the Bingerville region of Côte d’Ivoire, has been extensively used in the breeding programs of several countries. Both Deli and La Mé populations have been the subject of several generations of selection and inbreeding (Corley and Tinker 2016; Soh et al. 2017a). In the current RRS scheme, 100 to 150 individuals belonging to group A and group B are evaluated in A × B hybrid progeny tests. Statistical analysis using a pedigree-based mixed model accurately estimates the general combining ability (GCA, i.e., half their breeding value in hybrid crosses) of the progeny-tested individuals (Soh et al. 2017b). This breeding scheme enables an estimated annual genetic progress of 1% (Durand-Gasselin et al. 2010).
The development of new breeding approaches, combining large-scale high-throughput genotyping and statistical methods able to take advantage of these large amounts of data, is expected to further increase annual genetic progress. For quantitative and complex traits such as BN and BW, the most promising approach is currently genomic selection (GS) (Meuwissen et al. 2001). GS uses a mixed model approach that gives the genomic estimated genetic value (GEBV) of selection candidates usually without phenotypic data records, but genotyped at high marker density. The prediction model is calibrated with the phenotypic data records and the genotypes of individuals that constitute the “training set.” Existing literature on genomic selection in oil palm (Wong and Bernardo 2008; Cros et al. 2015a, b, 2017; Marchal et al. 2016; Kwong et al. 2017) indicates it has potential advantages over the current phenotypic RRS, due to the ability of GS to provide GEBV for immature individuals (for instance, plantlets at the nursery stage). These GEBV can be used to make a preselection before progeny tests, thereby increasing selection intensity (Cros et al. 2017). They can also be used to make the final selection directly (i.e., avoiding progeny tests), which reduces the generation interval, as the sexual maturity of oil palm is reached at 3 years old, while the results of the progeny tests are obtained when the progeny-tested individuals are 13 to 15 years old. In addition, if the number of selection candidates is higher than the number of individuals that are usually progeny tested in conventional RRS, this increases selection intensity.
In a previous study (Cros et al. 2015a), our team compared RRS and different reciprocal recurrent genomic selection (RRGS) schemes. This was done over four cycles and, in RRGS, the phenotypic evaluations (i.e., progeny tests, used for both training the GS model and for selection) were made at varying frequencies, from only in the first cycle to in every cycle. It was concluded that the best option was RRGS with progeny tests every other cycle, which appeared as a good compromise between increased annual genetic gain compared to RRS, low risk around the expected gain, increase in inbreeding and cost. In addition, such a breeding scheme made it possible to train the GS model using aggregated data from several cycles. This was not investigated in our previous study, but we expect this could increase selection accuracy and annual genetic progress, as GS accuracy is positively correlated with the size of the training data set (Lorenz et al. 2011; Grattapaglia 2014).
The goal of the present oil palm breeding in silico study was to compare two methods to train the GS model, i.e., using aggregated data from the two most recent breeding cycles versus data from the single last breeding cycle. For this purpose, we adapted the simulation program of Cros et al. (2015a) and used two oil palm breeding populations simulated based on the actual genetic data of current Deli and La Mé populations. The comparison was made in terms of genetic gain for bunch production in Deli × La Mé hybrids, and selection accuracy and additive variance in parental populations.
Material and methods
Simulation overview
Based on the known history of actual Deli and La Mé populations, we simulated two oil palm breeding populations, with a simulation procedure calibrated so that the genetic parameters in the simulated populations were close to the actual values obtained in empirical datasets and the literature. As the true number of QTLs (quantitative trait loci) (nQTL) and the percentage of pleiotropic QTLs (pQTL) were not known, we considered a range of values for these two parameters. We simulated nQTL = 100, 500, and 1000 QTLs per trait and pQTL = 60, 75, and 90%. Six initial breeding populations were generated for each combination of nQTL and pQTL and, for each combination, the simulation was launched five times, starting with random Deli and La Mé individuals. This led to a total of 270 replicates.
Using these simulated populations, we compared RRGS schemes over four breeding cycles using a GS model trained using aggregated data from the two most recent breeding cycles (Tr2Gen) or only data from the last breeding cycle (Tr1Gen). In addition, two RRGS schemes were compared. First, we defined a 2PT-2noPT scheme that started with two cycles including progeny tests, used to calibrate the GS model that made it possible to select among the progeny-tested individuals, and, if any, their non-progeny-tested sibs. The 2PT-2noPT scheme then ended with two cycles with no progeny tests, i.e., with selection only based on markers. Second, the PT-noPT scheme alternated one cycle with progeny tests and one cycle with selection based only on markers (Fig. 1). Tr2Gen and Tr1Gen were compared at each cycle among the four cycles investigated. In particular, we distinguished between within-cycle GS and across-cycle GS. Traditional RRS was also simulated and used as a benchmark method. The aim of all the breeding schemes is to improve the hybrid performance of interpopulation crosses for bunch production.

Reciprocal recurrent genomic selection breeding schemes between Deli and La Mé oil palm populations investigated in this study
This study was conducted with R software version 3.2.5 (R Core Team 2016). The scripts were adapted from the ones used in Cros et al. (2015a), where detailed information on the simulation process can be found. All the modifications to the original scripts are explained in the following paragraphs.
Simulation of the initial breeding populations
The simulated genome had a length of 17 M and 16 chromosomes, corresponding to the actual values in oil palm (Billotte et al. 2005). Prior to the simulation of the initial Deli and La Mé populations, i.e., the individuals used as a starting point in this study (corresponding to the “parental generation 0” in Fig. 1 in Cros et al. (2015a)), an equilibrium base population was simulated. The QTLs controlling BW and BN were assigned in this base population, assuming additive architecture. The base population was then divided into two independent populations which gave, after generations of selection and drift with population specific parameters based on their known history (see details in Cros et al. (2015a)), the initial Deli and La Mé breeding populations. In particular, a different selection regime was applied to obtain divergent evolution: increasing BW to create the initial Deli population and increasing BN for La Mé. As a result, the initial Deli and La Mé populations differed in allele frequencies at QTLs and SNPs. The mutation rate was 10−5 per base pair per meiosis, with mutations generating new SNPs (i.e., no causative mutations). Haplotypes and meiosis were simulated with the Haplosim package in R (Coster and Bastiaansen 2010).
The initial Deli and La Mé populations were calibrated on the following parameters: Weir and Cockerham fixation index (Fst) and complementarity for BW and BN between the two populations, linkage disequilibrium (LD) profiles, narrow-sense heritabilities (h2) and additive variances for BW and BN, and genetic correlations between BW and BN. The mean values and standard deviations obtained for these genetic parameters in the different combinations of nQTL and pQTL are summarized in Supplementary Table 1. The target values used to calibrate the simulations were obtained from real data of the Deli and La Mé breeding populations used in the breeding program of PalmElit and its partners, and from the literature. The target values are given in Supplementary Table 1. The data, methods of computations, and associated references used to obtain these target values can be found in Cros et al. (2015a), except for Fst and additive variances for which better values were obtained with more recent datasets. Thus, the target Fst used in the present study was computed using the SNPs with no missing data in the Deli and La Mé individuals of Cros et al. (2017), with the R package Geneland (Guillot et al. 2005). The value obtained was 0.55, i.e., 12.2% higher than the value used in Cros et al. (2015a), which had been computed from SSR data. Also, the target interpopulation additive variances used here were mean values obtained from pedigree-based mixed model analyses made on two datasets involving Deli × La Mé hybrid progeny tests (Cros et al. 2015b, 2017). These estimates of additive variances were associated with the individuals that appeared as founders in the pedigrees used in the analysis, i.e., with the “generation -2” in Cros et al. (2015a). The simulation was therefore calibrated so that the additive variance in “generation -2” of the simulated populations matched the actual values obtained with the real datasets.
Breeding schemes
Three breeding schemes were simulated: conventional RRS, 2PT-2noPT RRGS, and PT-noPT RRGS.
For RRS and RRGS, the progeny tests involved 120 individuals per parental population, with a mean number of 2.25 hybrid crosses per parent and 40 hybrid individuals per cross. This led to a total of 10,800 hybrid individuals per progeny test (Fig. 1). We considered that a breeding cycle including a progeny test required 20 years. In all breeding cycles (i.e., regardless of the existence of progeny test, and, for RRGS, regardless of the number of selection candidates), the 18 best individuals were selected in each parental population. The following generation of individuals was obtained by mating, in each parental population, the selected individuals. Mating was performed according to an incomplete diallel design in which one sixth of the 182 possible crosses were randomly made (i.e., 54 full-sib families produced).
In RRS, the selection candidates in a given cycle were the individual progeny tested in this cycle, and progeny tests were conducted in each cycle.
With RRGS, it was possible to avoid progeny tests in some generations. The first cycle necessarily included a progeny test as the phenotypic data used to train the GS model were collected on the hybrid individuals. In the 2PT-2noPT scheme, the two first cycles included progeny tests, whereas the two last cycles did not. This made it possible to compare training on one generation versus two in cycles 2, 3, and 4. In cycle 2, all the selection candidates had full-sibs among the individual progeny tested in the same cycle (within-cycle GS). By contrast, cycles 3 and 4 represented across-cycle GS, where the selection candidates were descendants (i.e., direct progenies, grand-children, or great grand-children, depending on the cycle) of the individual progeny tested to train the GS model (Supplementary Table 2). In the PT-noPT scheme, progeny tests were conducted every other cycle (i.e., in cycles 1 and 3, while cycles 2 and 4 only relied on GS). This made it possible to measure the effect of training on one generation versus two in cycles 3 (within-cycle GS) and 4 (across-cycle GS). A breeding cycle without a progeny test requires 6 years. As a consequence, with the PT-noPT and 2PT-2noPT GS schemes studied here, it only takes 52 years to complete the four breeding cycles, versus 80 years with RRS (− 35%).
In addition, with RRGS, the set of selection candidates could differ from that in RRS, as GS allows selection among individuals that have not been progeny tested. Here, we considered nc = 120, 250, and 500 selection candidates per population and per breeding cycle. The set of selection candidates could then include only progeny-tested individuals (in cycles with progeny tests and nc = 120), or only individuals that were not progeny tested, or a mixture of the two (in cycles with progeny tests and nc > 120).
Models for prediction of breeding values
For computational reasons, we used univariate models rather than the bivariate models used in Cros et al. (2015a). We thus predicted the breeding values for BN and BW for one trait after another.
For RRS and RRGS, the mixed model used to predict the GCAs of the Deli and La Mé individuals took the form:
where Y is the vector of the phenotypes of the hybrid individuals, μ is the overall mean, 1 is a column vector of 1 s, aD and aL are the vectors of GCA of Deli and La Mé parents, respectively, ZD and ZL their incidence matrices (with 0 s and 1 s, to connect the phenotypes to the parents of the corresponding cross), and e is the vector of residual effects. The random genetic effects followed the model of Stuber and Cockerham (1966) for hybrid crosses, with aD ~ N(0, \( {\sigma}_{a_D}^2 \)× ΓD) and aL ~ N(0, \( {\sigma}_{a_L}^2 \)× ΓL). \( {\sigma}_{a_D}^2 \) and \( {\sigma}_{a_L}^2 \) are the additive variances associated with the Deli and La Mé breeding populations, respectively; and ΓD and ΓL are the matrices of known constants used to define the covariance among GCAs of the Deli and La Mé, respectively. In RRS, we used ΓD = 0.5AD and ΓL = 0.5AL, with AD and AL, the genealogical relationship matrices computed from the pedigree of the corresponding parental population, with elements 2fxy, where fxy is the coefficient of coancestry between individuals x and y. In RRGS, matrices of additive relationships AD and AL were replaced by molecular relationship matrices GD and GL computed from parental genotypes, using observed allele frequencies (VanRaden 2007; Habier et al. 2007). This corresponded to the RRGS_PAR method described in Cros et al. (2015a). The errors e followed N(0, \( {\sigma}_e^2 \)× I), where \( {\sigma}_e^2 \) is the residual variance and I is the identity matrix. For RRGS, when training included two breeding cycles, a supplementary fixed effect related to the breeding cycle was included.
The genomic matrices GD and GL were computed with 2500 random non-causal SNPs with minor allele frequency (MAF) ≥ 5%. MAFs were computed separately for the two parental populations.
Variance parameters were estimated by restricted maximum likelihood (REML) and the solutions of the mixed models were obtained by resolving Henderson’s mixed model equations (Henderson 1975) using R-ASReml version 3.0 (Gilmour et al. 2009).
Analysis of results
For a given cycle (n), the genetic gain was defined as the difference between bunch production (BN × BW) by the hybrids between the progenies of the Deli and La Mé individuals selected at the end of the cycle (bn + 1) and bunch production by the hybrids between the Deli and La Mé individuals used as selection candidates at the beginning of the cycle (bn). This per cycle genetic gain was expressed as the percentage of bunch production by hybrid crosses at the beginning of the cycle (100 × (bn + 1 − bn)/bn). At the end of cycle 4, we also measured the cumulative genetic gain, which is expressed as the percentage of hybrid production in the initial generation (100 × (b4 − b0)/b0). The risk concerning the genetic gain (i.e., the variation in genetic gain among replicates in a given breeding scheme) was quantified by the coefficient of variation (CV) of the genetic gain per cycle of the 270 replicates. The annual genetic gain was computed as the genetic gain obtained after four breeding cycles divided by the number of years required to carry out the four cycles.
Selection accuracy was computed for BN and BW traits in the two parental populations as the Pearson correlation between the true and estimated GCAs. The additive variances were defined according to the quantitative genetic model of Falconer and Mackay (1996). The mean additive relationship between the training individuals and the selection candidates was computed from the pedigrees.
Two-tailed paired sample Wilcoxon tests were used to compare the Tr2Gen and Tr1Gen effect on four parameters: genetic gain after four cycles, genetic gain per cycle, additive variances, and the relationship between training parents and selection candidates. For selection accuracies, the comparison was made using paired t tests after Fisher’s Z transformation. An analysis of variance was performed to compare the annual genetic gain of RRS and of the PT-noPT/Tr2Gen GS breeding schemes, with multiple comparisons of breeding schemes using Tukey’s test.
Results
Genetic gain per cycle
RRGS with two-cycle training sets (Tr2Gen) performed better than RRGS with single-cycle training sets (Tr1Gen) in almost every cycle, and this was significant in 80% of the situations (Fig. 2). In the generations in which Tr1Gen and Tr2Gen were compared, the genetic gain per cycle with Tr2Gen was on average 10.7% higher than with Tr1Gen (increase ranging from 3.4 to 34.9%), with a mean genetic gain per cycle of 3.6% with Tr2Gen, versus 3.3% with Tr1Gen. In the case of within-cycle selection (generation 2 of 2PT-2noPT and generation 3 of PT-noPT), the genetic gain increased by an average of 6.9% (range 4.5 to 11.2%) with Tr2Gen and was always significant. The increase obtained with 120 candidates indicated that Tr2Gen was advantageous for the evaluation of progeny-tested individuals (i.e., the training parents). In the case of across-cycle selection, bigger increases in genetic gain per cycle were achieved, as it was on average 13.3% higher (range 3.4 to 34.9%, although not always significant). Tr2Gen was therefore also advantageous for selection among non-progeny-tested candidates.

Genetic gain per breeding cycle according to the number of generations used to train the GS model (Tr1Gen: one, Tr2Gen: two), breeding scheme (PT-noPT: GS with progeny tests every two generations, 2PT-2noPT: GS with two generations using progeny tests and two generations with no progeny tests), and the number of selection candidates. Genetic gain is expressed as a percentage of hybrid production in the previous generation. Values are means of 270 replicates. Significance of two-tailed paired sample Wilcoxon tests: *0.05 > P ≥ 0.01; **0.01 > P ≥ 0.001; ***P < 0.001; ns not significant
Another desirable feature of Tr2Gen over Tr1Gen was its ability to reduce the risk concerning genetic gain. Indeed, Tr2Gen reduced the CV of genetic gain per cycle (Supplementary Fig. S. 1). The CV in the generations in which Tr1Gen and Tr2Gen were compared was on average 66.1% with Tr2Gen, versus 75.3% with Tr1Gen, leading to a − 11.0% decrease (− 4.5 to − 31.1%).
Genetic gain after four cycles and annual genetic gain
Tr2Gen increased the genetic gain obtained after four cycles in the two breeding schemes 2PT-2noPT and PT-PT (Fig. 3). Tr1Gen led to an average genetic gain of 16.6 versus 17.4% with Tr2Gen. This corresponded to a 5.0% increase, ranging from 2.6 to 8.4%, depending on the breeding scheme and on the number of candidates, always highly significant (P < 0.001).

Genetic gain after four cycles according to the number of generations used to train the GS model (Tr1Gen: one, Tr2Gen: two), breeding scheme (PT-noPT: GS with progeny tests every two generations, 2PT-2noPT: GS with two generations using progeny tests and two generations with no progeny tests), and number of selection candidates. Annual genetic gain is expressed as a percentage of hybrid production in the initial generation (generation 0, i.e., the breeding population used as starting point for cycle 1) per year. Values are means of 270 replicates. Significance of paired t tests: ***P < 0.001
The genetic gain was significantly higher with PT-noPT than with 2PT-2noPT (P < 0.001). The genetic gain with PT-noPT was 5.7% higher than with 2PT-2noPT when Tr2Gen was used, and 9.7% higher when Tr1Gen was used. As expected, the genetic gain increased with the number of selection candidates.
The genetic gain of RRS after four cycles was 18.6%, which is similar to the highest genetic gain obtained with RRGS. However, as the number of years required to complete the four RRGS cycles was 35% lower than with RRS, the annual genetic gain of the GS schemes was finally much higher than with RRS, for all numbers of candidates, numbers of generations in the training set, and breeding schemes. PT-noPT with Tr2Gen, which was the best breeding scheme, enabled an annual genetic gain ranging from 37.6 to 57.5% over RRS, depending on the number of selection candidates used in GS (Table 1).
Selection accuracy
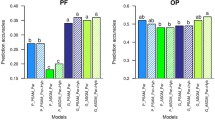
Two-cycle training sets increased selection accuracy for both BN and BW traits in Deli and La Mé parental populations, with an average increase of 4.9%, ranging from − 0.4 to 13.8%, depending on the cycle, trait, population, number of candidates, and breeding scheme (see Fig. 4 for the example of BW in Deli and Supplementary Fig. S. 2, Supplementary Fig. S. 3, and Supplementary Fig. S. 4 for the other results). In the case of within-cycle selection, accuracy increased by an average of 2.0% (range − 0.4 to 4.8%, with mean decreases observed for BN with 120 candidates in PT-noPT). The effect of the number of training generations on the selection accuracy of the progeny-tested individuals could be evaluated with 120 candidates in generation 2 of 2PT-2noPT and in generation 3 of PT-noPT. This indicated that, although the selection accuracy of progeny-tested individuals was already very high (> 0.9), on average, Tr2Gen further increased this value, with a mean increase of 0.49% (although it was not always better than Tr1Gen, as it ranged from − 0.36 to + 1.52%). For the non-progeny-tested selection candidates, Tr2Gen also increased selection accuracy compared to Tr1Gen, but with higher magnitude than for progeny-tested individuals. The increase was thus significant and, in across-cycle selection, reached 6.7% on average (range 3.0 to 14.3%), with the maximum value obtained when selection was applied two generations after training.

Selection accuracy for BW in Deli, according to the number of generations used to train the GS model (Tr1Gen: one, Tr2Gen: two), breeding scheme (PT-noPT: GS with progeny tests every two generations, 2PT-2noPT: GS with two generations using progeny tests and two generations with no progeny tests), and the number of selection candidates. Values are means of 270 replicates. Significance of paired t tests: ***P < 0.001; ns not significant
Additive variances
Two-cycle training sets also slowed down the decrease in additive variance over cycles for both BN and BW traits in Deli and La Mé parental populations (data not shown). However, the extra additive variance with Tr2Gen was small; on average, only 1.6% of the additive variance with Tr1Gen (ranging from − 0.5 to 5.6%) and Tr2Gen yielded a significantly higher additive variance in only about 50% of the situations observed (i.e., combinations of cycle, number of candidates, trait, parental population, and breeding scheme).
Relationship between training parents and selection candidates
The Tr2Gen strategy decreased the mean additive relationship between the training individuals and the selection candidates compared to Tr1Gen. In the Deli population, the decrease was on average 10.8% (range 5.7 to 17.7%, depending on the breeding scheme, cycle, and number of candidates). In the La Mé population, the mean decrease reached 26.6% (range 18.0 to 36.1%) (see Supplementary Table 3 for details).
Discussion
When selecting among Deli and La Mé parental populations for hybrid performance regarding bunch production, training the GS model with data aggregated from the two most recent breeding cycles (Tr2Gen) led to an average genetic gain per cycle 10.7% higher compared to training using only the single most recent cycle (Tr1Gen). This was the result of an increase in selection accuracy and, to a lesser extent, to a slower decrease in additive variance over cycles. The highest increases in genetic gain per cycle and in selection accuracy were obtained in across-cycle selection, although Tr2Gen was also advantageous for within-cycle selection, and even for progeny-tested individuals. After four cycles, Tr2Gen had a cumulative genetic gain on average 5% higher than Tr1Gen, and a lower risk concerning the genetic gain. In addition, alternating one cycle with progeny tests with one cycle with only GS (PT-noPT breeding scheme) was a more efficient way to benefit from the advantages offered by Tr2Gen, compared to alternating two cycles of progeny tests and two cycles of GS alone (2PT-2noPT). Finally, over the four breeding cycles, combining the PT-noPT scheme and the Tr2Gen training method led to a large increase in annual genetic gain, ranging from 37.6 to 57.5%, compared to RRS, depending on the number of selection candidates used in GS.
Our results confirmed the simulation study by Denis and Bouvet (2013) in eucalyptus and the empirical results obtained by Auinger et al. (2016) in rye, which also showed that accumulating data over cycles to train the GS model was beneficial. Although our increase in accuracy could be considered weak in comparison to doubling the size of the training set, Auinger et al. (2016) obtained a similar result. Thus, they reported that Tr2Gen increased across-cycle GS accuracy by 5 to 20%, depending on the trait, which is comparable with our 4.9% increase. However, in the simulation scenarios of Denis and Bouvet (2013) that were close to our study (i.e., their scenarios with lowest dominance to additive variance ratios (0.1)), much higher increases in GS accuracy were noted when using two-cycle training sets. Although the GS accuracy they obtained using a one-cycle training set to predict the breeding values in the following generation was close to ours (approximately 0.45 with H2 = 0.1 and 0.70 for H2 = 0.6, versus 0.70 in our study), with two-cycle training sets, GS accuracy increased by 60% for H2 = 0.1 and 25% with H2 = 0.6, versus only 4.9% here. There are three possible explanations for this discrepancy. First, the simulated initial generation of Denis and Bouvet (2013) had an effective size (Ne) of 100. By contrast, Ne was small in our study (< 10 in the oil palm populations used to calibrate our simulations (Cros et al. 2014, 2015b)) and in Auinger et al. (2016). As a result of these low Ne, the size of the training sets in Tr1Gen here and in Auinger et al. (2016) (208 lines) might have been close to their optimum, thus limiting the impact of doubling the training size. Indeed, in a canola population with Ne ≤ 11, Jan et al. (2016) showed that GS accuracy plateaued for almost all traits with 333 lines in the training set. In a maize population with small Ne, GS accuracy increased by only 20% when the training set was doubled from 172 to 344 lines and increased even less (7%) when the training set was again doubled (Albrecht et al. 2011). Second, Denis and Bouvet (2013) used the least recent cycle for training in Tr1Gen, while we used the most recent. In their study with Tr1Gen, there were therefore two generations between the training individuals and the selection candidates, versus only one in our study. This reduced the accuracy of Tr1Gen in their study compared to ours, and thus led to a relatively bigger advantage of Tr2Gen over Tr1Gen in Denis and Bouvet (2013) than in our study. Third, they showed that the higher dominance to additive variance ratio, the greater the benefit of using Tr2Gen. Thus, their simulation was more advantageous to Tr2Gen than was our simulation, where no dominance effects were considered.
The relatively low increase in GS accuracy obtained in the present study with Tr2Gen compared to Tr1Gen also resulted from the fact that aggregating data from two breeding cycles decreased the relationship between the training individuals and the selection candidates, which is detrimental to GS accuracy (see, for example, Pszczola et al. 2012; Daetwyler et al. 2013; Gowda et al. 2014; Lorenz and Smith 2015). The decrease in the relationship was expected from the composition of the training sets (Supplementary Table 2). The pattern of change over cycles in the mean relationship between the training individuals and the selection candidates resulted from the effect of selection and depended on selection intensity and selection accuracy, thus producing contrasting results depending on the selection method (GS or phenotypic selection) and on the number of candidates. Although breeding cycles in oil palm require many years when progeny tests are implemented, in the long-term, it will be possible to aggregate data from more than two cycles. This is of interest as the more cycles, the larger the size of the training set, which benefits GS accuracy. However, as we observed here, each time a new cycle is added, the oldest cycles become less related to the new selection candidates. Therefore, we can question the extent to which the oldest cycles remain useful in the training set, or if they may become detrimental to GS accuracy. However, the results of Neyhart et al. (2017) with rye simulated data suggest that this is not of concern in oil palm, nor in perennial crops in general. Indeed, they showed that aggregating even as many as 15 generations in the training set only decreased accuracy in a negligible way (0.02–0.04) compared to when aggregating only the most recent generations. For species with long breeding cycles where it will only be possible to aggregate a few generations in the training set, using all the available data is therefore reasonable, and we recommend that oil palm breeders use all data from past cycles to train the GS models.
We expect that the interest of cumulating data from several breeding cycles when implementing GS in this species will vary according to the trait. Indeed, Denis and Bouvet (2013) showed that low h2 and high proportion of dominance variance in total genetic variance increased the relative interest of cumulating data in the training set. Here, we focused on BN and BW, with a simulated h2 of around 0.4, but, for instance, the fruit to bunch ratio, another major component of oil palm yield, has a mean h2 of around 0.2 (Corley and Tinker 2016, p. 180). In addition, the proportion of dominance variance between crosses in total genetic variance between crosses, although generally low, is actually significant for some traits, with a value as high as 30% for the fruit to bunch ratio (Cros et al. 2017). Cumulating data from several cycles is therefore expected to generate a greater increase in genetic gain per cycle for fruit to bunch ratio. This would be of great interest, because for this trait, GS so far fails to reach better accuracy in non-progeny-tested individuals than a control PBLUP prediction model (where the genomic relationship matrices used in the mixed model are replaced by genealogical coancestries) (Cros et al. 2017), while Auinger et al. (2016) noted that cumulating data from several cycles had a negligible effect on PBLUP accuracy.
In the present study, the individuals of the parental populations that were genotyped and made up the training set were not phenotyped directly, as phenotypic data were collected on their hybrid progenies. Based on the experimental designs generally used in oil palm, this results in large datasets of phenotypic data with tens of thousands of hybrid individuals phenotyped. Thus, with progeny tests, in each generation, we disposed here of the phenotype of 10,800 hybrid individuals. Aggregating data from several generations therefore multiplies the size of the dataset, which slows down the mixed model analysis. In our study, using a 64-bit Linux on a 6-core Intel Xeon W3690 at 3.47 GHz machine with 24 Gb RAM, Tr2Gen increased the computation time required to run the mixed model analysis (time cumulated for the two traits) by 36 to 49%. This also increased computer memory requirements. This is problematic in a simulation study like ours, where the analyses are conducted many times due to the numerous replicates considered. However, in our study, the mean mixed model computation time cumulated for the two traits for GBLUP with Tr2Gen and 500 selection candidates was only 18 s, and therefore, cumulating data in the training set will not be a problem in practical breeding where the analyses are conducted a limited number of times.
Conclusion
When selecting among Deli and La Mé oil palm parental populations for hybrid performance in bunch production, aggregating data from the two most recent breeding cycles to train the GS model increased the selection response per cycle (+ 10.7%), mostly under the effect of increased selection accuracy (+ 4.9%), and despite a decrease in the relationship between the training individuals and the selection candidates. This method also reduced the risk concerning the expected genetic gain, another desirable feature for breeders. This study confirms the advantage of GS over conventional RRS, and we thus recommend that when making genomic predictions, oil palm breeders include all available data from past cycles in their training set.
References
Albrecht T, Wimmer V, Auinger H-J, Erbe M, Knaak C, Ouzunova M, Simianer H, Schön CC (2011) Genome-based prediction of testcross values in maize. Theor Appl Genet 123:339–350
Auinger H-J, Schönleben M, Lehermeier C, Schmidt M, Korzun V, Geiger HH, Piepho HP, Gordillo A, Wilde P, Bauer E, Schön CC (2016) Model training across multiple breeding cycles significantly improves genomic prediction accuracy in rye (Secale cereale L.). Theor Appl Genet 129:2043–2053. https://doi.org/10.1007/s00122-016-2756-5
Billotte N, Marseillac N, Risterucci A-M, Adon B, Brottier P, Baurens FC, Singh R, Herran A, Asmady H, Billot C, Amblard P, Durand-Gasselin T, Courtois B, Asmono D, Cheah SC, Rohde W, Ritter E, Charrier A (2005) Microsatellite-based high density linkage map in oil palm (Elaeis guineensis Jacq.). Theor Appl Genet 110:754–765. https://doi.org/10.1007/s00122-004-1901-8
Corley R, Tinker P (2016) Selection and breeding. In: The oil palm, 5th edn. Wiley-Blackwell, Chichester, UK, p. 138–207
Coster A, Bastiaansen J (2010) HaploSim: R package version 1.8.4. http://CRAN.R-project.org/package=HaploSim
Cros D, Sánchez L, Cochard B, Samper P, Denis M, Bouvet JM, Fernández J (2014) Estimation of genealogical coancestry in plant species using a pedigree reconstruction algorithm and application to an oil palm breeding population. Theor Appl Genet 127:981–994. https://doi.org/10.1007/s00122-014-2273-3
Cros D, Denis M, Bouvet J-M, Sanchez L (2015a) Long-term genomic selection for heterosis without dominance in multiplicative traits: case study of bunch production in oil palm. BMC Genomics 16:651
Cros D, Denis M, Sánchez L, Cochard B, Flori A, Durand-Gasselin T, Nouy B, Omoré A, Pomiès V, Riou V, Suryana E, Bouvet JM (2015b) Genomic selection prediction accuracy in a perennial crop: case study of oil palm (Elaeis guineensis Jacq.). Theor Appl Genet 128:397–410. https://doi.org/10.1007/s00122-014-2439-z
Cros D, Bocs S, Riou V, Ortega-Abboud E, Tisné S, Argout X, Pomiès V, Nodichao L, Lubis Z, Cochard B, Durand-Gasselin T (2017) Genomic preselection with genotyping-by-sequencing increases performance of commercial oil palm hybrid crosses. BMC Genomics 18:839. https://doi.org/10.1186/s12864-017-4179-3
Daetwyler HD, Calus MPL, Pong-Wong R, de los Campos G, Hickey JM (2013) Genomic prediction in animals and plants: simulation of data, validation, reporting, and benchmarking. Genetics 193:347–365. https://doi.org/10.1534/genetics.112.147983
Denis M, Bouvet J-M (2013) Efficiency of genomic selection with models including dominance effect in the context of Eucalyptus breeding. Tree Genet Genomes 9:37–51. https://doi.org/10.1007/s11295-012-0528-1
Durand-Gasselin T, Blangy L, Picasso C, de Franqueville H, Breton F, Amblard P, Cochard B, Louise C, Nouy B (2010) Sélection du palmier à huile pour une huile de palme durable et responsabilité sociale. OCL 17:385–392
Falconer D, Mackay T (1996) Introduction to quantitative genetics, 4th edn. Longman, Harlow, 464 p
Gallais A (2009) Hétérosis et variétés hybrides en amélioration des plantes. Quae éditions, Versailles, France, 376 p
Gascon JP, de Berchoux C (1964) Caractéristique de la production d’Elaeis guineensis (Jacq.) de diverses origines et de leurs croisements—application à la sélection du palmier à huile. Oléagineux 19:75–84
Gascon JP, Noiret JM, Bénard G (1966) Contribution à l’étude de l’hérédité de la production de régimes d’Elaeis guineensis Jacq.—application à la sélection du palmier à huile. Oléagineux 21:657–661
Gilmour AR, Gogel BJ, Cullis BR, Thompson R (2009) ASReml user guide release 3.0, Queensland Department of Primary Industries and Fisheries, Australia, 148 p
Gowda M, Zhao Y, Wurschum T et al (2014) Relatedness severely impacts accuracy of marker-assisted selection for disease resistance in hybrid wheat. Heredity 112:552–561
Grattapaglia D (2014) Breeding forest trees by genomic selection: current progress and the way forward. In: Genomics of plant genetic resources, Springer Netherlands. Tuberosa R, Graner A, Frison E, p. 651–682
Guillot G, Mortier F, Estoup A (2005) Geneland: a computer package for landscape genetics. Mol Ecol Notes 5:712–715. https://doi.org/10.1111/j.1471-8286.2005.01031.x
Habier D, Fernando RL, Dekkers JCM (2007) The impact of genetic relationship information on genome-assisted breeding values. Genetics 177:2389–2397. https://doi.org/10.1534/genetics.107.081190
Henderson CR (1975) Best linear unbiased estimation and prediction under a selection model. Biometrics 31:423–447
Jan HU, Abbadi A, Lücke S, Nichols RA, Snowdon RJ (2016) Genomic prediction of testcross performance in canola (Brassica napus). PLoS One 11:e0147769. https://doi.org/10.1371/journal.pone.0147769
Kwong QB, Ong AL, Teh CK, Chew FT, Tammi M, Mayes S, Kulaveerasingam H, Yeoh SH, Harikrishna JA, Appleton DR (2017) Genomic selection in commercial perennial crops: applicability and improvement in oil palm (Elaeis guineensis Jacq.). Sci Rep 7:2872. https://doi.org/10.1038/s41598-017-02602-6
Lorenz AJ, Smith KP (2015) Adding genetically distant individuals to training populations reduces genomic prediction accuracy in barley. Crop Sci 55:2657–2667. https://doi.org/10.2135/cropsci2014.12.0827
Lorenz AJ, Chao S, Asoro FG, et al (2011) Genomic selection in plant breeding: knowledge and prospects. In: Donald L. Sparks (ed) Advances in agronomy. Academic Press, , p. 77–123
Marchal A, Legarra A, Tisné S, Carasco-Lacombe C, Manez A, Suryana E, Omoré A, Nouy B, Durand-Gasselin T, Sánchez L, Bouvet JM, Cros D (2016) Multivariate genomic model improves analysis of oil palm (Elaeis guineensis Jacq.) progeny tests. Mol Breed 36:1–13. https://doi.org/10.1007/s11032-015-0423-1
Meunier J, Gascon J (1972) Le schéma général d’amélioration du palmier à huile à l’IRHO. Oléagineux 27:1–12
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Neyhart JL, Tiede T, Lorenz AJ, Smith KP (2017) Evaluating methods of updating training data in long-term genomewide selection. G3 GenesGenomesGenetics 7:1499–1510. https://doi.org/10.1534/g3.117.040550
Pszczola M, Strabel T, Mulder HA, Calus MPL (2012) Reliability of direct genomic values for animals with different relationships within and to the reference population. J Dairy Sci 95:389–400. https://doi.org/10.3168/jds.2011-4338
R Core Team (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org
Schnell FW, Cockerham CC (1992) Multiplicative vs. arbitrary gene action in heterosis. Genetics 131:461–469
Soh AC, Mayes S, Roberts JA (2017a) Oil palm breeding: genetics and genomics. CRC Press, Boca Raton 446 p
Soh AC, Mayes S, Roberts JA et al (2017b) Breeding plans and selection methods. In: Soh AC, Mayes S, Roberts JA (eds) Oil palm breeding: genetics and genomics. CRC Press, Boca Raton, pp 143–163
Stuber CW, Cockerham CC (1966) Gene effects and variances in hybrid populations. Genetics 54:1279–1286
USDA (2017) Oilseeds: world market and trade. Foreign Agricultural Service, Circular Series May 2017. https://apps.fas.usda.gov/psdonline/circulars/oilseeds.pdf
VanRaden PM (2007) Genomic measures of relationship and inbreeding. Interbull Bull 37:33–36
Wong CK, Bernardo R (2008) Genomewide selection in oil palm: increasing selection gain per unit time and cost with small populations. Theor Appl Genet 116:815–824. https://doi.org/10.1007/s00122-008-0715-5
Acknowledgments
We thank two anonymous reviewers for their helpful comments.
Funding
This work was partly funded by a grant from PalmElit SAS. It was also supported by the CIRAD-UMR AGAP HPC Data Center of the South Green Bioinformatics platform (http://www.southgreen.fr/) and by the CETIC (African Center of Excellence in Information and Communication Technologies).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cros, D., Tchounke, B. & Nkague-Nkamba, L. Training genomic selection models across several breeding cycles increases genetic gain in oil palm in silico study. Mol Breeding 38, 89 (2018). https://doi.org/10.1007/s11032-018-0850-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-018-0850-x