
Abstract
Increasing the rice productivity from the current 10 to 12 tons/ha to meet the demand of estimated 8.8 billion people in 2035 is posing a major challenge. Wild relatives of rice contain some novel genes which can help in improving rice yield. Spikelet per panicle (SPP) is a valuable trait for determining yield potential in rice. In this study, a major QTL for increasing SPP has been identified, mapped, and transferred from African wild rice O. longistaminata to O. sativa (L.). The QTL was mapped on the long arm of chromosome 2 in a 167.1 kb region flanked by SSR markers RM13743 and RM13750, which are 1.0 cM apart, and is designated as qSPP2.2. The QTL explained up to 30% of phenotypic variance in different generations/seasons and showed positive additive effect of allele contributed by O. longistaminata. In addition, O. longistaminata allele in qSPP2.2 contributed to increase in grains per panicle, but decrease in the tillers per plant. The 167.1 kb region contains 23 predicted genes. Based on the functional annotation, three genes, LOC_Os02g44860, LOC_Os02g44990, and LOC_Os02g45010, were selected as putative candidates for characterization. Sequence analysis of the three genes revealed functional variations between the parental lines for LOC_Os02g44990 and a variation in 5′UTR for LOC_Os02g45010 which will help further to identify putative candidate gene(s). This is the first yield component QTL to be identified, mapped, and transferred from O. longistaminata.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Global rice demand is estimated to rise from 723 million tons in 2015 to 763 million tons in 2020 and further increase to 852 million tons in 2035, an overall increase of 129 million tons in the next 20 years (Fischer and Edmeades 2010; Khush 2013). To produce 129 million tons of additional paddy rice by 2035, an increase in the yield potential of rice from the present ~ 10.0 tons/ha to more than 12.0 tons/ha is required. However, increasing competition for land, water, and energy has made it an uphill task to achieve this target in the next 20 years. Compounding the problem is the current practice of crossing elite lines which is expected to reduce genetic variability in the working germplasm, thus preventing discovery of novel traits to improve yield. One of the alternatives for increasing agriculture production is to identify subset of genes lost during the domestication process and use them in subsequent targeted breeding (Shakiba and Eizenga 2014).
The genus Oryza has 22 wild species, which contain numerous valuable genes/traits (Brar and Singh 2011; Shakiba and Eizenga 2014) for improving rice yield. This was first demonstrated by Khush et al. (1977) by introgressing a gene for resistance to grassy stunt virus from O. nivara to cultivated rice. Since then, wild species have been widely used for introgression of several agronomically important traits such as tolerance to biotic and abiotic stresses, diversification of cytoplasmic male sterility sources, and yield enhancing components (Dalmacio et al. 1995, 1996; Shakiba and Eizenga 2014; Bhatia et al. 2017). The exemplary study to map yield enhancing QTLs yld1.1 and yld2.1 from O. rufipogon (Xiao et al. 1998) gave incremental emphasis to use wild relatives of rice for increasing the rice productivity. Since then, QTLs for several yield component traits have been identified and mapped in O. rufipogon (McCouch et al. 2007; Imai et al. 2013), O. nivara (Ma et al. 2016), O. glumaepatula (Brondani et al. 2002), O. grandiglumis (Yoon et al. 2006), and O. minuta (Balkunde et al. 2013). In addition to yield components, heterotic loci associated with yield potential have also been introgressed from wild Oryza species (Luo et al. 2011; Gaikwad et al. 2014).
The grain yield of rice can be divided into four major components: productive tiller number per plant, spikelet per panicle, percent spikelet fertility, and grain weight. Among these, spikelet number per panicle (SPP) is the most important trait contributing directly to rice yield. Several QTLs for SPP in rice have been identified from wild relatives of rice such as O. nivara (Onishi et al. 2007), O. glumaepatula (Brondani et al. 2002), O. rufipogon, O. minuta, and O. grandiglumis (Shakiba and Eizenga 2014). Besides, several genes for grain number (fertile spikelets per panicle) have been cloned that offers potential for breeding rice with high yield (Ikeda et al. 2013; Huo et al. 2017).
O. longistaminata (A. Chev. et Roehr.), the most widely distributed Oryza species in the African continent, exhibits high levels of genetic diversity in different populations (Melaku et al. 2013) and has considerably diverged from O. sativa and other “AA” genome species (Wambugu et al. 2013). This genetically diverse species has served as a source of economically important traits such as disease resistance (Song et al. 1995), perenniality-associated genes Rhz2, Rhz3 (Hu et al. 2003), and drought tolerance (Liu et al. 2004). Despite harboring tremendous useful variability, O. longistaminata has not been used for transfer of yield and yield component traits into cultivated rice.
Most of the complex quantitative traits present in the wild species are cryptic and need to be introgressed into established lines for further mapping. Advanced mapping populations like backcross inbred lines (BILs) are more suitable in this case than recombinant inbred lines (RILs) and double haploids (DHs) due to associated problems of wild weedy traits (Jacquemin et al. 2013). Once a desirable BIL has been selected after screening, mapping populations like advanced alien introgression lines (AILs) or near isogenic lines (NILs) can be developed for mapping/cloning of the trait. The AILs (usually referred as alien introgression lines), also sometimes referred as ILs, are the result of introgressing small chromosomal segments from the donor into the recurrent parent by consecutive backcrossing and repeated selfing to identify the chromosomal regions introgressed from the donor parent (Ali et al. 2010). Realizing the importance of wild species for increasing the yield potential of cultivated rice, a total of 1780 alien introgression lines referred as BILs were generated by crossing 70 accessions of six “AA” genome species: O. glaberrima Steud., O. barthii A. Chev., O. nivara Sharma & Shastry, O. rufipogon Griff., O. longistaminata A. Chev. & Roehr., and O. glumaepatula Steud. with PR114 and Pusa44, two elite cultivars of Oryza sativa L. (Bhatia et al. 2017). All the BILs showed significant variation for yield and yield component traits based on evaluation in three different seasons. In the present study, one of the BIL possessing significantly higher spikelet per panicle derived from O. longistaminata was used to generate advanced AILs by crossing with recurrent parent PR114. Here, we report the mapping and transfer of a 167.1 kb QTL region for SPP from O. longistaminata to O. sativa and identification of the underlying putative candidate genes of SPP in the QTL region.
Materials and methods
Population development and phenotyping
At Punjab Agricultural University (PAU), India, a number of alien introgression lines possessing variation for yield component traits have been generated utilizing six different “AA” genome Oryza species in the background of two different recurrent parents O. sativa cvs. PR114, Pusa44 (Bhatia et al. 2017). One of the introgression lines (IL1792) in BC2F6 derived from crossing O. longistaminata IRGC104301 and PR114 showed significantly higher spikelet per panicle (mean value of 183.1) as compared to PR114 (mean value of 143.1). This BC2F6 introgression line (IL1792) was backcrossed to PR114 to generate BC3F1 and selfed to generate BC3F2. The BC3F2 was advanced by continuous selfing to develop advanced alien introgression lines (AILs).
A total of 474 advanced AILs were phenotyped in BC3F3 in year 2011 and BC3F6 to BC3F8 in years 2013–2015. The advanced AILs were evaluated in a randomized block design (RBD) in paired rows with 10 plants/row in two replications. The plant-to-plant and row-to-row distance was 15 cm and 20 cm, respectively, in 0.75-m2 plot size. The introgression line IL1792, recurrent parent PR114, and three commercially recommended varieties PAU201, PR121, and PR122 were included as standard checks. Data for various agronomic traits namely days to 50% flowering (DF), plant height (PH), tillers per plant (TN), panicle size (PS), spikelets per panicle (SPP), fertile grains per panicle (GP), sterile spikelets per panicle (SSPP), spikelet fertility (SF), 1000-grain weight (TGW), and plot yield (PY) in both the replications were recorded each year from five randomly chosen plants, excluding the border plants. Data for primary branches per panicle were recorded only in 2013. Data for year 2014 was not used for QTL mapping due to poor crop stand because of adverse climatic conditions. Data for 2013 and 2015 are presented in Table S1.
Statistical analysis
Analysis of variance (ANOVA) for RBD for various agronomic traits was carried out using the software CPCS-I (Cheema and Singh 1990) and Pearson’s correlation analysis was performed using SigmaPlot 11.0.
DNA extraction and marker analysis
DNA was isolated from fresh leaves of the recurrent parent (PR114), the introgression line (IL1792), the donor parent (O. longistaminata acc. IRGC104301), and a bulk of 10 plants of advanced AILs following the standard CTAB method (Saghai-Maroof et al. 1984). Simple sequence repeat (SSR) markers (Temnykh et al. 2001; McCouch et al. 2002) were used for parental polymorphism and genotyping the mapping population. PCR was performed with a total reaction volume of 20 μl using the following conditions: initial denaturation at 94 °C for 4 min, followed by 35 cycles of 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min, and final product extension at 72 °C for 7 min. PCR products were separated in a 2.5% agarose gel and visualized using Gel Documentation System (UVP Imaging System). For parental polymorphism and determining the introgressions from O. longistaminata, SSR markers chosen randomly at 5–10 cM intervals were applied on PR114, IL1792, and O. longistaminata acc. IRGC104301. Graphical genotype for the introgression line IL1792 was generated to determine the introgressed segments from wild species using the software GGT version 2.0 (Berloo 2008). Markers showing polymorphism between PR114 and IL1792 were used for genotyping advanced AILs and identification of QTLs.
Linkage map construction and QTL analysis
Segregation ratios for each of the marker loci were performed using a Chi-square test, with significant deviations from expected ratios reported at p ˂ 0.01. A linkage map was constructed using software package MAPMAKER/EXP version 3.0 (Lander et al. 1987). All the markers were allocated to linkage groups by pairwise analysis with a threshold LOD score of 3.0, recombination fraction of 0.4, and “Kosambi” mapping function. The WinQTL Cartographer version 2.5 (Wang et al. 2010) was used to map QTLs using composite interval mapping. The experiment wise threshold of composite interval mapping was obtained by doing 1000 permutations at p < 0.01. Markers used as cofactors in the CIM model were selected by forward and backward regression analysis using a significance level of p < 0.05 and a window size of 10 cM. The proportion of observed phenotypic variance attributable to a particular QTL was estimated by the coefficient of determination (R2) using maximum likelihood for composite interval mapping. Initially, advanced AILs in BC3F3 generation were used to identify QTLs, which were later confirmed and narrowed down using data of BC3F6 and BC3F8 generations.
Identification of candidate gene(s) in the QTL region for SPP
The QTL region for SPP bracketed by the flanking markers was analyzed in the rice genome annotation project (RGAP) database (http://rice.plantbiology.msu.edu/cgi-bin/gbrowse/rice/) and InterPro (https://www.ebi.ac.uk/interpro/) to identify putative candidate genes. Primers were designed using the software Perl Primer (http://perlprimer.sourceforge.net/) to amplify the entire putative candidate genes. The primer pairs were designed to amplify a 1 kb region with 200 bp overlap with other primer pair. The amplicons were purified using QIAquick® Gel extraction kit (QIAGEN India Pvt. Ltd.) as per manufacturer’s protocol, sequenced using ABI Bigdye Terminator v3.1 chemistry and run on ABI Sequencer 3730XL.
The amplicon sequences were then extracted from chromatograms using CHROMAS Lite 2.1.1 (http://technelysium.com.au/). Further, the sequences were trimmed to remove poor quality reads on both ends. Reads were assembled using DNA Baser v.4.16.0 (http://www.dnabaser.com/) to generate contigs. Alignment of the gene sequences for both the parents was done using ClustalX 2.1 to detect SNPs. SNPs were then manually curated by analyzing and comparing chromatogram files with ClustalX alignment files. Q-value ≥ 30 was used as threshold for defining SNPs.
Results
Evaluation for yield and yield component traits in advanced AILs
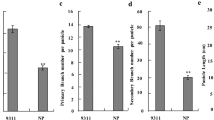
Variation in yield and yield contributing traits was observed in advanced AILs at BC3F3, BC3F6, and BC3F8 generations during years: 2011, 2013, and 2015, respectively (Table 1; Figs. S1-S3). Analysis of variance for RBD revealed significant variation for all the traits in all the generations. Although variation was observed for number of traits in the two parents IL1792 and PR114 over different years, the differences in SPP and TN were the most promising and observable over 3 years (Table 1). IL1792 had a higher number of spikelet per panicle but had fewer tillers per plant compared to PR114 (Table 1, Fig. 1).

Plant type of donor parent O. longistaminata acc. IRGC104301B, introgression line IL1792, and the recipient parent PR114 (a); variation in spikelets per panicle in introgression line IL1792, PR114, and selected BC3F3 progenies (b); frequency distribution for spikelets per panicle (c) and tillers per plant (d) in 474 advanced alien introgression lines (AILs) in year 2013. Mean of parental lines (PR114 and IL 1792) is indicated by arrows. Skewness (Sk) and kurtosis (Kt) values for each trait are presented at the top
Correlations of yield component traits between years
Significant positive correlation was observed between the 2 years (2013 and 2015) for all the traits except SSPP, indicating the stability of these traits over multiple years (Table S2). The highest significant correlation was observed for PH followed by DF between the years. There was a significant positive correlation observed for SPP, FGP, and TN. The trait PB was also positively correlated with SPP and FGP.
Parental polymorphism and introgression in IL1792
A total of 423 SSR markers, spanning the 12 linkage groups, were amplified in PR114, IL1792, and O. longistaminata acc. IRGC104301 for parental polymorphism and to examine the extent of introgression from O. longistaminata acc. IRGC104301 into IL1792. Out of these, 249 SSR markers showed an overall 58.9% polymorphism between O. sativa cv. PR114 and O. longistaminata acc. IRGC104301. Percent polymorphism varied from 48% on chromosome 1 to 73.3% on chromosomes 6 and 11 (Table S3). Out of the 249 polymorphic SSR markers, 42 SSR markers (16.9%) showed introgression in IL1792 (Table S3). Percent introgression varied from zero on chromosome 7 to 36.4% on chromosome 6. Introgression from O. longistaminata occurred at 29 positions throughout the genome (Fig. 2).

Graphical genotype of IL1792 generated based on 249 polymorphic SSR markers between O. sativa cv. PR114 and O. longistaminata acc. IRGC104301B. The regions in blue are the PR114 and the red regions depict introgression from O. longistaminata acc. IRGC104301B. Markers and centi Morgan (cM) distance of each of the linkage group are based on Temnykh et al. (2001)
Identification and mapping of spikelet per panicle QTL in BC3F3
Initially, a random subset of 90 out of 474 advanced AILs in BC3F3 generation was analyzed with polymorphic SSR markers to identify target QTL regions. Of the 42 polymorphic SSR markers, 24 that showed explicit introgression from O. longistaminata were used for genotyping. Most of the markers showed segregation distortion skewed towards O. longistaminata allele. Single marker analysis with 24 SSR markers revealed the QTL region for SPP and TN on the long arm of chromosome 2. To narrow down this region, an additional 27 SSR markers were retrieved from the IRGSP map (IRGSP 2005). Only three SSR markers (RM13742, RM13750, and RM13781) showed introgression in IL1792 from O. longistaminata. The 90 BC3F3 progenies were genotyped with the additional three SSR markers and QTL analysis was repeated with a total of 27 SSR markers.
Single marker analysis with 27 SSR markers showed the introgressed region on chromosome 2, associated with several yield component traits (Table S4). QTLs for TN, PH, PS, SPP, and FGP were found within a 45 cM region between SSR markers RM526 and RM6 on chromosome 2, whereas a QTL for DF was identified between RM469 and RM225 on chromosome 6 (Table S4).
QTL mapping using the CIM approach identified QTLs for SPP, FGP, and TN in a 13.4 cM region between marker intervals RM13742-RM13781 on chromosome 2 (Table 2, Fig. 3). The O. longistaminata allele at these loci contributed to higher SPP and FGP, but reduced the TN. The QTL for PH was mapped to a 21.2 cM region flanked by the markers RM13750 and RM525. The O. longistaminata allele at this locus led to an increase in plant height. The QTL for DF mapped to the marker interval RM204-RM225 on chromosome 6 (Table 2). We designated the QTL region identified from O. longistaminata responsible for increasing SPP as qSPP2.2 (Fig. 3), because a QTL qSPP2 for SPP has already been mapped on chromosome 2 (Zhang et al. 2009).

Linkage maps showing the position of QTL qSPP2.2 on chromosome 2. a The linkage map showing the initial position of QTL as obtained in advanced alien introgression lines in BC3F3. b The linkage map showing final position of the QTL obtained after evaluation of advanced alien introgression lines in years 2013 and 2015 and saturating the region with additional markers
Enrichment and validation of qSPP2.2 region
To validate and enrich the QTL regions with more markers, the data of advanced AILs in BC3F6 and BC3F8 generations was used. We focused on the qSPP2.2 region that was responsible for an increase in SPP in introgression line IL1792. An expanded QTL region between RM526 and RM6 harboring qSPP2.2 was chosen based on a previous linkage map as shown in Fig. 3. An additional 22 SSR markers (Table S5) were selected between the expanded QTL region from the IRGSP (IRGSP 2005). In addition, 10 SSR markers were designed from three BAC sequences: P0684A08, OJ1725_H08, and OJ1493_H11 spanning the target region of chromosome 2 (Table S6). Among the 28 SSR markers (including 22 additional SSR markers and 6 SSR markers from the QTL region in the linkage map flanked by RM526 and RM6), only 10 markers, namely RM526, RM13742, RM13743, RM13750, RM13755, RM13756, RM13779, RM13781, RM525, and RM6, showed polymorphism between PR114 and IL1792. Likewise, out of the ten SSR markers designed from BAC sequences, only one marker, A-SSR-4, was polymorphic between PR114 and IL1792. The 11 polymorphic markers were used for genotyping the 474 advanced AILs (Fig. S4, Table S7). Chi-square analysis showed segregation distortion for all 11 markers in favor of the O. longistaminata allele (Table S8). There were also more heterozygotes observed than expected (Fig. S4). A linkage map of 11 markers was generated using genotypic data of 474 advanced AILs in BC3F6 generation. The 11 markers spanned a map distance of 9.5 cM (Fig. 3) which is smaller than the linkage map generated based on BC3F3 progenies and reported by Temnykh et al. (2001). However, the order of the markers is the same as given in IRGSP (2005). The difference in recombination distances could be due to segregation distortion. The 3.4 cM target region bracketed by SSR markers RM13742 and RM13781 was enriched with six additional SSR markers.
Composite interval mapping was performed using genotypic and phenotypic data from 2013 to 2015. For QTL analysis, heterozygotes were considered as missing data points. In the year 2013, QTLs for SPP, FGP, and TN were localized in a narrower region spanning marker interval of RM13743-RM13750 (Fig. 3) explaining 11.3% of phenotypic variance. As observed in the BC3F3, the O. longistaminata allele at this locus contributed to higher SPP and FGP and reduced TN. In addition, the QTL for FGP and TN were also detected in the marker interval RM525-RM6 (Table 2). Similar results were obtained for 2015, where an additional QTL for SPP was identified in marker interval RM525-RM6, but did not harbor the QTL for TN (Table 2). We chose to further analyze the marker interval RM13743-RM13750 that was consistently identified in all the years.
Identification of candidate gene(s) in the qSPP2.2 region
The qSPP2.2 region flanked by RM13743 and RM13750 contains 23 annotated genes (Table S9) on the Nipponbare reference genome (http://rice.plantbiology.msu.edu/cgi-bin/gbrowse/rice/). Of 23 genes, three encoded for transposon proteins and were not considered for further analysis. The remaining 20 genes were analyzed to identify putative candidate genes using BLASTP and InterPro (https://www.ebi.ac.uk/interpro/) search for each gene (data not shown) along with other information provided by RGAP (http://rice.plantbiology.msu.edu/). Based on their functional annotations, three genes, LOC_Os02g44860, LOC_Os02g44990, and LOC_Os02g45010, were considered for sequence analysis that could be candidates for SPP. The LOC_Os02g44860 is a GDSL-like lipase/acylhydrolase, associated with cytokinin (CK) signaling which is known to be an important regulator of plant growth and development (Hirose et al. 2007). The effect of CK on grain number in rice was revealed after cloning the Gn1a gene (Ashikari et al. 2005), which encodes a cytokinin oxidase/dehydrogenase (OsCKX2). The LOC_Os02g44990 encodes an OsFBDUF13-F-Box and DUF domain containing protein. The F-box functions in regulating various developmental processes in plants, like photomorphogenesis, circadian clock regulation, self-incompatibility, floral meristem, and floral organ identity determination (Jain et al. 2007). The APO1 (an ortholog of Arabidopsis UFO gene in rice) gene encodes an F-box protein and positively regulates spikelet number by suppressing the precocious conversion of inflorescence meristems to spikelet meristems (Ikeda et al. 2009), whereas the D3 gene encoding an F-box leucine-rich repeat is a negative regulator of tillering in rice (Ishikawa et al. 2005). The LOC_Os02g45010 encodes for an ethylene responsive related protein that functions in spikelet meristem identity (Chuck et al. 2002). InterPro identified a helix-loop-helix DNA binding domain in LOC_Os02g45010 that is reported to be involved in regulating the axillary meristem formation and consequently changing the grain number in rice (Komatsu et al. 2003).
The three genes were sequenced in the parents, PR114, and IL1792 using overlapping primers (Table S10) and analyzed for nucleotide variations. The sequences are submitted to NCBI GenBank under the accession numbers KT264280, KT264281, KT264282, KT264283, KT264284, and KT264285. The collinearity of three genes in O. longistaminata was analyzed using BLAST analysis. Sequences of three genes amplified from PR114 and IL1792 including Nipponbare were BLAST analyzed on O. longistaminata genome sequence available at Gramene (www.gramene.org). In addition, one upstream or downstream functional gene was also included in BLAST analysis. BLAST analysis showed collinearity of the region in O. longistaminata (Table S11).
Comparison of gene sequences amplified from PR114 and IL1792 revealed number of variations at nucleotide level. A total of 66 nucleotide variations were detected between PR114 and IL1792 for LOC_Os02g44860 (Table S12). Only two SNPs were found in exons, with the rest in introns. Two large deletions of 8 and 6 bp were observed in PR114 in the intronic region. Sequence comparison for gene LOC_Os02g44990 revealed only five nucleotide variations, with four of them in the exon. For the gene LOC_Os02g45010, PR114 and IL1792 had variations at 19 positions, but the exonic region was identical in both parents. A six-bp deletion (GCGGCG), 10 bp upstream of the initiation codon, was present in the 5′UTR of LOC_Os02g45010 in PR114. The comparison of the protein sequences showed that out of the six exonic SNPs for all three genes, five were non-synonymous leading to change in the amino acid and only one was synonymous (Table S13).
The protein sequence for LOC_Os02g44860 showed an amino acid change at position 21 from valine (V) in PR114 to alanine (A) in IL1792 and at position 380 from leucine (L) in PR114 to V in IL1792 PR114 (Table S13). For LOC_Os02g44990, variations in amino acid sequence were observed at three positions: 157, 268, and 367 (Table S13). The A/C SNP detected in the exonic region resulted in a change in amino acid from aspartic acid (D) in PR114 to (A) in IL1792 at position 157. The amino acids A and Serine (S) in PR114 at positions 268 and 367, respectively, were replaced by threonine (T) in IL1792. For LOC_Os02g45010, the SNPs in the parental lines did not show any variation in the amino acid.
Discussion
Interspecific populations particularly from wild × cultivated crosses uncover vast allelic diversity of yield component traits that can be utilized in improving the rice yield (Bhatia et al. 2017). O. longistaminata is the most distinct in “AA” genome wild species with strong rhizomes and long anthers (Vaughan et al. 2008). The species have been utilized for identification of exemplified bacterial blight resistant gene Xa21 (Song et al. 1995) that has been introduced into several elite cultivars all over the world. In addition, it is also an important reservoir of yield component traits (Gichuhi et al. 2016; Bhatia et al. 2017), which is still untapped. The introgression line IL1792 derived from O. longistaminata in the background of recurrent parent PR114 showed higher spikelet per panicle and revealed introgressions at 29 genomic locations with 42 out of 249 polymorphic SSR markers. Thus, any phenotypic differences between PR114 and IL1792 will be primarily due to the introgressed segments. Significant variation for yield component traits such as SPP, FGP, PB, and TN was observed between IL1792, PR114, and among their progenies indicating that introgressions from O. longistaminata have the potential to improve important traits in cultivated rice.
In the present study, the qSPP2.2 QTL for increasing SPP has been identified on chromosome 2 between SSR markers RM13743 and RM13750 which are 1.0 cM apart. The qSPP2.2 was captured in early backcross segregating generations and validated in advanced backcross homozygous lines. We tried to enrich the qSPP2.2 region with more number of markers exploiting recombinations based on selfing of heterozygous segments. However, application of additional physical map-based SSRs and newly designed markers could not significantly narrow down the qSPP2.2 region that identified in early backcross segregating generation. To further fine map the qSPP2.2 region, there is a need to generate large F2 population of advanced AIL with least introgression for qSPP2.2 region and recurrent parent.
Several SSR markers showed distorted segregation in the advanced AILs with O. longistaminata alleles being favored over PR114 alleles. Segregation distortion of markers has been reported as a common phenomenon in both intraspecific and interspecific crosses in rice and might be due to segregation distortion loci (Xu et al. 1997; Xu 2008). These loci are subject to gametic selection, zygotic selection, or both, and their distorted segregation causes the observed markers to deviate from the Mendelian ratio. Several QTLs controlling SPP have been mapped to different rice chromosomes, of which two QTL have been mapped to chromosome 2 (Bai et al. 2012). The QTL qSPP2 reported by Zhang et al. (2009) on chromosome 2 was mapped to a 5.8 cM interval between markers MRG2762 and RM3515. The markers RM13743 and RM13750 which flank qSPP2.2 in the present study map between two other SSR markers: RM526 and RM525. Based on genetic map published by McCouch et al. (2002), RM526 and RM525 map at recombination positions 109.3 and 125.9 cM, respectively, whereas RM3515, which is linked to qSPP2 (Zhang et al. 2009) maps to 95.2 cM on chromosome 2. Similarly, a grain yield QTL named as qGY2.1 has been fine mapped on chromosome 2, between the SSR markers RM5897 and RM262 (He et al. 2006), which map to 32.8 and 81.4 cM positions (McCouch et al. 2002). Thus, the QTL qSPP2.2 might be different from qSPP2 and qGY2.2 identified from cultivated rice. In addition, a number of genes that primarily regulate SPP has been cloned and characterized (Ikeda et al. 2013), of which the F-box gene, LP/EP3, has been identified on long arm of chromosome 2 (Piao et al. 2009; Li et al. 2011). However, qSPP2.2 is present on short arm of chromosome 2.
Besides SPP, QTLs for FGP and TN were also detected in the qSPP2.2 region. The O. longistaminata allele of qSPP2.2 for SPP and FGP exhibited a positive additive effect, whereas it showed negative additive effect for tiller number. Both SPP and FGP were also negatively correlated with tiller number based on phenotypic analysis. Similar results were reported in few other studies. In one of study, the QTL qSPP7 was identified for tiller number and spikelet per panicle, where the Minghui 63 allele of qSPP7 showed opposite effects on TN and SPP (Xing et al. 2008). Similarly, transgenic plants carrying OsSPL14/IPA1 showed reduced tillers, stronger culms, and increased panicle branches and grain yield (Jiao et al. 2010).
Out of 23 genes present in qSPP2.2 region, three genes were identified based on domain analysis and their possible role in controlling SPP in plants. The three putative candidate genes were sequenced in PR114 and IL1792 in order to find causal SNP and possibility to develop further markers to narrow down the region in future. Sequence comparison of the three putative candidate genes revealed several SNPs and InDels in PR114 and IL1792, but few variations at the protein level. There were two amino acid substitutions in LOC_Os02g44860, three in LOC_Os02g44990, and no variation in LOC_Os02g45010 between PR114 and IL1792. There was a change from valine (Val 21) and leucine (Leu 380) to alanine (Ala 21) and valine (Val 380), respectively, in IL1792 for Os02g44860. All these amino acids, i.e., alanine, valine, and leucine, are polar in nature and are involved in hydrogen bond formation. Thus, these substitutions might not lead to any changes in the properties or function of the protein. However, the variation in PR114 and IL1792 for LOC_Os02g44990 caused changes in the amino acid properties. Negatively charged aspartate (Asp 157) changed to hydrophobic alanine (Ala 157). Likewise, a substitution from hydrophobic (Alanine) to polar (Threonine) amino acid was observed at position 268. Single amino acid substitutions are capable of causing dramatic changes in protein structure or function. Thus, the amino acid substitutions observed in LOC_Os02g44990 might alter the protein function. On the other hand, a 6-bp (GCGGCG) deletion at 10 bases upstream of the initiation codon was observed in PR114 for LOC_Os02g45010. The region might be a functional site for cis regulatory elements that have a pronounced effect on the translation efficiency of the genes (Kim et al. 2014). Therefore, we speculate the possibility of LOC_Os02g44990 and LOC_Os02g45010 as potential candidate genes for the QTL qSPP2.2 controlling SPP. However, further expression analysis of these genes may validate their role in controlling SPP in plants.
One of the unanswered questions in plant breeding is the observed negative correlation between grains per panicle and tiller number. It is not clear how this relationship operates. Is it a pleiotropic effect or different genes that are tightly linked? The advanced introgression lines used in the study are uniform over a large part of the genome with little background noise. Hence, it could be an excellent genetic material for studying the molecular basis of the negative correlation between grain number and tiller number traits. Selective silencing of the putative candidate genes introgressed from O. longistaminata can lead to identification of the gene(s) responsible for SPP and/or TN. Regardless, we have shown the importance of O. longistaminata as a source of yield and yield component traits. The QTL qSPP2.2 mapped and transferred in the present study is a novel QTL, because (1) it differs from two other QTLs identified on chromosome 2 in previous studies and (2) the QTL has been identified from O. longistaminata, an African wild species that has shown considerable divergence in the ancestry from other “AA” genome wild species (Wambugu et al. 2013). To validate the commercial utility of qSPP2.2 allele transferred from O. longistaminata, we have already started introgressing this QTL into Basmati rice which has a low number of spikelets per panicle. The F1 plants showed a higher number of grains per panicle compared to the Basmati cultivars, and significant segregation has been observed in the backcross generations (Kuldeep Singh—personnel communication). Since the QTL qSPP2.2 has been transferred into an elite background, it has already found its way in Breeders’ crossing blocks.
References
Ali ML, Sanchez PL, Yu S-B, Lorieux M, Eizenga GC (2010) Chromosome segment substitution lines: a powerful tool for the introgression of valuable genes from Oryza wild species into cultivated rice (O. sativa). Rice 3:218–234
Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M (2005) Cytokinin oxidase regulates rice grain production. Science 309:741–745
Bai X, Wu B, Xing Y (2012) Yield-related QTLs and their applications in rice genetic improvement. J Integr Plant Biol 54:300–311
Balkunde S, Le HL, Lee HS, Kim DM, Kang JW et al (2013) Fine mapping of a QTL for the number of spikelets per panicle by using near-isogenic lines derived from an interspecific cross between Oryza sativa and Oryza minuta. Plant Breed 132:70–76
Berloo RV (2008) GGT 2.0: versatile software for visualization and analysis of genetic data. J Hered 99:232–236
Bhatia D, Joshi S, Das A, Vikal Y, Sahi GK, Kumari N, Kaur K, Singh K (2017) Introgression of yield component traits in rice (Oryza sativa ssp indica ) through interspecific hybridization. Crop Sci 57:1–17
Brar DS, Singh K (2011) Rice. In: Kole C (ed) Wild crop relatives: genomics and breeding resources. Springer, Berlin, pp 321–365
Brondani C, Rangel PHN, Brondani RPV, Ferreira ME (2002) QTL mapping and introgression of yield-related traits from Oryza glumaepatula to cultivated rice (Oryza sativa) using microsatellite markers. Theor Appl Genet 104:1192–1203
Cheema MS, Singh B (1990) A user's Manual to CPCS1: a computer programme package for the analysis of commonly used experimental design. Punjab Agricultural University, Ludhiana, pp 1–10
Chuck G, Muszynski M, Kellogg E, Hake S, Schmidt RJ (2002) The control of spikelet meristem identity by the branched silkless1 gene in maize. Science 298:1238–1241
Dalmacio R, Brar DS, Ishii T, Sitch LA, Virmani SS, Khush GS (1995) Identification and transfer of a new cytoplasmic male sterility source from Oryza perennis into indica rice (O. sativa). Euphytica 82:221–225
Dalmacio RD, Brar DS, Virmani SS, Khush GS (1996) Male sterile line in rice (Oryza sativa) developed with O. glumaepatula cytoplasm. Int Rice Res Notes 21:22–23
Fischer RA, Edmeades GO (2010) Breeding and cereal yield progress. Crop Sci 50:85–98
Gaikwad KB, Singh N, Bhatia D, Kaur R, Bains NS, Bharaj T, Singh K (2014) Yield-enhancing heterotic QTL transferred from wild species to cultivated rice Oryza sativa L. PLoS One 9:e96939
Gichuhi E, Himi E, Takahashi H, Zhu S, Doi K, Tsugane K, Maekawa M (2016) Identification of QTLs for yield-related traits in RILs derived from the cross between pLIA-1 carrying Oryza longistaminata chromosome segments and Norin 18 in rice. Breed Sci 66:720–733
He G, Luo X, Tian F, Li K, Zhu Z, Su W, Qian X, Fu Y, Wang X, Sun C, Yang J (2006) Haplotype variation in structure and expression of a gene cluster associated with a quantitative trait locus for improved yield in rice. Genome Res 16:618–626
Hirose N, Makita N, Kojima M, Kamada-Nobusada T, Sakakibara H (2007) Over expression of a type-a response regulator alters rice morphology and cytokinin metabolism. Plant Cell Physiol 48:523–539
Hu FY, Tao DY, Sacks E, Fu BY, Xu P, Li J, Yang Y, McNally K, Khush GS, Paterson AH, Li ZK (2003) Convergent evolution of perenniality in rice and sorghum. Proc Natl Acad Sci U S A 100:4050–4054
Huo X, Wu S, Zhu Z, Liu F, Fu Y, Cai H, Sun X, Gu P, Xie D, Tan L, Sun C (2017) NOG1 increases grain production in rice. Nat Commun 8:1497. https://doi.org/10.1038/s41467-017-01501-8
Ikeda K, Yasuno N, Oikawa T, Iida S, Nagato Y et al (2009) Expression level of ABERRANT PANICLE ORGANIZATION1 determines rice inflorescence form through control of cell proliferation in the meristem. Plant Physiol 150:736–747
Ikeda M, Miura K, Aya K, Kitano H, Matsuoka M (2013) Genes offering the potential for designing yield-related traits in rice. Curr Opin Plant Biol 16:1–8
Imai I, Kimball JA, Conway B, Yeater KM, McCouch SR, McClung A (2013) Validation of yield-enhancing quantitative trait loci from a low-yielding wild ancestor of rice. Mol Breed 32:101–120
IRGSP (2005) The map-based sequence of the rice genome. Nature 436:793–800
Ishikawa S, Maekawa M, Arite T, Onishi K, Takamure I, Kyozuka J (2005) Suppression of tiller bud activity in tillering dwarf mutants of rice. Plant Cell Physiol 46:79–86
Jacquemin J, Bhatia D, Singh K, Wing RA (2013) The international Oryza map alignment project: development of a genus-wide comparative genomics platform to help solve the 9 billion-people question. Curr Opin Plant Biol 16:147–156
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, Kapoor S, Tyagi AK, Khurana JP (2007) F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143:1467–1483
Jiao Y, Wang Y, Xue D, Wang J, Yan M, Liu G, Dong G, Zeng D, Lu Z, Zhu X, Qian Q, Li J (2010) Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 42:541–544
Khush GS (2013) Strategies for increasing the yield potential of cereals: case of rice as an example. Plant Breed 132:433–436
Khush GS, Ling KC, Aquino RC, Aguiero VM (1977) Breeding for resistance to grassy stunt in rice In: Proc. 3rd Intern. Congr. SABRAO, Canberra, Australia, pp 3–9
Kim Y, Lee G, Jeon E, Sohn E, Lee Y et al (2014) The immediate upstream region of the 5'-UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res 42:485–498
Komatsu K, Maekawa M, Ujiie S, Satake Y, Furutani I, Okamoto H, Shimamoto K, Kyozuka J (2003) LAX and SPA: major regulators of shoot branching in rice. Proc Natl Acad Sci U S A 100:11765–11770
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Li M, Tang D, Wang K, Wu X, Lu L, Yu H, Gu M, Yan C, Cheng Z (2011) Mutations in the F-box gene LARGER PANICLE improve the panicle architecture and enhance the grain yield in rice. Plant Biotechnol J 9:1002–1013
Liu L, Lafitte R, Guan D (2004) Wild Oryza species as potential sources of drought-adaptive traits. Euphytica 138:149–161
Luo X, Wu S, Tian F, Xin X, Zhae X et al (2011) Identification of heterotic loci associated with yield-related traits in Chinese common wild rice (Oryza rufipogon Griff.). Plant Sci 181:14–22
Ma X, Fu Y, Zhao X, Jiang L, Zhu Z, Gu P, Xu W, Su Z, Sun C, Tan L (2016) Genomic structure analysis of a set of Oryza nivara introgression lines and identification of yield-associated QTLs using whole-genome resequencing. Sci Rep 6:27425
McCouch SR, Teyetlman L, Xu Y, Lobos KB, Clare K et al (2002) Development and mapping of 2240 new SSR markers for Rice (Oryza sativa L.). DNA Res 9:199–207
McCouch SR, Sweeney M, Li J, Jiang H, Thomson M, Septiningsih E, Edwards J, Moncada P, Xiao J, Garris A (2007) Through the genetic bottleneck: O. rufipogon as a source of trait-enhancing alleles for O. sativa. Euphytica 154:317–339
Melaku G, Haileselassie T, Feyissa T, Kiboi S (2013) Genetic diversity of the African wild rice (Oryza longistaminata Chev. Et Roehr) from Ethiopia as revealed by SSR markers. Genet Resour Crop Evol 60:1047–1056
Onishi K, Horiuchi Y, Ishigoh-Oka N, Takagi K, Ichikawa N, Maruoka M, Sano Y (2007) A QTL cluster for plant architecture and its ecological significance in Asian wild rice. Breed Sci 57:7–16
Piao R, Jiang W, Ham TH, Choi MS, Qiao Y, Chu SH, Park JH, Woo MO, Jin Z, An G, Lee J, Koh HJ (2009) Map-based cloning of the ERECT PANICLE 3 gene in rice. Theor Appl Genet 119:1497–1506
Saghai-Maroof M, Soliman KM, Jorgensen RA, Allard R (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci U S A 81:8014–8018
Shakiba E, Eizenga GC (2014) Unraveling the secrets of rice wild species. In Yan W, Bao J (eds) Rice germplasm, genetics and improvement. pp 1–58. https://doi.org/10.5772/58393
Song WY, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, Gardner J, Wang B, Zhai WX, Zhu LH, Fauquet C, Ronald P (1995) A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270:1804–1806
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Vaughan DA, Lu B-R, Tomooka N (2008) The evolving story of rice evolution. Plant Sci 174:394–408
Wambugu PW, Furtado A, Waters DLE, Nyamongo D, Henry R (2013) Conservation and utilization of African Oryza genetic resources. Rice 6:29
Wang S, Basten CJ, Zeng Z-B (2010) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC, http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Xiao J, Li J, Grandillo S, Yuan L, Tanksley SD et al (1998) Identification of traits improving quantitative trait loci alleles from wild rice relative Oryza rufipogon. Genetics 150:899–909
Xing YZ, Tang WJ, Xue WY, Xu CG, Zhang Q (2008) Fine mapping of a major quantitative trait loci, qSSP7, controlling the number of spikelets per panicle as a single Mendelian factor in rice. Theor Appl Genet 116:789–796
Xu S (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208
Xu Y, Zhu L, Xiao J, Huang N, McCouch SR (1997) Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid, and recombinant inbred populations in rice (Oryza sativa L.). Mol Gen Genet 253:535–554
Yoon DB, Kang KH, Kim HJ, Ju HG, Kwon S et al (2006) Mapping quantitative trait loci for yield components and morphological traits in an advanced backcross population between Oryza grandiglumis and the O. sativa japonica cultivar Hwaseongbyeo. Theor Appl Genet 112:1052–1062
Zhang Y, Luo L, Liu T, Xu C, Xing Y (2009) Four rice QTL controlling number of spikelets per panicle expressed the characteristics of single Mendelian gene in near isogenic backgrounds. Theor Appl Genet 118:1035–1044
Acknowledgements
Financial support for development of alien introgression lines was provided by Dept. of Biotechnology, Ministry of Science and Technology, Govt., of India; vide its grant letter No. BT PR2159/AGR/07/107/2000. Wild species germplasm including O. longistaminata acc. 104301 was provided by IRRI, Philippines. Corresponding author expresses his gratitude to both DBT for financial support and IRRI for germplasm. We are also grateful to Dr. Ajay Kohli IRRI, Philippines for discussions on identifying the putative candidate genes in the QTL region. We would like to thank Ross Zhan, Department of Botany and Plant Pathology, Purdue University, Indiana for devoting considerable time and effort in editing the language of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kaur, A., Sidana, K., Bhatia, D. et al. A novel QTL qSPP2.2 controlling spikelet per panicle identified from Oryza longistaminata (A. Chev. et Roehr.), mapped and transferred to Oryza sativa (L.). Mol Breeding 38, 92 (2018). https://doi.org/10.1007/s11032-018-0843-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-018-0843-9