
Abstract
Panicle length (PL) is an important trait that determines panicle architecture and strongly affects grain yield and quality in rice. However, this trait has not been well characterized genetically, and its contribution to yield improvement is not well understood. Characterization of novel genes related to PL is of great significance for breeding high-yielding rice varieties. In our previous research, we identified qPL5, a quantitative trait locus for PL. In this study, we aimed to determine the exact position of qPL5 in the rice genome and identify the candidate gene. Through substitution mapping, we mapped qPL5 to a region of 21.86 kb flanked by the molecular marker loci STS5-99 and STS5-106 in which two candidate genes were predicted. By sequence analysis and relative expression analysis, LOC-Os05g41230, which putatively encodes a BRASSINOSTEROID INSENSITIVE 1-associated receptor kinase 1 precursor, was considered to be the most likely candidate gene for qPL5. In addition, we successfully developed a pair of near-isogenic lines (NILs) for qPL5 in different genetic backgrounds to evaluate the genetic effects of qPL5. Agronomic trait analysis of the NILs indicated that qPL5 positively contributes to plant height, grain number per panicle, panicle length, grain yield per plant, and flag leaf length, but it had no influence on heading date and grain-size-related traits. Therefore, qPL5 and the markers tightly linked to it should be available for molecular breeding of high-yielding varieties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ensuring food security for an ever-increasing world population mostly depends on increasing grain yield in crop plants (Gupta et al. 2006). Rice (Oryza sativa L.) is an important staple crop consumed daily by a large percentage of the global population. However, the current pace of the yield increase in rice is insufficient to meet the needs of a world population predicted to reach 10 billion in 30 years (Hickey et al. 2019). Breeding practice shows that the cultivation of rice varieties with high and stable yields offers the opportunity to satisfy that demand. At present, molecular design breeding is showing great potential for the selection of excellent varieties, and the most crucial requirement is to identify a subset of genes/quantitative trait loci (QTLs) that control grain yield-related traits and to elucidate the interactions and genetic effects among these genes/QTLs (Marathi et al. 2012; Tian et al. 2015).
Panicle architecture, one of the most important morphological traits in rice that determines grain yield, consists of various quantitative characters such as panicle length (PL) and the number of primary branches (PBN) and secondary branches (SBN) (Crowell et al. 2016). PL has been defined as an important yield-related agronomic trait, and the genetic factors that control PL have been widely studied. During the past decade, based on the identification of mutants, several genes for PL have been cloned. SP1 encodes a possible peptide transporter that regulates the elongation of the branch resulting in shorter panicle length (Li et al. 2009). DEP2 encodes a plant-specific protein with unknown functional domains that regulates the rapid elongation of the axis and thus controls panicle length (Li et al. 2010). DEP3 is predicted to encode a patatin-like phospholipase A2 superfamily domain-containing protein that regulates the formation of vascular bundles, leading to changes in panicle length and grain size (Qiao et al. 2011). LP encodes a Kelch repeat-containing F-box protein that may be involved in modulating cytokinin contents in plant tissues, resulting in changes to panicle length; this indicates that the levels of some plant hormones, such as cytokinin, are tightly associated with PL regulation (Li et al. 2011). These results have broadened our understanding of the genetic mechanisms underlying PL regulation. However, mutations in these genes result in pleiotropic phenotypes, and almost all of them are unfavorable and impractical. Thus, it is necessary to mine novel genetic factors for PL in nature germplasm.
Like other grain yield-related traits, PL is also a canonical complex trait controlled by major and minor QTLs (Liu et al. 2011). At present, many QTLs for PL have been extensively characterized in rice, and at least 250 QTLs have been identified on the 12 chromosomes (http://www.gramene.org/). LP1, a major QTL for PL, was mapped to a small region of ~ 90 kb on chromosome 9 (Chr. 9) (Liu et al. 2016). qPL6, a QTL controlling PL, was fine-mapped to an interval of ~ 25 kb on Chr. 6 (Zhang et al. 2015). Low-resolution mapping delimited PL6-5, a QTL responsible for PL, to a ~ 1.3 Mb interval on Chr. 6 (Sun et al. 2017). qPL8, a novel QTL for PL, was mapped to a ~ 278 kb region on Chr. 8 (Zhang et al. 2021). Also, it is notable that several QTLs for PL have been cloned. DEP1 encodes a phosphatidylethanolamine-binding protein-like domain protein that regulates meristematic activity, resulting in changes in PL and grain number (GN) (Huang et al. 2009). IPA1, a gene that encodes OsSPL14 (SOUAMOSA PROMOTER BINDING PROTEIN-LIKE 14), is regulated by the microRNA (miRNA) OsmiR156 in vivo, and the mutant allele of IPA1 disrupts OsmiR156-directed regulation of OsSPL14, resulting in rice plants with an ideal plant architecture and with increased PL and enhanced grain yield (Jiao et al. 2010).
As mentioned above, although some genes/QTLs involved in PL regulation have been fine mapped or cloned, the underlying genetic mechanisms for PL regulation are unclear. Moreover, favorable alleles that could be directly employed in molecular marker-assisted selection of high-yielding varieties are still rare. It is therefore worthwhile to conduct a continued search for favorable alleles that are involved in the control of PL. In our previous study, a novel QTL controlling PL on Chr. 5, designated qPL5, was identified using a set of chromosomal segment substitution lines (CSSLs) (Xu et al. 2019). In that study, we performed fine-mapping of qPL5 and predicted the candidate gene for qPL5. Also, the genetic effects of qPL5 on the main agronomic traits were evaluated, and the utilization potential of qPL5 in genetic improvement of yield potential was discussed. Thus, our research not only lays the foundation for cloning qPL5, but also provides a new gene source for high-yielding rice breeding programs.
Materials and methods
Plant materials and population development
In our previous study, a population of CSSLs (named N1-125) that carry one or several chromosomal segments from the indica variety ‘9311’ in the japonica cultivar ‘Nipponbare’ (NP) has been successfully developed and widely used for gene mapping (Gao et al. 2020; Xu et al. 2017; Zhang et al. 2011). To precisely map qPL5, N58, a CSSL that carries a single substituted segment covering the location of qPL5, was selected from the CSSL population to cross with the recipient parent NP in the Spring of 2017. From this we developed the F2, F3, and F4 segregating populations. The substitution lines (SLs) with ‘9311’-type homozygous genotypes were obtained from the recombinants in the F2 and F3 populations, and these SLs were grown for phenotypic evaluation from 2018 to 2021, and were used for fine mapping of qPL5.
To evaluate the genetic effects of qPL5, we developed two near-isogenic lines (NILs) carrying the ‘9311’ allele of qPL5. The NIL in the NP background, a curved panicle-type japonica variety, called NP-qPL5, was developed from the gene mapping population. Using molecular marker-assisted selection, we developed a NIL named KWY3-qPL5 in the ‘Kangwuyujing3’ (KWY3) background, an erect panicle-type japonica variety, using N58 as the donor and KWY3 as recipient through constant backcrossing followed by self-pollination (Supplemental Figure S1).
Field planting
The F2, F3, and F4 segregating populations were grown in the experimental fields of Yangzhou University (YZU) in Jiangsu and Hainan provinces of China during 2018–2020. The parental lines and the newly-developed SLs were planted in the experimental fields of YZU and Jiudian (JD) (Yangzhou, Jiangsu Province, China). The NILs were planted in a randomized block design with two replications in the experimental field of YZU. The parental lines and the SLs were planted in 4-row plots with 10 plants per row in 2019–2021. NP and NP-qPL5, KWY3, and KWY3-qPL5, were grown in 20-row plots with 15 plants per row in the summer of 2021. The growing density was 15 × 25 cm for a line X row.
Trait measurements
The main agronomic traits, such as PL, plant height (PH), heading date (Hd), tiller number per plant (TN), grain number per panicle (GN), PBN, SBN, thousand-grain weight (TGW), grain length (GL), grain width (GW), grain yield per plant (GYPP), and flag leaf length (FLL) were assessed. The method used for phenotypic evaluation was previously described (Wang et al. 2017; Xu et al. 2017; Zhang et al. 2015). Briefly, the panicle architecture related traits, such as PL, GN, PBN, and SBN were recorded from six main panicles that were defined as the panicle from the tallest tiller at maturity. To measure the PH, TN and FLL, we selected six plants from the middle of the row in each line for phenotypic evaluation. Hd is defined as the day from sowing to 50% panicle appearance. GYPP was measured as the mean grain weight from 50 individual plants. In addition, 100 fully filled grains harvested from at least 10 individuals were used for measurement of GL, GW, and TGW using the MICROTEK ScanMaker i800 flat-bed scanner.
DNA extraction, PCR amplification, and molecular marker development
Genomic DNA was isolated from fresh leaves harvested from individual rice plants using the CTAB method with slight modifications (Murray et al. 1980). PCR amplifications were performed in 20 μl reaction volumes containing 10 µl 2 × Taq Master Mix (Dye Plus) (Vazyme, Nanjing, China), 2 µl of 0.2 µmol/L primer, 20 ng of template DNA, and 6 µl ddH2O. The PCR-amplified products were separated by agarose gel (3.0%) electrophoresis, and the agarose gels were photographed under UV light.
The novel InDel molecular markers (Supplemental Table 1) that were used in the mapping of qPL5 were designed using Primer 6.0 software based on genomic sequence variations between the indica and japonica rice subspecies (http://www.ncbi.nlm.nih.gov/). The dCAPS markers were designed with the dCAPS Finder 2.0 program (http://helix.wustl.edu/dcaps/).
The genetic background of KWY3-qPL5 was determined with the 40 k rice SNP-array, a whole-genome single nucleotide polymorphism (SNP) array with 40,000 SNP and InDel markers (Greenfafa, Wuhan). In brief, a DNA pool consisting of three KWY3-qPL5 plants was constructed and genotyped.
Quantitative real-time (qRT)‑PCR
qRT-PCR assays for characterizing the expression patterns of annotated genes were performed at different stages of panicle development in NP-qPL5 and NP. Developing panicles at four stages (≤ 0.5 cm, 1 cm, 2 cm, and 5 cm) were selected as one biological replicate from the panicle initiation stage to the mature pollen formation stage (Zhou et al. 2018). Total RNA was extracted from the different panicle developmental stages using the Total RNA Isolation Kit (Vazyme, Nanjing, China), and the RNA was reverse transcribed into cDNA using the Fast Quant RT Kit (with gDNase) (Novoptotein, Suzhou, China). The amplification conditions consisted of one cycle of 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, then 95 °C for 15 s and 60 °C for 1 min, with a melting curve from 60 °C to 95 °C. The expression of each gene was compared between NP and NP-qPL5 using the 2−ΔΔCt method (Livak and Schmittgen 2013). The names and sequences of the primers used are given in Supplemental Table 1.
QTL mapping and statistical analysis
The inclusive composite interval mapping (ICIM) method was performed to identify QTLs using ICI-Mapping 4.0 software with the aid of the genotypes and phenotypes of 234 F2 plants, in which the genotype of the ‘9311’-type, NP-type, heterozygous type, and the absence of marker type were assigned the values 2, 0, 1, and − 1, respectively.
Data analysis
The analysis of variance (ANOVA) package in SPSS 15.0 (IBM) was used for statistical analyses. Significant differences were determined at the p < 0.05 and p < 0.01 levels.
Results
Genetics validation of qPL5
In our previous study, qPL5 was preliminarily mapped to a ~ 2.71 Mb region on Chr. 5, and there are three CSSLs (N22, N57, and N58) that harbor qPL5 (Xu et al. 2019). In order to validate qPL5, we performed map-based cloning. Re-sequencing data from the CSSL population showed that N58 carries only a single ‘9311’ chromosomal segment on Chr. 5 (from 18,695,860 bp to 24,855,730 bp) encompassing qPL5 in the NP genetic background (Fig. 1A and Supplemental Fig. 2A and B). Based on this information, STS5-1, STS5-6, and STS5-12, three Indel molecular markers that map to loci within this target region, were used to genotype N58, and the substitution segment in N58 was further confirmed (data not shown). ANOVA of PL between NP and N58 revealed a significant difference, with N58 having longer panicles than NP (Fig. 1B–D). Also, GN and PH were obviously increased in N58 compared with NP (Fig. 1B, E, and F). There was no significant difference in Hd between N58 and NP (Fig. 1B, G). These results indicate that N58 carries qPL5 on the introduced chromosomal DNA segment from ‘9311’. Thus, N58 was selected to develop the segregating population for validating and fine mapping qPL5.

Phenotypic differences between ‘Nipponbare’ (NP) and N58. A Graphical genotype of N58. The position of the introgressed chromosomal segment from ‘9311’ is shown in red. B Mature plants of NP and N58. C Panicle phenotypes. D–G Comparisons of panicle length (PL), grain number per panicle (GN), heading date (Hd), and plant height (PH) between NP and N58. Error bars represent the mean value ± SE (n = 6 plants). ** and * indicate significant differences at p < 0.01 and p < 0.05, respectively, as determined by ANOVA. Scale bars = 5 cm in B and C

Genetic validation of qPL5. A Frequency distribution of PL within the F2 segregating population derived from NP and N58. B Localization of the QTL for PL detected in the F2 segregating population to a region of ~ 910 kb on chromosome 5. C Comparison of PL between genotyped plants homozygous for the NP (NP-Homo) and ‘9311’ (‘9311’-Homo) alleles and the NP/’9311’ heterozygote (Heter), which were selected from the F2 segregating population. Error bars represent the mean value ± SE (n = 40 individuals). p values were calculated using one-way ANOVA
To confirm the location of qPL5, 234 individuals in the N58/NP F2 population were randomly selected for genotyping using seven polymorphic markers that map to loci distributed in the qPL5 mapping region, and PL in these plants was evaluated. Panicle length in this population showed a continuous distribution, indicating that PL is a complex trait (Fig. 2A). A major-effect QTL that explains 23.21% of the phenotypic variance for PL was detected and localized to a ~ 910-kb region flanked by the marker loci STS5-6 and STS5-10 (Fig. 2B), and this QTL should be qPL5. Moreover, the PLs of the heterozygous plants were significantly longer than in homozygous NP plants but shorter than in 9311 homozygous plants, indicating that qPL5 is a partially dominant gene (Fig. 2C).
Substitution mapping of qPL5
To narrow down the mapped region containing qPL5, 3600 F2 plants from the N58/NP population were genotyped with the STS5-6 and STS5-10 InDel markers. As a result, we identified 65 recombinant individuals at these two marker loci. Using an additional six polymorphic markers that map between the STS5-6 and STS5-10 loci, the 65 recombinants were divided into nine groups, from which nine types of SLs were obtained (I–IX) (Fig. 3A, B). Progeny testing of the SLs showed that plants in groups I–III and V–VII had significantly increased PL compared with NP. In contrast, no significant differences in PL were observed between NP and plants in groups IV, VIII, and IX. Thus, we were able to reduce the qPL5 interval to a genomic region of ~ 132.6 kb flanked by the STSN5-9 and STS5-33 marker loci (Fig. 3B).

Fine mapping and candidate gene analysis of qPL5. A Physical location of the ‘9311’ donor chromosomal segment on Chr. 5 in N58. B, C Primary mapping (B) and fine mapping (C) of qPL5 based on the genotypes (left) and phenotypes (right) of the substitution lines (SLs). D Two open reading frames (ORFs) were identified within the 21.86-kb target region. E Comparison of the genomic sequences of the two ORFs from NP and N58. F, G Comparison of the expression levels of LOC_05g41230 (F) and LOC_05g41240 (G) in NP and NP-qPL5. Black and white rectangles represent homozygous ‘9311’ and NP genotypes, respectively, in A − C. ∆ and ▼ represent nucleotide deletions and insertions, respectively. Error bars represent the mean value ± SD (n = 6 individuals in each substitution line). Gene expression levels are presented as ratios of the expression of the individual ORFs to that of the rice UBQ gene. Means ± SD were obtained from three technical replicates and two biological replicates. ** and * indicate significant differences at p < 0.01 and p < 0.05, respectively, as determined by one-way ANOVA
To clarify the precise position of the qPL5 locus, 15 individuals with heterozygous genotypes at qPL5 in the F2 population were selected to develop an F3 population. Similarly, two molecular markers, STS5-9 and STS5-33, were initially used to identify the genotypes of 5,000 individuals, and a total of 52 recombinants were obtained at these two marker loci. Another seven polymorphic molecular markers that map between the STS5-9 and STS5-33 loci were then developed and used to detect the recombinants. We obtained six groups of recombinants, in which there were six types of SLs (X-XV). Progeny testing showed that plants of types X, XI, and XIII had longer panicles than did NP. In contrast, no significant differences in PL were observed between NP and types XII, XIV, and XV of the SLs (Fig. 3C). Finally, the location of qPL5 was narrowed down to a genomic region of 21.86 kb that is flanked by molecular marker loci STS5-99 (24.147 Mb) and STS5-106 (24.168 Mb) based on the reference sequence of the NP genome (Fig. 3C).
Identification of the candidate gene for qPL5
According to the information of the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu), two predicted open reading frames (ORFs), LOC_Os05g41230 and LOC_Os05g41240, are present within this 21.86 kb interval. These genes are predicted to encode a BRASSINOSTEROID INSENSITIVE 1-associated receptor kinase 1 precursor (BAK1) and a Myb-like DNA-binding domain containing protein, respectively (Fig. 3D). To identify the candidate gene for qPL5, the genomic sequences containing 2 kb of the promoter regions and the full coding regions of those two ORFs were sequenced to examine the sequence variation between the parental lines NP and N58. As a result, 12 base substitutions and two insertions in the promoter region, as well as two base substitutions in exons of LOC_Os05g41230, were identified between the two parental lines. Moreover, the variations in exon 4 and 6 were predicted to cause amino acid changes from valine to glycine and serine to asparagine, respectively (Fig. 3E). A sequence comparison of LOC_Os05g41240 identified six variations in the promoter region (Fig. 3E). Additionally, to analyze the relative expression of the two ORFs, NILs differing only in the qPL5 region were developed in the NP genetic background, which carries a ~ 910-kb chromosomal segment introgressed from ‘9311’ flanked by marker loci STS5-10 and STS5-6. The NILs carrying the ‘9311’ and NP alleles in the qPL5 region were designated as NP-qPL5 and NP, respectively. The results showed that the mRNA levels of LOC_Os05g41230 were all higher in NP-qPL5 than in NP at the different developmental stages of the young panicles. In contrast, the relative expression level of LOC_Os05g41240 did not show a significant difference between the two NILs (Fig. 3F). Taken together, LOC_Os05g41230 is predicated to be the most likely candidate gene for qPL5.
qPL5 increases panicle length and grain yield per plant
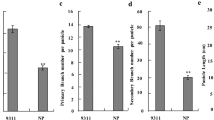
To clarify the genetic effects of qPL5, several agronomic traits in the NIL and receipt parent were evaluated. Like the phenotype of N58, PL in NP-qPL5 was significantly longer (by 10.41%) than in NP (Fig. 4A, B, and E, Supplemental Table 2). We also found that SBN and GN were significantly increased in NP-qPL5 compared with NP (Fig. 4F, G, Supplemental Table 2). The average increases in SBN and GN were 26.46% and 24.42%, respectively. Additionally, PH and FLL were both higher in NP-qPL5 than in NP, with average increases of 3.97% and 9.13%, respectively (Fig. 4A, D, I, and J, Supplemental Table 2). There were no significant differences in Hd, PBN, and grain size-related traits such as GL, GW, and TGW, between NP-qPL5 and NP (Fig. 4C, Supplemental Table 2). More importantly, compared with NP, GYPP in NP-qPL5 was increased by 15.13% although the TN was slightly decreased in NP-qPL5 (Fig. 4H and K, Supplemental Table 2).

Phenotypic characteristics of Nipponbare (NP) and the NIL, NP-qPL5. A–D Gross morphology of individuals at maturity (A), individual panicles (B), grain size (C) and flag leaf (D). Scale bars = 10 cm (A), 2 cm (B, D) and 0.5 cm (C). E–K Comparisons of PL (panicle length), SBN (secondary branch number), GN (grain number per panicle), GYYP (grain yield per plant), PH (plant height), FLL (flag leaf length), and TN (tiller number) between NP and NP-qPL5. Error bars represent the mean value ± SE of three biological replicates (n = 50 plants in H, n = 6 plants for the other traits). P values were calculated by one-way ANOVA. ** and * indicate significant differences at P < 0.01 and P < 0.05, respectively
Considering that erect panicle architecture varieties are used widely in japonica production, we developed another NIL in the genetic background of KWY3, an elite erect-panicle architecture variety (Zhang et al. 2009). We obtained five BC5F3 lines (named HN1001-1005), and the genetic background of HN1001 was determined using the RICE 40 K SNP-array (Supplemental Fig. 1). The results showed that 97.60% of the KWY3 genome was recovered in HN1001, and only two introgression segments located on Chr. 5 and Chr. 11 were identified, one of contained qPL5 (Supplemental Fig. 3). Thus, HN1001 could be considered to be a NIL for qPL5 and was designated KWY3-qPL5. Compared with KWY3, KWY3-qPL5 showed significant increases in PL, GN, SBN, PH, and FLL, while TN was slightly decreased (Fig. 5A–J, Supplemental Table 2). In addition, there were no significant differences in Hd, GL, GW, and TGW (Fig. 5A and Supplemental Table 2). Eventually, we found that GYPP in KWY3-qPL5 was increased by 2.28% compared with KWY3 (Fig. 5G, Supplemental Table 2).

Gross morphology of KWY3 (left) and KWY3-qPL5 (right). A Mature plant phenotypes. B, C The appearance of panicles and flag leaves. D–J Comparisons of PL (panicle length), SBN (secondary branch number), GN (grain number per panicle), GYYP (grain yield per plant), PH (plant height), FLL (flag leaf length), and TN (tiller number) between KWY3 and KWY3-qPL5. Error bars represent the mean value ± SE of three biological replicates (n = 50 plants in G; n = 6 plants for the other traits). p values were calculated by one-way ANOVA. ** and * indicate significant differences at p < 0.01 and p < 0.05, respectively. Scale bars = 15 cm (A) and 2 cm (B, C)
Overall, these data indicate that qPL5 has a noticeable genetic effect on PL and most of the panicle-related traits but with no obvious impacts on grain size related traits and Hd, which shows that qPL5 could be potentially useful in the breeding of high-yielding rice varieties.
Discussion
Increased productivity has long been one of the most important breeding targets in rice. To date, many QTLs for yield-related traits have been identified, and some of them have been used to develop varieties with high grain yield (Feng et al. 2017; Kim et al. 2018; Miura et al. 2011; Wang et al. 2020). Although PL has been reported to be positively associated with rice yield by regulating several panicle architecture related traits (Zhang et al. 2021), only a few PL-related genes/QTLs have been cloned or fine-mapped, and the molecular mechanisms underlying PL control are unknown. In our previous research, we identified qPL5, a PL QTL on Chr. 5, that leads to significantly increased PL in the NP genetic background (Xu et al. 2019). In this study, the location of qPL5 was successfully narrowed down to a 21.86-kb region of chromosome 5, and LOC_Os05g41230 was predicated to be the most likely candidate gene for qPL5. We also evaluated the genetic effect of qPL5 in different genetic backgrounds and showed that the boost in grain yield that resulted from incorporating the ‘9311’ allele of qPL5 is due to drastic changes in panicle architecture and related traits.
PL is a typical complex quantitative trait that is controlled by multiple quantitative loci and is significantly influenced by environmental factors, which increases the difficulty of identifying PL QTL (Liu et al. 2011; Xing et al. 2002). Previous studies have shown that advanced genetic populations, NILs or CSSLs, show great potential in mapping QTLs, especially QTLs with minor genetic effects (Shibaya et al. 2016; Uga et al. 2007). In our previous study, we found qPL5 that explained ~ 10% of the phenotypic variation for PL in the CSSL population, indicating that qPL5 might be a minor-effect QTL (Xu et al. 2019). To avoid ‘noise’ due to the genetic background, N58, the CSSL carrying only one chromosomal segment introgressed from ‘9311’, was selected as a parent to develop the segregating populations used in this study, and qPL5 was found to explain 23.21% of the phenotypic variation in the N58/NP F2 population. These results not only show that PL can be affected by genetic background, but also indicate that N58 is an ideal parent for fine mapping qPL5. Furthermore, to increase the accuracy of phenotypic characterization, we developed a set of introgression lines surrounding the qPL5 locus and localized qPL5 to an interval of 21.86 kb flanked by the molecular marker loci STS5-99 (~ 24.147 Mb) and STS5-106 (~ 24.168 Mb) on Chr. 5. At present, there are 16 reported PL QTLs on Chr. 5, but most have not been further characterized or fine mapped. Among these reported QTLs, one called qPL5-1 has been detected in a region defined by molecular marker loci RM18626 and RM3089 in a CSSL population derived from the indica variety ‘Guangluai 4’ as recipient and NP as the donor, and this region overlaps the location of qPL5. Thus, further study will be required to determine whether qPL5 and qPL5-1 are the same QTL. Nonetheless, our estimate for the location of qPL5 is more precise.
The analysis of ORFs in the mapping region predicted two putative ORFs, LOC_Os05g41230 and LOC_Os05g41240, that encode BAK1 (Brassinosteroid insensitive 1-associated kinase 1) and a Myb-like DNA-binding domain-containing protein, respectively. In a previous study, the rice BAK1 homolog, OsI-BAK1, was reported to interact with BRI1 for brassinosteroid perception and signal transduction to control the genetic regulation of PL, and the expression level of Osl-BAK1 was positively correlated with PL (Khew et al. 2015). Sequence comparison showed that there are some variations in both the promoter region and the coding sequence of LOC_Os05g41230. In addition, the relative expression level of LOC_Os05g41230 was higher in young panicles of NP-qPL5 compared to NP. Thus, we speculate that LOC_Os05g41230 is the most likely candidate gene for qPL5. Of course, the candidate gene must be subjected to verification by positional cloning and transformation.
Rice yield is a complex trait that is ultimately determined by three main factors: TGW, TN, and GN (Xing and Zhang 2010). Development of a NIL population is an appropriate strategy for confirming and evaluating the genetic effects of a QTL (Benson et al. 2015; Ding et al. 2011; Zhou et al. 2018). In the current study, we developed NILs in different genetic backgrounds, and the genetic backgrounds were quantified using the rice 40 k SNP array. Agronomic trait analysis of the NILs showed that qPL5 did positively contribute to GN (grain number per panicle) but that its contribution to TN (tiller number) was negative. The GYPP (grain yield per plant) was significantly increased in the NILs, and this resulted from the increased GN. We noted that there were obvious differences in the relative increase in GYYP between NP/NP-qPL5 (~ 15.13%) and KWY3/KW3-qPL5 (~ 2.28%). This can be explained by the fact that KWY3 is an elite, high-yielding japonica variety with good-quality grain, and our result is consistent with results of previous studies showing that it is difficult to sharply increase grain yields in high yielding varieties (Zhang et al. 2009, 2014). In addition, qPL5 can cause significant changes in PH (plant height) and FLL (flag leaf length), similar to the genetic effects of qPL8 and Ghd7.1 (Liu et al. 2013; Tang et al. 2018; Zhang et al. 2021). Furthermore, there were no significant differences in Hd (heading date) and grain-size-related traits between the NILs and their recipient parental lines, and this is different from the genetic effects of the cloned genes PT2, OsGRF4, and DTH8/Ghd8 that control the regulation of PL (Sun et al. 2016; Xue et al. 2008; Yan et al. 2011). In breeding practice, PH is negatively correlated with lodging resistance, and we might need to pyramid qPL5 with other genes controlling PH to outweigh the genetic effect of qPL5 in future rice breeding (Wang et al. 2016). Meanwhile, we will evaluate the quality traits of NILs, which would provide a comprehensive evaluation of the breeding value of qPL5. Nevertheless, we conclude that qPL5 should be a valuable gene for breeding high-yield japonica varieties, and these results lay the foundation for gene cloning and breeding utilization of qPL5 in the future.
Data availability
All data supporting the conclusions of this article are provided within the article (and its Additional files).
References
Benson JM, Poland JA, Benson BM, Stromberg EL, Nelson RJ (2015) Resistance to gray leaf spot of maize: genetic architecture and mechanisms elucidated through nested association mapping and near-isogenic line analysis. PLoS Genet 11(3):e1005045
Crowell S, Korniliev P, Falcao A, Ismail A, Gregorio G, Mezey J, McCouch S (2016) Genome-wide association and high-resolution phenotyping link Oryza sativa panicle traits to numerous trait-specific QTL clusters. Nat Commun 7:10527
Ding XP, Li XK, Xiong LH (2011) Evaluation of near-isogenic lines for drought resistance QTL and fine mapping of a locus affecting flag leaf width, spikelet number, and root volume in rice. Theor Appl Genet 123:815–826
Feng XM, Wang C, Nan JZ, Zhang XH, Wang RS, Jiang GQ, Yuan QB, Lin SY (2017) Updating the elite rice variety Kongyu 131 by improving the Gn1a locus. Rice 10:35
Gao YH, Xu ZP, Zhang LJ, Li S, Wang SG, Yang HL, Liu XL, Zeng DL, Liu QQ, Qian Q, Zhang BC, Zhou YH (2020) MYB61 is regulated by GRF4 and promotes nitrogen utilization and biomass production in rice. Nat Commun 11:5219
Gupta PK, Rustgi S, Kumar N (2006) Genetic and molecular basis of grain size and grain number and its relevance to grain productivity in higher plants. Genome 49:565–571
Hickey LT, Hafeez AN, Robinson H, Jackson SA, Leal-Bertioli SCM, Tester M, Gao CX, Godwin ID, Hayes BJ, Wulff BBH (2019) Breeding crops to feed 10 billion. Nat Biotechnol 37:744–754
Huang XZ, Qian Q, Liu ZB, Sun HY, He SY, Luo D, Xia GM, Chu CC, Li JY, Fu XD (2009) Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet 41:494–497
Jiao YQ, Wang YH, Xue DW, Wang J, Yan MX, Liu GF, Dong GJ, Zeng DL, Lu ZF, Zhu XD, Qian Q, Li JY (2010) Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 42:541-U536
Khew CY, Teo CJ, Chan WS, Wong HL, Namasivayam P, Ho CL (2015) Brassinosteroid insensitive 1-associated kinase 1 (OsI-BAK1) is associated with grain filling and leaf development in rice. J Plant Physiol 182:23–32
Kim SR, Ramos JM, Hizon RJM, Ashikari M, Virk PS, Torres EA, Nissila E, Jena KK (2018) Introgression of a functional epigenetic OsSPL14(WFP) allele into elite indica rice genomes greatly improved panicle traits and grain yield. Sci Rep-Uk 8:3833
Li F, Liu WB, Tang JY, Chen JF, Tong HN, Hu B, Li CL, Fang J, Chen MS, Chu CC (2010) Rice dense and erect panicle 2 is essential for determining panicle outgrowth and elongation. Cell Res 20:838–849
Li M, Tang D, Wang KJ, Wu XR, Lu LL, Yu HX, Gu MH, Yan CJ, Cheng ZK (2011) Mutations in the F-box gene larger panicle improve the panicle architecture and enhance the grain yield in rice. Plant Biotechnol J 9:1002–1013
Li SB, Qian Q, Fu ZM, Zeng DL, Meng XB, Kyozuka J, Maekawa M, Zhu XD, Zhang J, Li JY, Wang YH (2009) Short panicle1 encodes a putative PTR family transporter and determines rice panicle size. Plant J 58:592–605
Liu E, Liu Y, Wu GC, Zeng SY, Thi TGT, Liang LJ, Liang YF, Dong ZY, She D, Wang H, Zaid IU, Hong DL (2016) Identification of a candidate gene for panicle length in rice (Oryza sativa L.) via association and linkage analysis. Front Plant Sci 7:596
Liu TM, Li LZ, Zhang YS, Xu CG, Li XH, Xing YZ (2011) Comparison of quantitative trait loci for rice yield, panicle length and spikelet density across three connected populations. J Genet 90:377–382
Liu TM, Liu HY, Zhang H, Xing YZ (2013) Validation and characterization of Ghd7.1, a major quantitative trait locus with pleiotropic effects on spikelets per panicle, plant height, and heading date in rice (Oryza sativa L.). J Integr Plant Biol 55:917–927
Livak KJ, Schmittgen TD (2013) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408
Marathi B, Guleria S, Mohapatra T, Parsad R, Mariappan N, Kurungara VK, Atwal SS, Prabhu KV, Singh NK, Singh AK (2012) QTL analysis of novel genomic regions associated with yield and yield related traits in new plant type based recombinant inbred lines of rice (Oryza sativa L.). BMC Plant Biol 12:137
Miura K, Ashikari M, Matsuoka M (2011) The role of QTLs in the breeding of high-yielding rice. Trends Plant Sci 16:319–326
Murray MG, Thompson CL, Wendel JF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:432–4326
Qiao Y, Piao R, Shi J, Lee SI, Jiang W, Kim BK, Lee J, Han LZ, Ma WB, Koh HJ (2011) Fine mapping and candidate gene analysis of dense and erect panicle 3, DEP3, which confers high grain yield in rice (Oryza sativa L.). Theor Appl Genet 122:1439–1449
Shibaya T, Hori K, Ogiso-Tanaka E, Yamanouchi U, Shu K, Kitazawa N, Shomura A, Ando T, Ebana K, Wu J, Yamazaki T, Yano M (2016) Hd18, encoding histone acetylase related to arabidopsis FLOWERING LOCUS D, is involved in the control of flowering time in rice. Plant Cell Physiol 57:1828–1838
Sun PY, Zhang WH, Wang YH, He Q, Shu F, Liu H, Wang J, Wang JM, Yuan LP, Deng HF (2016) OsGRF4 controls grain shape, panicle length and seed shattering in rice. J Integr Plant Biol 58:836–847
Sun ZZ, Yin XL, Ding J, Yu D, Hu M, Sun XW, Tan YN, Sheng XB, Liu L, Mo Y, Ouyang N, Jiang BB, Yuan GL, Duan MJ, Yuan DY, Fang J (2017) QTL analysis and dissection of panicle components in rice using advanced backcross populations derived from Oryza Sativa cultivars HR1128 and ‘Nipponbare’. Plos One 12:e0175692
Tang XX, Gong R, Sun WQ, Zhang CP, Yu SB (2018) Genetic dissection and validation of candidate genes for flag leaf size in rice (Oryza sativa L.). Theor Appl Genet 131:801–815
Tian YH, Zhang HY, Xu PZ, Chen XQ, Liao YX, Han BL, Chen XB, Fu XD, Wu XJ (2015) Genetic mapping of a QTL controlling leaf width and grain number in rice. Euphytica 202:1–11
Uga Y, Nonoue Y, Liang ZW, Lin HX, Yamamoto S, Yamanouchi U, Yano M (2007) Accumulation of additive effects generates a strong photoperiod sensitivity in the extremely late-heading rice cultivar “Nona Bokra.” Theor Appl Genet 114:1457–1466
Wang JX, Sun J, Li CX, Liu HL, Wang JG, Zhao HW, Zou DT (2016) Genetic dissection of the developmental behavior of plant height in rice under different water supply conditions. J Integr Agr 15:2688–2702
Wang SS, Chen RK, Chen KY, Liu CY, Kao SM, Chung CL (2017) Genetic mapping of the qSBN7 locus, a QTL controlling secondary branch number per panicle in rice. Breeding Sci 67:340–347
Wang Y, Zhai LY, Chen K, Shen CC, Liang YT, Wang CH, Zhao XQ, Wang S, Xu JL (2020) Natural sequence variations and combinations of GNP1 and NAL1 determine the grain Number per panicle in rice. Rice 13:14
Xing YZ, Tan YF, Hua JP, Sun XL, Xu CG, Zhang Q (2002) Characterization of the main effects, epistatic effects and their environmental interactions of QTLs on the genetic basis of yield traits in rice. Theor Appl Genet 105:248–257
Xing YZ, Zhang QF (2010) Genetic and molecular bases of rice yield. Annu Rev Plant Biol 61:421–442
Xu ZP, Li SC, Zhang CQ, Zhang BC, Zhu KZ, Zhou YH, Liu QQ (2017) Genetic connection between cell-wall composition and grain yield via parallel QTL analysis in indica and japonica subspecies. Sci Rep-Uk 7:12561
Xu ZP, Miao YX, Chen ZA, Gao HL, Wang RX, Zhao DS, Zhang BC, Zhou YH, Tang SZ, Zhang HG, Liu QQ (2019) Identification and fine mapping of qGN1c, a QTL for grain number per panicle, in rice (Oryza sativa). Mol Breeding 39:129
Xue WY, Xing YZ, Weng XY, Zhao Y, Tang WJ, Wang L, Zhou HJ, Yu SB, Xu CG, Li XH, Zhang QF (2008) Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40:761–767
Yan WH, Wang P, Chen HX, Zhou HJ, Li QP, Wang CR, Ding ZH, Zhang YS, Yu SB, Xing YZ, Zhang QF (2011) A Major QTL, Ghd8, plays pleiotropic roles in regulating grain productivity, plant height, and heading date in rice. Mol Plant 4:319–330
Zhang H, Zhao Q, Sun ZZ, Zhang CQ, Feng Q, Tang SZ, Liang GH, Gu MH, Han B, Liu QQ (2011) Development and high-throughput genotyping of substitution lines carring the chromosome segments of indica 9311 in the background of japonica Nipponbare. J Genet Genomics 38:603–611
Zhang HG, Bo L, Zhu ZB, Cui XF, Tang SZ, Liang GH, Gu MH (2009) Improving resistance of a good-quality japonica variety Wuyujing 3 to rice stripe virus via molecular marker-assisted selection. Chin J Rice Sci
Zhang HG, Zhu GY, Feng ZQ, Ming XU, Jian-An JI, Pei Y, Qian K, Tang SZ, Ming-Hong GU (2014) Analysis on yield and quality of the late-maturity medium japonica rice varieties released in Jiangsu Province in the last 30 years. Chin J Rice Sci 28:327–334
Zhang L, Wang JJ, Wang JM, Wang LY, Ma B, Zeng LJ, Qi YB, Li Q, He ZH (2015) Quantitative trait locus analysis and fine mapping of the qPL6 locus for panicle length in rice. Theor Appl Genet 128:1151–1161
Zhang L, Zou YT, Bian Z, Xie D, Yeilaghi H, Fan XL, Zhao DS, Liu QQ (2021) Fine mapping and candidate gene prediction of the quantitative trait locus qPL8 for panicle length in rice. Phyton-Int J Exp Bot 90:789–802
Zhou Y, Tao YJ, Yuan Y, Zhang YZ, Miao J, Zhang R, Yi CD, Gong ZY, Yang ZF, Liang GH (2018) Characterisation of a novel quantitative trait locus, GN4-1, for grain number and yield in rice (Oryza sativa L.). Theor Appl Genet 131:637–648
Funding
This study was financially supported by the Jiangsu Province Government (Grant Nos. BK20200927, BE2022336, BE2021301-1, and PAPD program), and the National Natural Science Foundation (Grant Nos. 32001517 31771913 and 3207150123).
Author information
Authors and Affiliations
Contributions
ZX and HZ analyzed the data and drafted the manuscript. ZX performed the phenotypic evaluation and data analysis with assistance from ML, YD, XL, and RW. ZC and XZ participated in the construction of the SLs. ST participated in the design of the study. QL and HZ designed the study and revised the manuscript. All of the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflicts of interest
The authors declare no conflicts of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, Z., Li, M., Du, Y. et al. Characterization of qPL5: a novel quantitative trait locus (QTL) that controls panicle length in rice (Oryza sativa L.). Mol Breeding 42, 70 (2022). https://doi.org/10.1007/s11032-022-01339-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-022-01339-z