
Abstract
A total of 26,685 unutilized public domain expressed sequence tags (ESTs) of Arachis hypogaea L. were analyzed to give a total of 4442 EST-SSRs, in which 517 ESTs contained more than one simple sequence repeat (SSR). Of these EST-SSRs, 2542 were mononucleotide repeats (MNRs), 803 were dinucleotide repeats (DNRs), 1043 were trinucleotide repeats (TNRs), 40 were tetranucleotide repeats (TtNRs), six were pentanucleotide repeats (PNRs) and eight were hexanucleotide repeats (HNRs). Out of these 4442 EST-SSRs, only 1160 were found to be successful in non-redundant primer design; 1060 were simple SSRs, while the remaining 100 were compound forms. Among all the motifs, MNRs were abundant, followed by TNRs and DNRs. The AAG/CTT motif was the most abundant (~33 %) TNR, while AG/CT was the most abundant DNR. For redundancy and novelty, a stringent criterion deploying three different strategies was used and a total of 782 novel EST-SSRs were added to the public domain of peanut. These novel EST-SSR markers will be useful for qualitative and quantitative trait mapping, marker-assisted selection and genetic diversity studies in cultivated peanut as well as related Arachis species. A subset of 30 novel EST-SSRs was further randomly selected for validation and genotyping studies with eight well-known cultivars and 32 advanced breeding lines (ADBX lines, ADBY lines and ADBZ lines) from Odisha state, India. The number of polymorphic markers among accessions of A. hypogaea was low; however, a set of informative EST-SSR markers detected considerable levels of genetic variability in peanut cultivars and uncharacterized breeding lines collected from Odisha. The 30 newly developed EST-SSRs from Arachis spp. showed ~97 % amplification in Cicer arientinum and 93 % in pigeon pea. Thus, the EST-SSRs developed in this study will be a very useful asset for genetic analysis, comparative genome mapping, population genetic structure and phylogenetic inferences among wild and allied species of Arachis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Arachis hypogaea L., commonly known as peanut or groundnut, is the only allotetraploid species (2n = 4x = 40) in one of the nine intra-generic taxonomic sections of the genus Arachis. This species is probably derived from a unique cross between wild diploid species of A. duranensis having the A-genome and A. ipaensis having the B-genome, resulting in a natural hybrid followed by spontaneous chromosome duplication (Kochert et al. 1991; Seijo et al. 2007). However, this species behaves genetically as diploid (Stalker and Moss 1987) with equal amounts of contribution from the A and B genomes (Singh et al. 1996). Peanut is generally divided into two subspecies, hypogaea and fastigiata, based on growth habit and the presence or absence of flowers on the main axis. Subspecies hypogaea is again divided into two varieties, var. hirsuta and var. hypogaea, and subspecies fastigiata into four varieties, var. aequatoriana, var. fastigiata, var. peruviana and var. vulgaris based on inflorescence, pod and seed characteristics. Peanut is mainly grown in the semiarid tropics of more than 100 countries with an annual production of 35.5 million tonnes (FAO 2009), India being the second largest producer after China. Crop productivity has been adversely affected by several abiotic and biotic stresses in the tropics. Peanut is an excellent cash crop for domestic as well as international trade and is used for direct consumption, in the confectionary industry, in cooking as vegetable oil, and as a source for protein feed in the animal industry.
A few reports have revealed a large amount of morphological and agronomic variations among accessions of cultivated peanuts, with low levels of polymorphism using markers like RFLP, RAPD and AFLP (Kochert et al. 1991; He and Prakash 1997; Subramanian et al. 2000). To date (June 3, 2014), a total of 15,125 simple sequence repeats (SSRs) were available in the public domain of the Kazusa marker database (http://marker.kazusa.or.jp/Peanut) along with five genetic maps and a consensus map; these do not have sufficient very useful markers for genetic studies. Thus, there is great need for development of more novel SSR markers. Recently, expressed sequence tag (EST)-SSRs have received much attention due to the increasing numbers of ESTs in the public domain for various plants (Scott et al. 2000; Kantety et al. 2002; Varshney et al. 2002). However, EST-SSRs can be rapidly developed by data mining and are well known as SSRs due to their existence in transcribed regions of the genome. Thus, many researchers are currently trying to develop gene-based genetic maps that may help to identify functions of candidate genes and increase the efficiency of marker-assisted selection.
With the development of peanut EST projects, a large number of available EST sequence data have been generated. These data offer an opportunity to identify SSRs in ESTs by data mining. The present study aimed to investigate the available non-redundant ESTs for identification and development of SSRs and their utilization for genetic structure and diversity analysis of available well-known peanut cultivars and breeding lines across the Indian state of Odisha (Odisha-wide). These developed novel EST-SSRs enriched the current resource of molecular markers, which can be useful for qualitative and quantitative trait mapping, marker-assisted selection and genetic diversity studies in cultivated peanut as well as their wild relatives.
Materials and methods
EST-SSR database search and removal of redundancy
A total of 29,503 A. hypogaea EST sequences were retrieved from the NCBI database (search up to January 22, 2014). From these, a total of 3187 ESTs had already been used for designing EST-SSRs in the Kazusa database and the remaining 26,685 ESTs (equivalent to 16.2 Mb) were taken into consideration in designing EST-SSRs. For EST-SSR development, we analyzed all these sequences by tandem repeat finder and then sequences were assembled by CAP3 program (Huang and Madan 1999) to remove the redundant ones. After pre-processing, SSR-containing sequences were identified by a PERL script-based program, MISA software (MIcroSAtellite identification tool, http://pgrc.ipk-gatersleben.de/misa/).
Primer design
All SSR-containing non-redundant ESTs were utilized for primer design following standard parameters by using Primer3 software (Rozen and Skaletsky 1999) considering the following criteria: (a) target amplicon size of 100–500 bp, (b) optimal annealing temperature of 55–60 °C, (c) average GC content of 50–60 % and (d) primer length of 18–24 bp. Putative functions of EST-SSR loci were assigned by comparison with the non-redundant sequence database at NCBI using BLASTX 2.2.17 software (Altschul et al. 1997).
Plant materials and genomic DNA isolation
Eight potential Indian cultivars of A. hypogaea L. and 32 breeding lines, established at Orissa University of Agriculture and Technology (OUAT), Bhubaneswar, were used in this study (Table 1). In addition, four important allied species of Arachis, namely Caesalpinia pulcherrima, Cicer arientinum, pigeon pea and Pisum sativum were used in our study to check the cross-genera transferability of the identified EST-SSRs. All the potential cultivars and breeding lines of A. hypogaea were grown in the experimental gardens and greenhouse facility of OUAT, Bhubaneswar, in 2011. Genomic DNA was isolated from the leaves of each plant using the CTAB method (Williams et al. 1990).
PCR amplification for EST-SSR polymorphism
The polymorphism and transferability study of 30 developed primer pairs was carried out following the method of Sim et al. (2009). The primers were commercially synthesized (ABI, USA, Gene Identification Pvt. Ltd., USA, and MWG Pvt. Ltd., Bangalore, India) with forward primers having the fluorescent label FAM/HEX/VIC/NED. The details of these new markers, viz., locus designation, primer sequences, repeat motifs, allele attributes, PIC estimates and Genbank accession numbers, are summarized in Supplementary data S6. Each 20-µl PCR reaction mix contained 50 ng of DNA, 2 µl of 10× PCR buffer, 2 mM MgCl2, 0.2 mM of each dNTPs, 1 µM of each primer and 1 U of Taq DNA polymerase (Invitrogen, USA). A touchdown PCR program was used consisting of an initial denaturation at 95 °C for 2 min, followed by 15 cycles of denaturation at 95 °C for 20 s, annealing at 65 °C for 50 s and extension at 72 °C for 50 s. The annealing temperature decreased by 1 °C after every cycle till it reached 55 °C. The next 25 cycles consisted of denaturation at 95 °C for 20 s, annealing at 55 °C for 50 s and extension at 72 °C for 50 s, followed by a final extension at 72 °C for 7 min. The amplified products were run on a capillary-based 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA), and the products were precisely sized for major, comparable and conspicuous peaks using Gene Mapper v4.0 (Applied Biosystems).
Primer redundancy analysis
All EST-SSRs were compared with Kazusa microsatellite databases using its BLAST server. The designed primers (forward and reverse) for various EST-SSRs were used to compare with the downloaded reference primer sequences in order to remove duplications/redundancies. This analysis tool is built around the pair-wise comparison algorithm with the restriction of no gap and no mismatch.
Statistical and genetic analysis
The allelic data for 40 genotypes of A. hypogaea including the breeding lines established at OUAT, Bhubaneswar, Odisha, were used to calculate different statistical and genetic parameters. Effective allele number was estimated as the reciprocal of homozygosity (Hartl and Clark 1989), and Shannon index was measured for gene diversity (Shannon and Weaver 1949). Observed heterozygosity (H o) was calculated as the fraction of heterozygous genotypes over the total number of genotypes. Expected heterozygosity (H e) was calculated based on the probability that two individuals taken at random from a given sample would possess different alleles at a locus (Nei and Li 1979) and according to the following formula:
Two-locus linkage disequilibrium (LD) was estimated as Burrow’s composite measure of linkage disequilibrium between pairs of loci and χ 2 tests for significance (Weir 1979) using these codominant markers.
The polymorphism information content (PIC) is a measure of the effectiveness of a given DNA marker for detecting polymorphism. The PIC value for each EST-SSR marker was calculated by using the standard formula (Botstein et al. 1980):
where n is the total number of alleles detected for a microsatellite marker, p i is the frequency of the ith allele, and p j is the frequency of the (i + 1)th allele in the set of analyzed genotypes.
The bi-allelic polymorphic data were also tested for Hardy–Weinberg equilibrium (HWE) using Fisher’s exact test and Markov chain algorithm with a forecasted chain length of 10,000,000 and 100,000 dememorization steps, and the linkage disequilibrium (LD) test was performed using 1000 permutations. All these estimates were calculated using Power Marker version 3.25, Popgene version 1.32, Arlequin version 3.1 and Darwin version 5.0.158. The discriminatory power of each microsatellite locus was calculated by estimating sib-based and unbiased corrected p i estimates, and the cumulative power of discrimination was calculated as products of p i s of successive informative markers arranged in decreasing order as described by Waits et al. (2001).
Cross-taxa transferability was calculated as the proportion of primers showing successful amplification vis-a-vis all the primers tested, whereas primer conservation was calculated as the proportion of the species displaying successful amplification vis-a-vis all the tested markers.
In addition, amplification products of ten EST-SSRs with parental lines and breeding lines of Arachis were sequenced to check the repeat motif conservation and variations (SNPs/indels) in their flanking region, if any. The final edited sequences belonging to each locus were compared with respective original EST-SSR sequences using CLUSTALX (www.ftp-igbmc.u-strasbg.fr/pub/ClustalX) to ascertain the target domain/SSR conservation.
The SSR data were used to ascertain the generic relationships/affinities between the cultivated genotypes and breeding lines using cluster analysis based on genetic distance values. Initially, 100 bootstrap distance matrices were generated using a bi-allelic microsatellite data analysis tool and Nei’s genetic distance measure. From these distance data, neighbor-joining (NJ) trees were generated for each matrix separately using Mega 4.1 with the ‘neighbor’ command, which was followed by the generation of consensus trees.
Genetic structure analysis
Hierarchical partitioning of genetic diversity among and within the natural and the ex situ populations was examined by the Bayesian clustering model built into the software STRUCTURE version 2.3 (Pritchard et al. 2000). STRUCTURE was used to estimate the number of natural populations (K) and the proportion of assignment of individuals from each of the assumed populations to each of the inferred natural genetic clusters. An admixture model with correlated allele frequencies was used to infer the value of K with prior population information. All analyses were performed with a burn-in period of 100,000 and a Markov chain Monte Carlo (MCMC) replication number set up to 1,000,000. In order to estimate the real value of K and the ancestry membership of each individual in the inferred cluster, the program was run 20 times for each K, ranging from 1 to 8. The value of K was estimated using the method described by Evanno et al. (2005). The output from STRUCTURE analysis was the proportion of ancestry membership of each individual of the population in the inferred cluster calculated by averaging the membership coefficient obtained from 20 runs of the real K.
Secondly, a principal coordinate analysis (PCA) was conducted, on the basis of similarity measures (S ij ) using DCENTER and EIGEN procedures in NTSYS version 2.02 (Rohlf 1998) to cluster all the accessions into major groups. To complement this factorial analysis, a hierarchical cluster analysis such as UPGMA was employed for dendrogram construction. This was done with the respective pair-wise Jaccard’s genetic similarities (GS) with the same package of NTSYS. Confidence limits of the UPGMA dendrogram were determined by computing 500 bootstrap replicates and constructing a majority-rule consensus tree based on bootstrap values of >50 %, by using WinBoot version 4.0 (Yap and Nelson 1996).
Results
Mining of functional SSRs in peanut ESTs
ESTs provide the opportunity to explore single, low-copy conserved sequence motifs for the mining of functional SSRs. Out of 29,503 ESTs, 2818 were utilized to develop 3187 EST-SSRs in the Kazusa database and 26,685 ESTs were left unutilized (Supplemental table S1). Thus, 26,685 ESTs (16.2 Mb) of more than 180 bp were mined with higher stringency to search 4442 SSRs to give a total of 3772 ESTs, in which 517 ESTs contained more than one SSR (Table 2). These 4442 EST-SSRs included 2542 mononucleotide repeats (MNRs), 803 dinucleotide repeats (DNRs), 1043 trinucleotide repeats (TNRs), 40 tetranucleotide repeats (TtNRs), six pentanucleotide repeats (PNRs) and eight hexanucleotide repeats (HNRs) (Table 2). Out of 4442 SSRs, 1160 were found to be successful in non-redundant primer design and the rest were unsuitable for further analysis, as those sequences did not fit the criteria of 200 bp whole length, a minimum of 50 bp of both flanking regions, and non-redundancy in primer sequences. Out of 1160 EST-SSRs, 1060 were simple SSRs, while the remaining 100 were in compound form (Table 2). A total of 37 different SSR motifs were identified in the unigene sets. The top 15 motifs with two complementary sequences represented 98 % of the functional SSRs, while 22 motifs accounted for 2 % of SSRs. Among all the motifs, MNRs were abundant (frequency 2542, ~57 %) followed by TNRs (frequency 1043, ~23.5 %) and DNRs (frequency 803, ~18 %) (Supplemental table S2, Supplemental figure S3; Fig. 1). Among MNRs, the A/T motif was the most common (~95 %) followed by C/G (5 %). Similarly, among TNRs, the AAG/CTT motif was the most abundant (~33 %) followed by 12 % AAT/ATT and ~10 % AAC/GTT (Supplemental figure S2). However, functional SSR data in the present study represented a smaller number of DNRs than TNRs (Fig. 1). The 3372 SSR-containing ESTs represented approximately 16.2 Mb of peanut, having 4442 functional SSRs, thereby suggesting an average frequency of SSRs of ~1/3.6 kb and/or 1/6 non-redundant ESTs (Table 2).

Graphical frequency distribution of various nucleotide repeats in SSR sequences identified in public domain ESTs. a Representation of total various repeat motifs and b representation of various repeat motif with its number
Putative functions of the ESTs containing SSRs
EST-SSR markers were explored for their potential utility in the study of the peanut structural genome; 1160 EST sequences (1060 simple and 100 compound SSRs) were compared with the NR as well as TAIR database using BLASTX. Out of 1160 EST sequences, only 206 (~18 %) showed homology to a large number of annotated proteins in the TAIR database, while 238 (20 %) showed homology to annotated proteins in the NR database. Thus, a large number of ESTs (~80 %) when searched for putative function resulted in no hit or a hypothetical protein. This above-mentioned specific transcript of peanut was not yet characterized for its putative function. Information on these EST-SSRs as well as their putative function are presented in Supplemental tables S4 and S5 to give an overview of functional categories of EST-SSRs. Gene ontology (GO) analysis was recorded for the 20 % of sequences that had homology to known proteins; GO analysis of EST-SSRs was divided into the broadest functional groups of molecular function, biological process and cellular component categories.
SSR redundancy and novelty with respect to public domain database
The verification of redundancy against existing sequence data was performed by deploying three different strategies using BLASTN search. In the first strategy, we performed BLAST hits of the complete sequence against the public domain database; in the second, we used 50 bp immediately upstream and downstream of the repeat motif; and in the third, we used forward and reverse primers. A total of 974 non-redundant novel EST-SSRs were obtained in the first strategy, while 1028 and 822 EST-SSRs were obtained in the second and third strategies, respectively. When the non-redundant SSRs obtained in the three strategies were compared with each other, 782 EST-SSRs found common to all three strategies were novel (Supplemental table S6, Supplemental figure S7) in addition to the EST-SSRs in the Kazusa database.
Validation of EST-SSRs, their polymorphism and cross-genera transferability
Out of 1160 primer pairs flanking a total of 1260 SSRs (1060 in simple and 200 in 100 compound forms) (Supplemental table S8), 30 randomly selected primer pairs were assayed across a set of eight cultivars of A. hypogaea to validate the amplification and further screened for their cross-transferability in other leguminous and allied species.
In the present study, 30 valid EST-SSR primer pairs were used for assessment of the polymorphism among eight cultivars of Arachis. Within cultivated peanut, 18 (~60 %) EST-SSRs exhibited polymorphism and 12 were found to be monomorphic (Supplemental table S9). Out of 12 monomorphic markers, two were DNRs, five were TNRs, two were PNRs and three were perfect compound markers. Similarly, out of these 18 polymorphic EST-SSRs, eight were DNRs, six were TNRs, two were PNRs, and two were compound forms (Supplemental Table S9). A total of 49 alleles were detected with 18 polymorphic SSRs, and the average number of alleles per polymorphic SSR marker was 2.72, with a range of 2–5 alleles, revealing low-to-medium allelic diversity. The PIC values ranged from 0.15 (NBRI_RS836) to 0.77 (NBRI_RS494) with an average value of 0.49. The greatest variation in SSR alleles was found in NBRI_RS494, which interacted with five alleles in eight cultivated peanut genotypes (Supplemental table S9).
The polymorphism of 30 peanut-derived EST-SSRs in 40 peanut cultivars and breeding lines was evaluated. The results revealed that 49 out of 74 EST-SSR loci (~66 %) were polymorphic (Supplemental table S8), with a total of 2009 alleles. The number of alleles detected among 32 advanced breeding lines ranged from two to five, with an average of 2.46 alleles per locus. A maximum of five alleles were observed for primer NBRI_RS494 (Supplemental table S10). The level of allelic diversity within peanut varieties was estimated using mean panmictic heterozygosity (H t; Nei and Li 1979) and Shannon’s information index (I) (Shannon and Weaver 1949; Lewontin 1972). Mean panmictic heterozygosity and mean Shannon’s information index were 0.34 ± 0.19 and 0.50 ± 0.27, respectively. The effective number of alleles was also estimated in all 40 peanut cultivars and breeding lines. These studied cultivars/breeding lines showed a low level of homozygosity, as reflected in the effective number of alleles (1.63 ± 0.41). The mean observed number of alleles was 1.89 ± 0.49 in the different genotypes of peanut in the present study.
To test cross-genera transferability, 30 SSRs were tested on a panel of four allied species of Arachis (C. pulcherrima, C. arientinum, P. pea and P. sativum) with each having a single elite genotype. The 30 newly developed EST-SSRs from Arachis spp. showed ~97 % amplification in C. arientinum, ~93 % in pigeon pea, and ~86 % in C. pulcherrima and P. sativum (Fig. 2). A maximum of ~97 % cross-genera transferability was noticed with C. arientinum followed by pigeon pea and C. pulcherrima (Fig. 2).

Graphical presentation of cross-genera transferability of 30 EST-SSRs in different allied species of Arachis
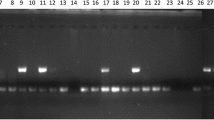
Subsequently, PCR amplification of ten EST-SSRs in six advanced breeding lines and two parental lines was sequenced to check the repeat motif conservation and variation in their flanking regions, if any. These sequence files were analyzed and unequivocally confirmed the motif conservation and presence of single nucleotide polymorphisms (SNPs) in the flanking region. For EST-SSR (titled NBRI_RS 492 in Supplemental table S8), peanut cultivar parent-4 AK 159, parent-7 TG 38B and one advanced breeding line ADBZ1 had seven dinucleotide repeat motifs CT, while advanced lines ADBY9, ADBY12, ADBY15, ADBY16 and ADBZ3 each had six dinucleotide repeat motifs. However, this EST-SSR was found to be different with regard to the flanking region as compared to SNPs (Supplemental figure S11).
Genetic affinities within varieties/advanced breeding lines/promising genotypes of peanut
To ascertain the useful attributes of the genetic markers in genetic diversity analysis, a total of 30 potential EST-SSRs were assayed with 40 peanut varieties/promising advanced breeding lines. The genetic distance (Nei et al. 1983) based on Jaccard’s similarity index was calculated with 54 polymorphic loci amplified by 30 EST-SSRs and varied from 0.07 to 0.52 with an average value of 0.27 (Supplemental table S12). All the eight parental cultivars showed an average genetic distance of 0.32 between each other, while the remaining 36 advanced breeding lines showed an average genetic distance of 0.26 between each other (Supplemental table S12). Of the eight parents, parent-8 Smruti showed the highest genetic distance with each advanced breeding line, while parent-3 R 2001-3 and parent-6 TAG24 showed the lowest genetic distance with the advanced breeding lines (Supplemental table S12). All the 40 genotypes of peanut cultivars/breeding lines fell into three well-supported major clusters: cluster I contained 12 genotypes including two parental lines, parent-1 AK 12–24 and parent-2 TG 26; cluster II contained 12 genotypes including one parental line, parent-3 R 2001-3; and cluster III contained 16 genotypes including five parental lines, parent-4 AK 159, parent-5 Devi, parent-6 TAG24, parent-7 TG 38B and parent-8 Smruti (Fig. 3).

Genetic relationship among the eight parental cultivars and 36 advanced breeding lines from Odisha based on NJ clustering using the data of 30 EST-SSRs
Population structure and genetic differentiation of Odisha-wide peanut cultivars
The model-based population structure analysis assuming the entire set of peanut cultivars as various sub-populations (K = 2 to K = 10) using 23 polymorphic SSR markers in STRUCTURE program grouped the 40 genotypes according to the value of K. Even with the assumption of a higher number of sub-populations, the number of sub-populations remained at two without much differentiation, representing many geographical locations (Supplemental figure S13). In most cases, the likelihood increased until the real K was reached and then eased off. On the other hand, the distribution of delta-K almost always showed a mode at the real K (Supplemental table S14). The Bayesian model of analysis of genetic structure of A. hypogaea in STRUCTURE revealed the value of K to be 2. The Evanno test detected two genetic clusters (K = 2) showing an average ancestry membership participation of >80 % to one of the two inferred clusters. The first cluster (I) contained 10 genotypes, while the second (II) contained only 27 genotypes. Only three genotypes showed an ancestry value lower than 70 %. Of these three, two genotypes (ADBY19 and ADBY1) showed an ancestry value of more than 50 % to inferred cluster I, while the third genotype (parent-2 TG 26) showed an ancestry value of more than 60 % to inferred cluster II (data not shown). The low ancestry memberships of these three genotypes of peanut signaled a weak population genetic structure with a strong admixture.
PCA analysis of genetic distance of SSR analysis revealed the same discrete clusters on the basis of their population identities (Supplemental figure S15). The first three axes in the PCA plot explained 21.31, 15.69 and 13.34 %, respectively. Parental cultivars such as parent-5 Devi, parent-6 TAG24, parent-7 TG 38B and parent-8 Smruti were clustered with few breeding lines while parent-1 AK 12–24, parent-2 TG 26 and parent-3 R 2001-3 with few breeding lines. Many breeding lines fell in between these parental clusters, having much similar genetic background. The parent-4 AK 159 clustered differently with eight breeding lines which revealed a genetic differentiation from many other breeding lines (Supplemental figure S15).
The percentage polymorphism (P), number of different alleles (N a), number of effective alleles (N e), number of private alleles, heterozygosity (H) and Shannon index (I) were estimated for all genotypes/breeding lines sampled from Odisha state. The observed number of alleles (N a) and effective number of alleles (N e) varied from 1 to 4 and 1.00 to 2.00, respectively. Nei’s as well as Levene’s expected heterozygosity (H) varied from 0 to 0.55 with an estimated mean value of 0.34 ± 0.19, although a small difference (~0.006) was seen between two methods. The Shannon index (I) varied from 0 (NBRI_RS1053, NBRI_RS666, NBRI_RS667, and NBRI_RS1074) to 0.85 (NBRI_RS10520), while the mean estimate was 0.23 ± 0.07.
Result of analysis of molecular variance (AMOVA) from the data revealed 94 % genetic differentiation within cultivars/breeding line, and 6 % among breeding lines and parental cultivars. with a negative Fst of −0.25 %. There was a negative Fst (−0.44 %) in the breeding lines, while an Fst of 0.52 % was found in the parental cultivars.
Discussion
SSR frequency and distribution
In this study, a total of 4442 SSR regions were identified in 26,685 potential non-redundant ESTs, of which 1160 ESTs were found successful with non-redundant primers. Previously, Koilkonda et al. (2012) reported the discovery of 1254 SSR regions in 10,102 non-redundant peanut ESTs, while Guo et al. (2009) reported 856 SSR regions in 6888 non-redundant peanut ESTs, and Liang et al. (2009) identified 881 SSR regions in 11,431 non-redundant ESTs. In this study, the frequency of SSR identification was more or less similar to these previous studies, as the SSR search criteria were different in all these studies. The frequcy of EST-SSRs was ~1/3.6 kb for peanut public domain ESTs, which was higher than those of wheat (~1/15.6 kb) (Kantety et al. 2002), barley (~1/6.3 kb) (Thiel et al. 2003), Arabidopsis thaliana (~1/13.83 kb), tomato (~1/11.1 kb), cotton (~1/20.0 kb), soybean (~1/7.4 kb) and poplar (~1/14.0 kb) (Cardle et al. 2000). However, it was lower than coffee (~1/1.56 kb) (Aggarwal et al. 2007) and rubber ~1/2.5 kb (Feng et al. 2009). The distribution, frequency and abundance level of EST-SSRs may fluctuate due to SSR search criteria, the size of the database and SSR development tools (Varshney et al. 2005). For example, La Rota et al. (2005) found that, in rice, the frequency of EST-SSRs was reduced from 50 to 1 % when SSR search criteria changed from 12 to 30 bp. In this study, we have also shown that changing the SSR search criteria resulted in frequency fluctuation of EST-SSRs in peanut, i.e., if the criteria were MNR ≥ 10, DNR ≥ 6, TNR to HNR ≥ 5, the EST-SSR frequency would be ~1/1.36 kb.
To date, DNRs and TNRs have mostly been reported in plants, but the dominant repeat motifs were different. For example, TNRs were the leading repeat motif in A. thaliana, wheat, rice, corn, soybean (Cardle et al. 2000), sugarcane, grapes (Cordeiro et al. 2001), barley (Thiel et al. 2003) and citrus (Chen et al. 2006), while DNRs were dominant in apricot and peach (Jung et al. 2005) and coffee (Aggarwal et al. 2007). Our findings confirm that in peanut TNRs (54.9 %) were the leading repeat motif, followed by DNRs (42.2 %), without considering the MNRs. In addition, AAG/CTT was the predominant TNR, which was consistent with the report of Zhao et al. (2012) and Koilkonda et al. (2012). Observations of AG/CT, as the leading repeat motif of DNRs in peanut, found it to be similar to that in citrus (Liu et al. 2013), rice (Miyao et al. 1996), pea (Gong et al. 2010), coffee (Aggarwal et al. 2007), proso millet (Rajput et al. 2014) and Cassava (Vásquez and López 2014). The number of PNRs and HNRs in our study was low and is possibly related to the stringent searching criteria. In fact, PNRs and HNRs could be obtained when the searching criteria were reduced (data not shown). This also verified an earlier viewpoint that EST-SSR distribution and frequency were related to the search criteria (Varshney et al. 2005).
The GC/CG repeat motif was not found in peanut EST-SSRs in our study, which is consistent with the results in other plants (Gao et al. 2003; Nicot et al. 2004; Jung et al. 2005). However, the GC/CG repeat motif was reported at 1 % or even lower frequency in coffee EST-SSRs (Aggarwal et al. 2007; Zhao et al. 2012). Comparison of the length of SSRs revealed that the mean length of dinucleotide SSRs was significantly longer than that of trinucleotide SSRs for EST-SSRs. This finding was consistent with observations in barley (Ramsay et al. 2000), sugarcane (Cordeiro et al. 2001) and soybean (Song et al. 2004).
Novel EST-SSR molecular markers
During the past two decades, much effort has been made to develop genetic and genomic tools in cultivated peanut, such as the construction of BAC libraries (Yüksel and Paterson 2005), cDNA libraries (Guo et al. 2009; Koilkonda et al. 2012) and genetic linkage maps (Moretzsohn et al. 2009; Varshney et al. 2009; Qin et al. 2012) and the development of DNA markers (He et al. 2005; Gimenes et al. 2007; Wang and Guo 2007; Cuc et al. 2008; Gautami et al. 2009; Macedo et al. 2012). In addition, a large amount of transcriptome sequencing of peanut has been performed on various tissues in different sequencing platforms without sufficient work on the development and application of SSR markers in peanut genetic and breeding studies. This present study added a total of 782 novel EST-SSRs to the previous 3187 public domain EST-SSRs in the Kazusa database (http://marker.kazusa.or.jp/Peanut/marker/list/EST-SSR), which contained a total of 15,125 SSRs of A. hypogaea. This novel search criterion in our study was stringent as whole sequence, primer sequence and both 50-bp flanking sequences were used following the published literature (Jena et al. 2012). Thus, these novel 782 EST-SSR markers could enrich the current resource of molecular markers for the peanut community and would be useful for qualitative and quantitative trait mapping, marker-assisted selection and genetic diversity studies in cultivated peanut as well as related Arachis species.
Polymorphism analysis and cross-genera transferability
The average PIC value amplified by 30 primer pairs across the cultivated species was 0.38, which represented the medium polymorphism of our developed EST-SSRs. These results indicated that the 30 markers would be useful for genetic and breeding studies in peanut. Only eight (~27 %) of 30 SSRs in eight cultivars amplified monomorphic bands, while the remaining 22 (~73 %) showed polymorphism. Among all the polymorphic primer pairs, eight (80 %) were DNRs (out of 10), seven (63 %) were TNRs (out of 11), and two (50 %) were PNRs (out of four), while the rest were compound type. This result confirmed that higher-order repeat motifs have lower polymorphism than lower-order repeat motifs. In our study, all the five compound imperfect SSRs showed polymorphism among eight peanut cultivars and the lower-order repeat motifs were predominant among all the polymorphic SSR loci. One possible explanation why the higher-order repeat motifs were less polymorphic is their lower slippage possibilities during the process of DNA replication.
Recent studies showed that the cross-genera transfer efficiency of EST-SSRs was higher than that of genomic SSRs. Liewlaksaneeyanawin et al. (2004) compared the transferability of 14 Pinus taeda EST-SSRs from public domain EST databases and 99 traditional microsatellite markers (including seven genomic SSRs) and found that EST-SSRs had higher transfer rates than the traditional microsatellite markers. Peakall et al. (1998) used the SSR markers developed from the soybean genome amplifying across Glycine max and the results showed that 65 % of the markers could be amplified in cross-species, but only 3–13 % in cross-genera. Similar results were obtained in coffee (Aggarwal et al. 2007). Transferability of EST-SSRs between closely related genera has been reported in many other crop species such as Actinidia chinensis, Oryza, Arabidopsis, apricot and grape (Huang et al. 1998; Chen et al. 2002; Clauss et al. 2002; Decroocq et al. 2003). In the study by Gao et al. (2005), the transfer rate decreased slightly in the more closely related species and became lower in the most distantly related species in the genus. According to Liewlaksaneeyanawin et al. (2004), the success of transferability decreased as the evolutionary distance between the source and target species increased.
In our study, all of the randomly selected 30 EST-SSR markers developed from peanut could be amplified in many allied genera, such as ~97 % in Cicer, ~93 % in Cajanus and ~86 % in Caesalpinia and Pisum. Our results show that the transfer rate decreased from 97 % in C. arientinum and 93 % in Cajanus cajan to ~86 % in C. pulcherrima and P. sativum, and seemed to indicate that C. pulcherrima and P. sativum had a more distant relationship with A. hypogea then C. arientinum and Cajanus cajan.
Evaluation of genetic diversity using the novel EST-SSR markers developed and genetic relationships of peanut cultivars/breeding lines
About 100 improved peanut cultivars have been released in India since 1905; 48 % of these resulted from selection from landraces and 45 % from hybridization followed by selection, and the remaining 7 % from mutation breeding (Nigam 2000). The NJ clustering pattern resulted in three clusters: the first cluster comprised parent-1 AK 12–24 and parent-2 TG 26 together with 10 breeding lines, while the second cluster comprised parent-3 R 2001-3 and 11 breeding lines. The cluster III comprised five parents (parent-4 AK 159 to parent-8 Smruti) and 11 breeding lines. Of the eight parents, parent-8 Smruti exhibited the highest genetic distance with each advanced breeding line, while parents parent-3 R 2001-3 and parent-6 TAG24 exhibited the lowest genetic distance with the advanced breeding lines (Supplemental table S9). Thus, all the eight parental lines of Odisha, India, showed various genetic backgrounds and were suitable for breeding programs. Some of the genetic distance estimates between cultivars and breeding lines were found to be high, and thus it is important for plant breeders to evaluate genetic diversity comprehensively not only in various cultivars but also in the advanced uncharacterized breeding lines. AMOVA analysis revealed that, of the total genetic variance, 6 % was attributed to among parental cultivar and breeding lines, while 94 % was within parental cultivar and breeding line, which indicated that there were obvious differences in peanut cultivars from different breeding lines in Odisha.
When we compared genetic diversity estimates of the present Odisha-wide cultivars and breeding lines with the published literature, we found many deviations. Tshilenge-Lukanda et al. (2012), with the help of ISSRs, reported genetic distances among the groundnut accessions varying from 11 to 37 %, and thus the accessions analyzed were different but genetically closely related. Similarly, Dwivedi et al. (2001) showed genetic dissimilarities of the same plant materials values ranging from 1.2 to 41 %. Other molecular studies detected up to 52 % genetic dissimilarities with AFLPs (He and Prakash 1997) and 56 % with SSRs (Mace et al. 2006) using different groundnut collections. The present study also revealed a moderate level of intra-species variability of 27 % on average for self-pollinating species like groundnut.
Compared with previous reports, the genetic diversity of the Odisha peanut cultivars/breeding lines was at a moderate level, as reflected by the average number of alleles per locus (2.73) and the gene diversity index (0.5). Other researchers have reported averages of 2.2–8.1 alleles per locus in various peanut collections (Kottapalli et al. 2007; Wang et al. 2011; Ren et al. 2014). Wang et al. (2011) carried out a study of a USA peanut collection and found an average of 8.1 alleles per locus. Similarly, Kottapalli et al. (2007) studied 168 accessions of peanut germplasm from 42 countries on five continents with SSRs and identified 4.29 alleles per locus. In a Chinese core collection, the number of polymorphic alleles of var. hypogaea germplasm was 4.0, while that of breeding lines was 2.2 (Jiang et al. 2010). In a similar recent study of Chinese cultivars, the allele number per locus was similar to that of breeding lines (Jiang et al. 2010). This indicates that there would be less genetic diversity in the breeding lines (released for cultivation) than the landraces. The present study on Odisha (India) peanut cultivars also revealed a similar pattern of low level of diversity among breeding lines in comparison to cultivars. In fact, the breeding lines and cultivars (outside of landraces) had the same origins from crossing.
In this study, the gene diversity index was 0.23, which was higher than those of previous peanut collections (0.15–0.18) (Kottapalli et al. 2007; Wang et al. 2011). This is probably due to the inclusion of 32 breeding lines in addition to eight cultivars, making the whole analysis system as a synthetic one with unrelatedness. Although allelic diversity and gene diversity index could be used as indicators of genetic variation, such values are relative and depend on the number of polymorphic loci and the relatedness of genotypes analyzed. However, in this study, the allelic diversity and gene diversity index showed that there was a higher level of genetic diversity in Odisha peanut cultivars than landraces, which was consistent with the results reported in rice (Zhang et al. 2011; Zhao et al. 2009), wheat (Chen et al. 2012) and soybean (Li et al. 2010). Thus, it is necessary to introduce more peanut germplasm into Odisha peanut cultivars to broaden their genetic diversity.
Validation of cross-genera amplicons
Cross-species/genera transferability of SSR markers has been previously reported (Davierwala et al. 2001). The results of amplification of expected products by 30 EST-SSRs in eight peanut cultivars and four related species and inter-genera provided clear evidence for the conservation and transferability of the EST-SSR markers in A. hypogaea. It also supported the fact that SSR markers can be obtained through transfer amplification (Varshney et al. 2005). However, compared to the RefEST, insertions and deletions were detected in the regions of SSR motifs in intra-species, inter-species and inter-genera, while point mutations or deletions in flanking regions were found more often in inter-genera and less in inter-species. Gutiérrez and Goyache (2005) found that the variations in the sequences in Medicago truncatula are mainly due to the diversification of the number of repeat motifs in the SSR region combined with insertions and base substitutions. Xie et al. (2006) showed that allelic size variation in almond resulted exclusively from differences in the structures of repeat motifs which involved interruptions or occurrences of new motif repeats in addition to varying numbers of AG/CT repeats. Guo et al. (2006) reported that no correlation was found between the repeat motif type and cross-species amplification. No positive pattern of allelic variation and transferability and their definite relationship to the repeat motif type for the number of repeats was confirmed in our study.
Population structure of peanut cultivars/breeding lines in Odisha
To identify the true optimal number of subsets (K) in STRUCTURE, LnP(D) and ΔK were chosen (Chen et al. 2012; Patzak et al. 2012). The K value that provides the maximum likelihood, called LnP(D) in STRUCTURE, is generally considered to be the optimal number of subdivisions (Pritchard et al. 2000). In this study, the distribution of L(K) did not show a clear mode for the true K in Odisha peanut cultivars. Thus, another ad-hoc quantity (ΔK) was used to overcome the difficulty in interpreting the real K values (Evanno et al. 2005). The highest value of ΔK for the 40 peanut accessions is ΔK = 2. Clustering bar plots with K = 2 are shown in Supplemental figure S13. At K = 3, 4 and 5, all 40 accessions are divided into three. Four and five subpopulations in which more or fewer breeding accessions made various subgroups and thus there was genetic differentiation in the breeding lines with various level admixtures. Analysis of these data identified the major substructure groups when the number of populations was set at two with the highest value of ΔK, which was inconsistent with the clustering results based on genetic distance (Fig. 3). As shown in Fig. 3, most of the accessions were clearly classified into one of the three subpopulations. PCA based on the marker genotypes revealed three distinct clusters for the entire population (Supplemental figure S15). Furthermore, the neighbor-joining tree showed three clusters within the peanut cultivars, which was fairly consistent with the structure-based membership assignment for most of the cultivars.
A moderate genetic base had been reported for peanut cultivars in Odisha, probably because of high selection pressure for good grain quality and repeated use of the same-origin parents with proven yield ability in the breeding program, resulting in significant genetic erosion of the local peanut gene pool (Yu 2010, 2011).
Conclusions
The results of this study reveal that the peanut accessions analyzed are closely related despite the high level of molecular variation detected using SSR markers. The genetic diversity is greater in cultivars than in breeding lines. The present study showed the utility of EST-derived microsatellite markers for the detection of polymorphisms among cultivated peanut and breeding line accessions and for genetic relationship analysis between A. hypogaea accessions. A total of 782 novel EST-SSR markers were developed and added to the public domain for genetic analysis of peanut. The number of polymorphic markers among accessions of A. hypogaea was still low, corroborating the notion that cultivated peanut presents a relatively reduced variation at the DNA level. However, a set of informative EST-SSR markers in the present study detected considerable levels of genetic variability in the Odisha peanut cultivar and uncharacterized breeding line collections. Based on this data, the genetic background of breeding lines could be assessed. The identification of similarity groups could be useful for the selection of parental plants to be used in the breeding programs. The 30 newly developed EST-SSRs from Arachis spp. showed ~97 % amplification in C. arientinum, ~93 % in pigeon pea, and ~86 % in C. pulcherrima and P. sativum (Fig. 3). A maximum of ~97 % cross-genera transferability was seen with C. arientinum followed by pigeon pea and C. pulcherrima (Fig. 3). Marker transferability ranged from 97 % (C. arientinum) to 86 % (C. pulcherrima) in different genera, while it was not all checked in other species of Arachis. The possibility of using microsatellite markers developed for one species in genetic evaluation of other species greatly reduces the cost of the analysis, since the development of microsatellite markers is still expensive and time-consuming. The EST-SSR markers developed in this study could be very useful for genetic analysis of wild and allied species of Arachis, including comparative genome mapping, population genetic structure and phylogenetic inferences among species.
References
Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, Singh L (2007) Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet 114(2):359–372
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32(3):314–331
Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, Waugh R (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156(2):847–854
Chen X, Cho Y, McCouch S (2002) Sequence divergence of rice microsatellites in Oryza and other plant species. Mol Genet Genomics 268(3):331–343
Chen C, Zhou P, Choi YA, Huang S, Gmitter FG (2006) Mining and characterizing microsatellites from citrus ESTs. Theor Appl Genet 112(7):1248–1257
Chen X, Min D, Yasir TA, Hu YG (2012) Genetic diversity, population structure and linkage disequilibrium in elite Chinese winter wheat investigated with SSR markers. PLoS One 7(9):e44510
Clauss MJ, Cobban H, Olds TM (2002) Cross-species microsatellite markers for elucidating population genetic structure in Arabidopsis and Arabis (Brassicaeae). Mol Ecol 11(3):591–601
Cordeiro GM, Casu R, McIntyre CL, Manners JM, Henry RJ (2001) Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci 160(6):1115–1123
Cuc LM, Mace ES, Crouch JH, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8(1):55
Davierwala AP, Reddy APK, Lagu MD, Ranjekar PK, Gupta VS (2001) Marker assisted selection of bacterial blight resistance genes in rice. Biochem Genet 39(7–8):261–278
Decroocq V, Fave MG, Hagen L, Bordenave L, Decroocq S (2003) Development and transferability of apricot and grape EST microsatellite markers across taxa. Theor Appl Genet 106(5):912–922
Dwivedi SL, Gurtu S, Chandra S, Yuejin W, Nigam SN (2001) Assessment of genetic diversity among selected groundnut germplasm. Plant Breed 120(4):345–349
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Feng SP, Li WG, Huang HS, Wang JY, Wu YT (2009) Development, characterization and cross-species/genera transferability of EST-SSR markers for rubber tree (Hevea brasiliensis). Mol Breed 23(1):85–97
Gao L, Tang J, Li H, Jia J (2003) Analysis of microsatellites in major crops assessed by computational and experimental approaches. Mol Breed 12(3):245–261
Gao LZ, Zhang CH, Jia JZ (2005) Cross-species transferability of rice microsatellites in its wild relatives and the potential for conservation genetic studies. Genet Res Crop Evol 52(7):931–940
Gautami B, Ravi K, Narasu ML, Hoisington DA, Varshney RK (2009) Novel set of groundnut SSR markers for germplasm analysis and interspecific transferability. Int J Integr Biol 7(2):100–106
Gimenes MA, Hoshino AA, Barbosa AVG, Palmieri DA, Lopes CR (2007) Characterization and transferability of microsatellite markers of the cultivated peanut (Arachis hypogaea). BMC Plant Biol 7(1):9
Gong Y, Xu S, Mao W, Hu Q, Zhang G, Ding J, Li Y (2010) Developing new SSR markers from ESTs of pea (Pisum sativum L.). J Zhejiang Univ Sci B 11(9):702–707
Guo W, Wang W, Zhou B, Zhang T (2006) Cross-species transferability of G. arboreum-derived EST-SSRs in the diploid species of Gossypium. Theor Appl Genet 112(8):1573–1581
Guo B, Chen X, Hong Y, Liang X, Dang P, Brenneman T, Holbrook C, Culbreath A (2009) Analysis of gene expression profiles in leaf tissues of cultivated peanuts and development of EST-SSR markers and gene discovery. Int J Plant Genomics. doi:10.1155/2009/715605
Gutiérrez JP, Goyache F (2005) A note on ENDOG: a computer program for analysing pedigree information. J Anim Breed Genet 122(3):172–176
Hartl DL, Clark AG (1989) Principles of population genetics, 2nd edn. Sinauer Associates Inc, Sunderland, MA
He G, Prakash CS (1997) Identification of polymorphic DNA markers in cultivated peanut (Arachis hypogaea L.). Euphytica 97(2):143–149
He G, Meng R, Gao H, Guo B, Gao G, Newman M, Pittman RN, Prakash CS (2005) Simple sequence repeat markers for botanical varieties of cultivated peanut (Arachis hypogaea L.). Euphytica 142(1–2):131–136
Huang X, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res 9(9):868–877
Huang WG, Cipriani G, Morgante M, Testolin R (1998) Microsatellite DNA in Actinidia chinensis: isolation, characterisation, and homology in related species. Theor Appl Genet 97(8):1269–1278
Jena SN, Srivastava A, Rai KM, Ranjan A, Singh SK, Nisar T, Srivastava M, Bag SK, Mantri S, Asif MH, Yadav HK, Tuli R, Sawant SV et al (2012) Development and characterization of genomic and expressed SSRs for levant cotton (Gossypium herbaceum L.). Theor Appl Genet 124(3):565–576
Jiang HF, Ren X-P, Zhang XJ, Huang JQ, Lei Y, Yan L-Y, Liao B-S, Upadhyaya HD, Holbrook CC (2010) Comparison of genetic diversity based on SSR markers between peanut mini core collections from China and ICRISAT. Acta Agron Sin 36(7):1084–1091
Jung S, Abbott A, Jesudurai C, Tomkins J, Main D (2005) Frequency, type, distribution and annotation of simple sequence repeats in Rosaceae ESTs. Funct Integr Genomics 5(3):136–143
Kantety RV, Rota ML, Matthews DE, Sorrells ME (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol 4(5–6):501–510
Kochert G, Halward T, Branch WD, Simpson CE (1991) RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor Appl Genet 81(5):565–570
Koilkonda P, Sato S, Tabata S, Shirasawa K, Hirakawa H, Sakai H, Sasamoto S, Watanabe A, Wada T, Kishida Y, Tsuruoka H, Fujishiro T, Yamada M, Kohara M, Suzuki S, Hasegawa M, Kiyoshima H, Isobe S (2012) Large-scale development of expressed sequence tag-derived simple sequence repeat markers and diversity analysis in Arachis spp. Mol Breed 30:125–138
Kottapalli KR, Burow MD, Burow G, Burke J, Puppala N (2007) Molecular characterization of the US peanut mini core collection using microsatellite markers. Crop Sci 47(4):1718–1727
La Rota L, Mauricio Kantety RV, Yu JK, Sorrells ME (2005) Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley. BMC Genom 6(1):23
Lewontin RC (1972) The apportionment of human diversity. In: Evolutionary biology. Springer, US, pp 381–398
Li YH, Li W, Zhang C, Yang L, Chang RZ, Gaut BS, Qiu LJ (2010) Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol 188(1):242–253
Liang X, Chen X, Hong Y, Liu H, Zhou G, Li S, Guo B (2009) Utility of EST-derived SSR in cultivated peanut (Arachis hypogaea L.) and Arachis wild species. BMC Plant Biol 9(1):35
Liewlaksaneeyanawin C, Ritland CE, El-Kassaby YA, Ritland K (2004) Single-copy, species-transferable microsatellite markers developed from loblolly pine ESTs. Theor Appl Genet 109(2):361–369
Liu S, Li W, Wu Y, Chen C, Lei J (2013) De novo transcriptome assembly in chili pepper (Capsicum frutescens) to identify genes involved in the biosynthesis of capsaicinoids. PLoS One 8(1):e48156
Mace ES, Phong DT, Upadhyaya HD, Chandra S, Crouch JH (2006) SSR analysis of cultivated groundnut (Arachis hypogaea L.) germplasm resistant to rust and late leaf spot diseases. Euphytica 152(3):317–330
Macedo SE, Moretzsohn MC, Leal-Bertioli SCM, Alves DMT, Gouvea EG, Azevedo VCR, Bertioli DJ (2012) Development and characterization of highly polymorphic long TC repeat microsatellite markers for genetic analysis of peanut. BMC Res Notes 5(1):86
Miyao A, Zhong HS, Monna L, Yano M, Yamamoto K, Havukkala I, Takuji Minobe Y, Sasaki T (1996) Characterization and genetic mapping of simple sequence repeats in the rice genome. DNA Res 3(4):233–238
Moretzsohn MC, Barbosa AVG, Alves-Freitas DMT, Cristiane Teixeira C, Leal-Bertioli SCM, Guimarães PM, Pereira RW (2009) A linkage map for the B-genome of Arachis (Fabaceae) and its synteny to the A-genome. BMC Plant Biol 9(1):40
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76(10):5269–5273
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol 19(2):153–170
Nicot N, Chiquet V, Gandon B, Amilhat L, Legeai F, Leroy PI, Bernard M, Sourdille P (2004) Study of simple sequence repeat (SSR) markers from wheat expressed sequence tags (ESTs). Theor Appl Genet 109(4):800–805
Nigam SN (2000) Some strategic issues in breeding for high and stable yield in groundnut in India. J Oilseed Res 17(1):1–10
Patzak J, Paprštein F, Henychová A, Sedlák J (2012) Comparison of genetic diversity structure analyses of SSR molecular marker data within apple (Malus × domestica) genetic resources. Genome 55(9):647–665
Peakall R, Gilmore S, Keys W, Morgante M, Rafalski A (1998) Cross-species amplification of soybean (Glycine max) simple sequence repeats (SSRs) within the genus and other legume genera: implications for the transferability of SSRs in plants. Mol Biol Evol 15(10):1275–1287
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Qin H, Feng S, Chen C, Guo Y, Knapp S, Culbreath A, He G, Wang ML, Zhang X, Holbrook CC (2012) An integrated genetic linkage map of cultivated peanut (Arachis hypogaea L.) constructed from two RIL populations. Theor Appl Genet 124(4):653–664
Rajput SG, Plyler-Harveson T, Santra DK (2014) Development and characterization of SSR markers in proso millet based on switchgrass genomics. Am J Plant Sci 5(1):175
Ramsay L, Macaulay M, Ivanissevich SD, MacLean K, Cardle L, Fuller J, Edwards KJ, Tuvesson S, Morgante M, Massari A (2000) A simple sequence repeat-based linkage map of barley. Genetics 156(4):1997–2005
Ren Y, McGregor C, Zhang Y, Gong G, Zhang H, Guo S, Sun H, Cai W, Zhang J, Xu Y (2014) An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol 14(1):33
Rohlf FJ (1998) NTSYS-pc version 2.0. Numerical taxonomy and multivariate analysis system. Exeter software, Setauket, New York
Rozen S, Skaletsky H (1999) Primer3 on the WWW for general users and for biologist programmers. In: Bioinformatics methods and protocols. Humana Press, New York, pp 365–386
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ (2000) Analysis of SSRs derived from grape ESTs. Theor Appl Genet 100(5):723–726
Seijo G, Lavia GI, Fernández A, Krapovickas A, Ducasse DA, Bertioli DJ, Moscone EA (2007) Genomic relationships between the cultivated peanut (Arachis hypogaea, Leguminosae) and its close relatives revealed by double GISH. Am J Bot 94(12):1963–1971
Shannon CE, Weaver W (1949) The mathematical theory of communication. The University of Illinois, Urbana, pp 3–24
Sim SC, Yu JK, Jo Y, Sorrells ME, Jung G (2009) Transferability of cereal EST-SSR markers to ryegrass. Genome 52(5):431–437
Singh KP, Raina SN, Singh AK (1996) Variation in chromosomal DNA associated with the evolution of Arachis species. Genome 39(5):890–897
Song QJ, Marek LF, Shoemaker RC, Lark KG, Concibido VC, Delannay X, Specht JE, Cregan PB (2004) A new integrated genetic linkage map of the soybean. Theor Appl Genet 109(1):122–128
Stalker HT, Moss JP (1987) Speciation, cytogenetics, and utilization of Arachis species. Adv Agron 41:1–40
Subramanian V, Gurtu S, Rao RCN, Nigam SN (2000) Identification of DNA polymorphism in cultivated groundnut using random amplified polymorphic DNA (RAPD) assay. Genome 43(4):656–660
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106(3):411–422
Tshilenge-Lukanda L, Nkongolo KKC, Kalonji-Mbuyi A (2012) Epidemiology of the groundnut (Arachis hypogaea L.) leaf spot disease: genetic analysis and developmental cycles. Am J Plant Sci 3:582–588
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7(2A):537–546
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Varshney RK, Bertioli DJ, Moretzsohn MC, Vadez V, Krishnamurthy L, Aruna R, Nigam SN, Moss BJ, Seetha K, Ravi K (2009) The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor Appl Genet 118(4):729–739
Vásquez A, López C (2014) In silico genome comparison and distribution analysis of simple sequences repeats in Cassava. Int J Genomics. doi:10.1155/2014/471461
Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10(1):249–256
Wang Y, Guo X (2007) Development and characterization of EST-SSR markers in the eastern oyster Crassostrea virginica. Mar Biotechnol 9(4):500–511
Wang ML, Sukumaran S, Barkley NA, Chen Z, Chen CY, Guo B, Pittman RN, Stalker HT, Holbrook CC, Pederson GA (2011) Population structure and marker–trait association analysis of the US peanut (Arachis hypogaea L.) mini-core collection. Theor Appl Genet 123(8):1307–1317
Weir BS (1979) Inferences about linkage disequilibrium. Biometrics 1:235–254
Williams DE, Eisenman J, Baird A, Rauch C, Ness KV, March CJ, Park LS, Martin U, Mochizukl DY, Boswell HS (1990) Identification of a ligand for the c-kit proto-oncogene. Cell 63(1):167–174
Xie H, Sui Y, Chang FQ, Xu Y, Ma RC (2006) SSR allelic variation in almond (Prunus dulcis Mill.). Theor Appl Genet 112(2):366–372
Yap IV, Nelson RJ (1996) Winboot: a program for performing bootstrap analysis of binary data to determine the confidence limits of UPGMA-based dendrograms. International Rice Research Institute, Manila, pp 1–22
Yu SL (2010) Peanut varieties and their pedigree in China. Shanghai Science and Technology Press, Shanghai
Yu SL (2011) Peanut genetics and breeding in China. Shanghai Scientific and Technology Press, Shanghai
Yüksel B, Paterson AH (2005) Construction and characterization of a peanut HindIII BAC library. Theor Appl Genet 111(4):630–639
Zhang P, Li P, Li X, Liu X, Zhao X, Lu Y (2011) Population structure and genetic diversity in a rice core collection (Oryza sativa L.) investigated with SSR markers. PLoS One 6(12):e27565
Zhao W, Chung JW, Ma KH, Kim TS, Kim SM, Shin DI, Kim CH, Koo HM, Park YJ (2009) Analysis of genetic diversity and population structure of rice cultivars from Korea, China and Japan using SSR markers. Genes Genomics 31(4):283–292
Zhao W, Lee GA, Kwon SW, Ma KH, Lee MC, Park YJ (2012) Development and use of novel SSR markers for molecular genetic diversity in Italian millet (Setaria italic L.). Genes Genomics 34(1):51–57
Acknowledgments
We thank the Vice Chancellor, OUAT, Bhubaneswar, Odisha, for providing facilities to carry out this work; the SSR analysis bioinformatics team of CSIR-National Botanical Research Institute, Lucknow, for their rapid support; Vice Chancellor, Utkal University, Odisha, for his support extended to our collaborators.
Author contributions
Sushree Shivani Sardar was involved in collections of plant materials and breeding lines, DNA isolation and SSR genotyping. Kedareswar Pradhan coordinated collections of plant materials and breeding lines, DNA isolation, SSR genotyping and manuscript drafting. Ravi Prakash Shukla designed EST-SSRs primers and their comparison with existing public domain database. Ribha Saraswat was involved in peanut public domain data search and analysis for unutilized ESTs, designing of EST-SSRs and their primers. Anukool Srivastava was involved in repeat motif analysis and their frequency and comparison of EST–SSRs with existing public domain database and novelty of EST-SSRs. Satya Narayan Jena coordinated designing EST-SSRs, their primers, redundancy check, analysis for genetic relationship and population structuring pattern among breeding line and cultivars with SSRs and manuscript writing. Anath Bandhu Das was involved in amplification of EST-SSRs and their sequencing for number of repeat motif analysis, novel EST-SSRs annotation and cross-transferability analysis.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical standard
Authors have followed the ethical standards while preparing the present manuscript resulting from their investigations.
Additional information
Dr. Kedareswar Pradhan and Dr. Satya Narayan Jena have agreed to be shared corresponding authors as both have designed and coordinated work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental table S1
Details of number of public domain ESTs, utilized ESTs and studied ESTs (DOCX 14 kb)
Supplemental table S2
Frequencies of different subclasses of EST-SSRs measured in repeat number of count (DOCX 22 kb)
Supplemental figure S3
Frequencies of different subclasses of EST-SSRs found in 26,685 unutilized ESTs of Arachis (PPTX 74 kb)
Supplemental table S4
Details of protein homology search with NR database (XLSX 41 kb)
Supplemental table S5
Details of protein homology search with TAIR database (XLSX 63 kb)
Supplemental table S6
Unmatched SSR IDs with references to whole sequence, primer sequence and flanking region searches (XLSX 86 kb)
Supplemental table S7
Venn diagram of novel search of 1160 EST-SSRs with reference to whole sequence, primer sequence and 50-bp 3′ and 5′ flanking regions with public domain database (PPTX 94 kb)
Supplemental table S8
Details of non-redundant EST-SSRs, their primer pairs, EST sources, GenBank ID, repeat motif, Tm and expected size (XLSX 123 kb)
Supplemental table S9
Details of validation of 30 EST-SSRs with eight parental genotypes of Arachis of Odisha state (XLSX 18 kb)
Supplemental table S10
Allele-size data of 30 validated EST-SSRs in 40 genotypes/advanced breeding lines of peanut (XLSX 25 kb)
Supplemental table S11
Multiple alignment of size-variant fragments amplified from two peanut parents and advanced breeding lines for EST-SSR marker, NBRI RS 492, showing the presence of repeat motif (CT)n with seven units in both parents and ADBZ1, but six units in ADBY9, ADBY12, ADBY15, ADBY16, ADBZ3. The flanking region of EST-SSR revealed the presence of SNPs (PPTX 105 kb)
Supplemental table S12
Detailed pair-wise genetic distance of 40 cultivars/breeding lines of Arachis collected from Odisha state. (XLSX 52 kb)
Supplemental table S13
Population structure of 40 genotypes/advanced breeding lines estimated from 30 EST-SSRs using STRUCTURE program at K = 2 (PPTX 89 kb)
Supplemental table S14
Various values of ln′(K), |ln″(K)| and delta-K at different presumed K values in Evanno test (XLSX 11 kb)
Supplemental table S15
Principal coordinate analysis of 30 EST-SSRs profiles to resolve genetic relationships among the eight parents and 32 advanced breeding lines (PPTX 48 kb)
Rights and permissions
About this article

Cite this article
Sardar, S.S., Pradhan, K., Shukla, R.P. et al. In silico mining of EST-SSRs in Arachis hypogaea L. and their utilization for genetic structure and diversity analysis in cultivars/breeding lines in Odisha, India. Mol Breeding 36, 49 (2016). https://doi.org/10.1007/s11032-016-0466-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0466-y