
Abstract
By backcrossing and self-pollinating twice, a series of improved lines (BC2F3) of cotton derived from super-quality fiber property chromosome segment introgression lines were developed by molecular marker-assisted selection (MAS). One of these improved lines, 3326-7, in the background of Xinluzao (XLZ) 41, was transferred from an introgression line (IL088-A7-3) derived from a cross between Gossypium hirsutum acc. TM-1 and G. barbadense cv. Hai7124. This line has consistently demonstrated super-quality fiber properties including fiber length, strength, and fineness. To fine-map the clustered fiber quality quantitative trait loci (QTLs), we further crossed the homozygous line 3326-7 with its recurrent parent XLZ41 to produce BC3F2, BC3F3, and BC3F4 populations. A linkage group was constructed using 1248 BC3F2 plants, and QTL analysis using the fiber qualities of 229 recombinants confirmed the existence of qFL-chr.7 for fiber length, qFS-chr.7 for fiber strength, and qFM-chr.7 for micronaire value within the introgressed region, with R 2 values of 18.6, 29.4, and 26.3 %, respectively. The presence of qFL-chr.7, qFS-chr.7, and qFM-chr.7 led to an increase in fiber length and strength of approximately 2.0 mm and 3.0 cN/tex, respectively, and a decrease in micronaire of 0.8. Using substitution mapping with 229 BC3F2 recombinants and 207 BC3F3 and BC3F4 lines with informative recombination breakpoints in the target region, we anchored qFL-chr.7 for fiber length and qFS-chr.7 for strength to the same position, with a 0.36-cM interval between the two simple sequence repeat markers, NAU3735 and NAU845. Another QTL, qFM-chr.7, was mapped to the 0.44-cM interval between MGHES75 and NAU7445. These tightly linked QTLs may explain their positive relationship and have the potential to improve cotton fiber qualities through MAS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton is an important cash crop and the most widely planted renewable natural textile fiber source in the world. The improvement of cotton fiber quality has become critical due to improvements in spinning technology. The genus Gossypium comprises 45 diploid and five tetraploid species (Percival et al. 1999). Upland cotton, Gossypium hirsutum L., is the most important species, yielding approximately 90 % of the world’s cotton fiber. Another cultivated tetraploid species, G. barbadense L., is well known for its extra-long-staple fibers, and has much stronger and longer fibers and lower micronaire than G. hirsutum. The use of G. barbadense to introgress novel genetic variability into G. hirsutum elite cultivars is one of the most important challenges for cotton breeders. G. barbadense is a useful resource for fiber quality improvement. Unfortunately, the introduction of exotic genes into an elite genome can lead to a decrease in desirable agronomic characteristics due to linkage drag of genes with undesirable effects. Many loci associated with fiber quality have been identified using F2, BC1, and recombinant inbred lines (RILs) (Jiang et al. 1998; Ulloa and Meredith 2000; Kohel et al. 2001; Paterson et al. 2003; Mei et al. 2004; Lacape et al. 2005, 2010; Park et al. 2005; Zhang et al. 2005a, b, 2009a, b; Shen et al. 2005, 2006; He et al. 2007; Wu et al. 2009; Wang et al. 2012a, b; Sun et al. 2012; An et al. 2010; Qin et al. 2008; Shappley et al. 1998). The precision of estimates of the effects and position of fiber quality quantitative trait loci (QTLs) in these populations is limited due to interactions, as well as genetic and environmental factors.
Using molecular markers and suitable statistical methods (Tanksley 1993), the chromosomal locations of QTLs involved in characteristics of interest can be determined and selected, allowing the breeder to discard other regions with undesirable effects after relatively few generations of marker-assisted selection (MAS) (Young and Tanksley 1989). Near-isogenic lines (NILs) have been developed from diverse species (Tanksley and Nelson 1996; Bernacchi et al. 1998; Fray et al. 2000; Fridman et al. 2000; Brouwer and Clair. 2004; Xie et al. 2006; Thomson et al. 2006; Szalma et al. 2007; Zhang et al. 2009a, b; Ding et al. 2011; Chen et al. 2012). Despite the fact that these NILs usually contain only a small percentage of the donor genome, they are often modified for several traits, including some undesirable effects. It is necessary to reduce the extent of undesirable introgression in order to assess whether the undesirable effects are caused by linkage drag of other genes or by pleiotropic effects of a single locus. Breaking the undesirable linkage effects can render introgressions more useful for breeding programs. This process also has the added benefit of preventing the loss of desirable genes by recombination after the generations of breeding needed to incorporate them into the elite germplasm using MAS.
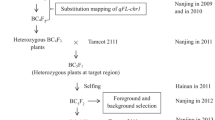
Wang et al. (2012a, b) developed a set of chromosome segment introgression lines (CSILs) from a cross between G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124. One of these CSILs, IL088-A7-3, contains the G. barbadense introgression of chromosome (Chr.) 7, a region previously shown to affect several traits including fiber length, strength, and micronaire. To improve the fiber quality of G. hirsutum cv. XLZ41, IL088-A7-3 was crossed twice with XLZ41. Seventeen homozygous plants were identified from 117 BC2F2 individuals from the cross of IL088-A7-3 × XLZ41 using the target region simple sequence repeat (SSR) markers NAU3028 and NAU2186. Among these BC2F2 individuals, 3326-7, which contains the target introgression of Chr. 7, shares approximately 89.5 % background similarity with the recurrent parent and exhibits significantly longer and stronger fibers and lower micronaire values than the recurrent parent (XLZ41). The main objectives of the current study were to determine the QTLs that might be responsible for the phenotypic effects modified in 3326-7 and to distinguish between pleiotropy and linkage in the genetic control underlying the traits affected by this region by analyzing a new set of recombinant subCSILs in order to enhance MAS efficiency. This study lays the foundation for map-based cloning of these important QTLs in the future.
Materials and methods
Plant materials
In our previous study, the clustered QTLs for fiber length, strength, and micronaire were confirmed in IL088-A7-3 with the target region (Wang et al. 2012a, b). To improve the fiber qualities of G. hirsutum cv. XLZ41, IL088-A7-3 was crossed and backcrossed with XLZ41 to produce the BC2 generation. Seventeen homozygous plants were identified from 117 BC2F2 individuals using the target region SSR markers NAU3028 and NAU2186 (Cao et al. 2014). To fine-map the clustered QTLs of fiber length, strength, and micronaire, one BC2F2:3 line, 3326-7, which is homozygous for the G. barbadense DNA in the target region, was selected from among the BC2F2:3 lines and crossed with XLZ41 to produce the BC3F2, BC3F3, and BC3F4 populations used as the mapping population in this study.
Phenotypic evaluation
A total of 1248 BC3F2 plants were grown in 2011 in Jiangpu Breeding Station at Nanjing Agricultural University (JBS/NAU), Nanjing (Jiangsu, the Yangtze River cotton-growing region in China) for fine mapping of the targeted QTLs. Of them, 207 recombinant plants were identified using SSR markers. These 207 recombinant BC3F3 seeds were divided into two parts; one was planted in Sanya, Hainan Province, in the winter of 2011 to generate BC3F4 lines, and another one was kept in the laboratory and planted in 2012 for QTL mapping in JBS/NAU. The BC3F4 lines harvested from Hainan were further planted in Shihezi (Xinjiang, the Northwestern cotton-growing region in China). The cotton plants were planted in rows 0.5 m wide and 5 m long (10 m2 per plot) at a planting density of approximately three plants/m2 in a completely randomized block design with two replications in field trials of BC3F3 plants in JBS/NAU in 2012 and Sanya in the winter of 2011. However, a field trial of BC3F4 plants in Shihezi in 2012 was carried out with the same completely randomized block design as in JBS/NAU with rows 0.5 m wide and 5 m long, but at a planting density of approximately 24 plants/m2.
The following yield traits were scored: boll weight (BW) and lint percentage (LP). For each plot, ten neighboring individuals were randomly selected to evaluate the number of bolls per plant (NB). A 50-boll sample was hand-harvested from the first position bolls near the middle nodes of plants in each plot. Each sample was weighed and ginned to determine boll weight (BW) and lint percentage (LP). The following fiber quality traits were evaluated using an HVI Spectrum tester: 2.5 % fiber span length (FL, mm), fiber strength (FS, cN/tex), and micronaire reading (FM). Trait means were calculated using SPSS v.17.0 software (SPSS, Chicago, IL, USA). Analysis of variance was performed using SAS/STAT software (SAS Institute, 1994).
DNA extraction and SSR genotyping
DNA was extracted from the fresh leaves of 1248 BC3F2 plants. DNA extraction was performed as described by Paterson et al. (1993). Sixteen SSR markers showing polymorphisms between XLZ41 and 3326-7 in the G. barbadense introgression region were used for genotyping of the 1248 BC3F2 plants. The polymorphic markers were used to conduct background screening of genetic background similarity (GBS) compared to the recipient. The genetic composition of the target genotype plants from the various levels of the CSILs was calculated according to the formula GBS = N/S × 100 % (Zhao et al. 2010), where S is the number of polymorphic markers between the donor parent (3326-7) and the recipient or recurrent parent (XLZ41), and N is the number of monomorphic markers between the selected plants and the recurrent parent.
QTL analysis
Genetic linkage and orientation of the SSR markers in the G. barbadense chromosomal segment on Chr. 7 were determined using Joinmap 3.0 (Ooijen and Voorrips 2002) with a logarithm of odds (LOD) value of 3.0. The conversion of recombination frequencies to centimorgans (cM) was calculated using the Kosambi function (Kosambi 1944). The graphic representation of the linkage groups was created using MapChart 2.2 (Voorrips 2006). QTLs were identified by composite interval mapping (Zeng 1994) using Windows QTL Cartographer 2.5 (Wang et al. 2010), and a LOD threshold of 2.5 was used in the F2 population. QTLs were fine-mapped by comparing the phenotypic means, and the significance of the difference was analyzed by Tukey’s multiple comparison test using substitution homozygous BC3F3 and BC3F4 lines.
Results
Genome-wide analysis of introgressed line 3326-7 with SSR markers
To identify the genetic background of the improved line 3326-7, which shares the greatest genetic similarity with the recipient parent (XLZ41), 630 SSR markers with an average intermarker distance of 5 cM that were evenly spaced on the 26 chromosomes of upland cotton were selected based on our linkage map (Guo et al. 2008) to screen polymorphisms between TM-1 and XLZ41; 57 polymorphic SSR markers showed such polymorphisms. A total of six G. hirsutum acc. TM-1 chromosome segments were detected in 3326-7, which showed 89.5 % background similarity to XLZ41. The TM-1 introgressed segments were distributed on Chrs. A1 (NAU3901), A3 (NAU1167), D5 (NAU2741), D11 (NAU3493), D4 (NAU3557), and D9 (NAU3100).
Trait performance of the parents, BC3F2, BC3F3 and BC3F4 populations
The distribution of the fiber qualities of the parents, BC3F2, BC3F3, and BC3F4 populations are shown in Table 1 and Fig. 1. The mean fiber length in the BC3F2, BC3F3, and BC3F4 populations in the four field trials was 31.4 mm (ranging from 29.7 to 33.8 mm), which was significantly higher (P < 0.05) than that of XLZ41 (30.7 mm). The mean fiber strength was 31.2 cN/tex, ranging from 29.0 to 34.1 cN/tex, which was significantly higher (P < 0.05) than that of XLZ41 (30.6 cN/tex), and the mean micronaire was 4.6 (ranging from 3.6 to 5.0), significantly lower (P < 0.05) than that of XLZ41 (5.0). These results suggest that the super-quality fiber alleles derived from G. barbadense cv. Hai7124 are stable.

Frequency distributions of fiber quality traits in four environments. E1, E2, E3, and E4 denote Hainan (Oct. 2011), JBS/NAU (Apr 2011), JBS/NAU (Apr 2012), and Shihezi (Apr 2012), respectively
The correlation coefficients between the three fiber qualities and two yield traits are shown in Table 2. Of all the investigated pairs, high positive correlations were found between fiber length and strength (r = 0.721), and between lint percentage and fiber micronaire (r = 0.712). Significant negative correlations were observed between fiber length and micronaire (r = −0.372) and between fiber strength and micronaire (r = −0.332), with no significant correlations observed in other pair-tests between fiber quality and yield traits.
QTL detection of fiber qualities by composite interval mapping
To enrich the number of markers within the target region, all SSRs in the vicinity of the target region were selected based on four other high-density maps (Guo et al. 2008; Lacape et al. 2009; Xiao et al. 2009; Yu et al. 2011). The 1248 BC3F2 individuals were genotyped using 16 SSRs showing polymorphisms between 3326-7 and XLZ41 in the G. barbadense introgression region, of which 229 recombinants were evaluated for fiber quality. The results of QTL analysis of fiber length, strength, and micronaire in the BC3F2, BC3F3, and BC3F4 generations derived from the cross between 3326-7 and XLZ41 are shown in Table 3.
QTL analysis revealed that there were significant peaks between markers NAU3735 and NAU845 on Chr. 7, with LR scores of 13.2 (LOD = 2.87) and 27.0 (5.87), and R 2 values (phenotypic variance explained by the QTL) of 18.6 and 29.4 % for qFL-chr.7 and qFS-chr.7, respectively. There was a significant peak between markers MGHES75 and NAU7445 in the target region, with an LR score of 22.3 (LOD = 4.85) and an R 2 value of 26.3 % (Table 3).
QTL analysis of BC3F3 lines derived from selfing each BC3F2 plant confirmed the qFL-chr.7 and qFS-chr.7 peaks between markers NAU3735 and NAU845 on Chr. 7, with LR scores of 22.0 and 22.8, and R 2 values of 25.0 and 28.4 %, respectively, which were consistent with the target intervals of these QTLs from the BC3F2 population. For qFM-chr.7, there was a significant peak between markers MGHES75 and NAU7445 in the target region, with a LR score of 24.6 and a R 2 value of 28.8 % (Table 3).
QTL analysis of BC3F4 lines derived from selfing each BC3F3 line further confirmed that qFL-chr.7, qFS-chr.7, and qFM-chr.7 did exist on Chr. 7 (Table 3).
The LOD distribution and confidence intervals for qFL-chr.7, qFS-chr.7, and qFM-chr.7 over three generations (BC3F2, BC3F3, and BC3F4) are shown in Fig. 2. In addition, a linkage map of the target region was constructed that spanned a distance of 7.57 cM. As populations vary, confidence intervals are likely to change. However, the overlapping marker intervals for NAU3735-NAU845 and MGHES75-NAU7445 were determined for qFL-chr.7 and qFS-chr.7, and for qFM-chr.7. The direction of additive effects was the same in G. barbadense (Table 3). These QTLs were confirmed to be in the same regions, as revealed by comparing the results of the QTL analysis with composite interval mapping from the BC3F2, BC3 F3, and BC3F4 populations.

LOD distribution and confidence intervals for different traits on Chr.7 over three generations. Black lines below the LOD distribution graph indicate the 1-LOD and 2-LOD QTL likelihood intervals, respectively. Trait legends from top to bottom. FL-F2: fiber length in the BC3F2 population; FS-F2: fiber strength in the BC3F2 population; FM-F2: fiber micronaire in the BC3F2 population; FL-F3: fiber length in the BC3F3 population; FS-F3: fiber strength in the BC3F3 population; FM-F3: fiber micronaire in the BC3F3 population; FL-F4: fiber length in the BC3F4 population; FS-F4: fiber strength in the BC3F4 population; FM-F4: fiber micronaire in the BC3F4 population
Fine mapping of fiber quality QTLs by substitution mapping
Of the 1248 BC3F2 plants subjected to genotyping with 16 SSR markers, a total of 229 recombinants were detected between markers CGR5372 and CGR6381. We constructed a high-resolution map with 1248 plants using published markers anchored in the target region (Fig. 3). According to their marker genotypes, 229 recombinants within the introgressed fragment in BC3F2 were segregated out. Of these recombinants, 207 were harvested to produce enough seeds to be planted in BC3F3 and BC3F4 for fine mapping of fiber quality QTLs. These recombinants could be further classified into 29 groups based on their breakpoints (Table 4). Using a substitution mapping strategy, we mapped qFL-chr.7, qFS-chr.7, and qFM-chr.7 to the same interval as the QTL interval that was identified based on mean comparisons of fiber qualities.

Fine mapping of qFL-chr.7, qFS-chr.7, and qFM-chr.7 using a substitution mapping strategy. High-resolution linkage map of the QTL region produced using 1248 BC3F2 plants. The number of recombinants between adjacent markers is indicated under the linkage map. Progeny testing of BC3F3 and BC3F4 homozygous recombinants delimited the qFL-chr.7 and qFS-chr.7 loci to the region between markers NAU3735 and NAU845, and the qFM-chr.7 locus to the region between markers MGHES75 and NAU7445. The 229 BC3F2 recombinants were grouped into 29 groups based on genotype. The numbers of recombinants in each group and phenotypic differences in mean fiber length, fiber strength, and micronaire between the group and the controls 3326-7 and XLZ41 in four environments are shown on the right. An “a” following the phenotypic value indicates that the mean phenotypic value of the recombinant was not significantly different from that of 3326-7 at a significance level of P < 0.05; a “b” indicates that the mean phenotypic value of the recombinant was not significantly different from that of XLZ41 at a significance level of P < 0.05. White portions of the graph represent homozygous XLZ41 DNA, black regions represent homozygous Hai7124 DNA, and grey portions represent regions where crossing-over occurred
In all 207 BC3F3 and BC3F4 lines from the 229 recombinants, homozygous and heterozygous recombinants were identified with all SSRs and evaluated for their fiber qualities. The number of recombinants in each group and the phenotypic differences in mean fiber length, strength, and micronaire between each group and the controls 3326-7 and XLZ41 over the four environments are presented in Fig. 3. To allow the fine mapping of qFL-chr.7 and qFS-chr.7, groups G1–G12 (carrying progressively reduced homozygous Hai7124 introgressed segments from marker CGR6381 to NAU3735) and G16–G18 (carrying progressively reduced homozygous Hai7124 introgressed segments from marker NAU4956 to NAU845) contained a common homozygous G. barbadense chromosomal region between NAU3735 and NAU845. Their mean fiber length and strength were similar to that of the donor parent, 3326-7, but significantly higher (P < 0.05) than that of the recipient parent, XLZ41, and other groups, such as groups G13–G15 (carrying progressively reduced homozygous Hai7124 introgressed segments from marker NAU1048 to CGR5372) and G19–G29 (carrying progressively reduced homozygous Hai7124 introgressed segments from marker BNL1597 to CGR6381), which lack an interval between NAU3735 and NAU845. These results indicate that qFL-chr.7 and qFS-chr.7 are located in the recombination region between markers NAU3735 and NAU845, which are 0.36 cM apart.
Based on the fiber performance and SSR anchoring of recombinants, we further fine-mapped qFM-chr.7. We found that groups G1–G3 (carrying progressively reduced homozygous Hai7124 introgressed segments from marker CGR6381 to NAU7445) and G16–G25 (carrying progressively reduced homozygous Hai7124 introgressed segments from marker NAU4956 to MGHES75) contained a common homozygous G. barbadense chromosomal region between MGHES75 and NAU7445. Their fiber micronaire values were similar to 3326-7, but significantly lower than those of XLZ41 (P < 0.05) and other groups that did not have a G. barbadense introgressed fragment between MGHES75 and NAU7445. Therefore, using pairwise comparisons between each recombinant family (groups G1–G4 and G27–G29) and the two parental controls, qFM-chr.7 was placed in a region upstream, between NAU2108 and MGHES75 (0.44 cM).
Discussion
Clustered QTLs for super-quality fiber properties have great potential for use in cotton breeding
We detected three QTLs for fiber quality that were localized to the same chromosomes in previous studies using SSR markers (Lacape et al. 2005; Zhang et al. 2009a, b; Sun et al. 2012; Xu et al. 2012). Lacape et al. (2005) identified STR-c7 for fiber strength and FIN-C7 for fiber micronaire on Chr. 7 from a cross between Guazuncho 2 (G. hirsutum) and VH8 (G. barbadense), which could be linked to markers CIR335 and CIR 393, respectively. Zhang et al. (2009a, b) identified three QTLs (FL2, FS2, and FF2) for fiber length, strength, and micronaire which clustered in a region between NAU3181 and CIR320 on Chr. 7, using 270 F2:7 recombinant inbred lines developed from upland cotton cultivars T586 and Yumian 1. Xu et al. (2012) also mapped two fiber micronaire QTLs (qFMIC-7-1 and qFMIC-7-2) on Chr. 7 by single marker analysis using BC2F2:3 and BC2F2:4 populations of Simian 2 × G. klotzschianum introgression lines. The QTLs qFMIC-7-1 and qFMIC-7-2 were mapped to positions on the chromosome that significantly correlate with NAU1362 and CGR5372, respectively. We could not correlate the fiber quality QTLs to those found in our studies due to a lack of common markers. Sun et al. (2012) identified qFS-C7-1, qFM-C7-1, and qFL-C7-1 in F2 and F2:3 populations using composite interval mapping, and detected qFS-C7-1, qFS-C7-2, qFL-C7-1, qFL-C7-2, and qFM-C7-1 in RIL populations using composite interval mapping of lines derived from an upland cotton cross between strains 0–153 and sGK9708. The mapped positions of qFL-C7-2 and qFS-C7-2 in an RIL population of upland cotton are located at intervals that overlap with those of qFL-chr.7 and qFS-chr.7 identified in the current study based on the shared markers NAU1048b and NAU3380, suggesting that this QTL for fiber length and strength might be an allele introgressed from the G. barbadense genome. In addition, our data indicate that the region near the qFL-chr.7, qFS-chr.7, and qFM-chr.7 loci did not show significant negative correlations between fiber properties and the yield-related traits investigated (Table 2). Therefore, the loci qFL-chr.7, qFS-chr.7, and qFM-chr.7 are of particular interest, as they can potentially be transferred into G. hirsutum to simultaneously improve yield and fiber quality. Based on all of these results, we can conclude that Chr.7 contains multiple genetic loci involved in fiber quality-related traits, with different allelic effects.
In this study, qFL-chr.7 and qFS-chr.7 were fine-mapped to the same region, 0.36 cM apart, and the location of qFM-chr.7 was narrowed down to a 0.44-cM region. This mapping result, which was primarily obtained due to the use of chromosome segment introgression lines, should be of great value for transferring these favorable alleles into cotton varieties. The QTL-CSIL populations produced in the present study revealed even richer information about these QTLs. In addition to the similar direction of the genetic effects to those of Hai7124 alleles, which increased trait values across different generations, the subCSIL population provided a more accurate estimation of the genetic effects of fiber length, strength, and micronaire due to the minimization of its genetic background noise. Obviously, these genetic effects in the F2 population were underestimated. In fact, several successful QTL cloning studies over the past decade have benefited greatly from the development of high-quality NILs (Yamamoto et al. 2000; Lin et al. 2003). The reliability of all detected QTLs was further confirmed by testing 207 subCSILs in four environments (Table 4). Results from the four field trials show that it is highly likely that the QTLs identified in this study are authentic.
We found that fiber quality QTLs form clusters, a phenomenon that was previously reported in cotton (Chen et al. 2008; He et al. 2007; Qin et al. 2008; Ulloa and Meredith 2000). There were undesirable and desirable linkage effects for fiber length, strength, and micronaire in different populations. We found that the target chromosomal interval was positively involved in the control of these fiber quality traits in the CSIL populations. Our results indicate that genes controlling fiber quality cluster on Chr. 7; the phenomenon of QTL clusters might explain the very high correlation of traits in breeding programs for improving fiber quality. Thus, this observation also confirms the findings of our previous study demonstrating that backcrossing combined with MAS can be used to simultaneously improve fiber length, strength, and micronaire in XLZ41.
Pleiotropy versus linkage loci controlling fiber quality QTL effects
Most agronomic traits related to fiber, such as lint yield, fiber length, strength, and fineness, are quantitatively inherited. Moreover, the frequency with which lint yield and fiber quality traits are unfavorably associated has greatly limited the efficiency of conventional cotton breeding (Meredith 1984). In recent decades, conventional breeding has led to the development of many high-yielding cotton varieties, but it has also inadvertently decreased fiber quality in these commercial varieties (Zhang et al. 2005a, b). Paterson et al. (1990) proposed substitution mapping as a method for fine-mapping QTLs. This method, which has been used to characterize QTLs in diverse species (Grandillo et al. 1996; Tuinstra et al. 1998; Lyman and Mackay 1998), is a powerful tool for distinguishing between pleiotropy and linkage, eliminating linkage drag, and providing a basis for map-based cloning of QTLs (Tanksley et al. 1995; Alpert and Tanksley 1996). The 3326-7 introgression region has been confirmed to improve fiber length, strength, and micronaire simultaneously. In previous studies, only backcross populations were evaluated, whereas in the present study, we analyzed both homozygous and heterozygous introgressions, thus enabling estimation of the mode of gene action. Furthermore, fiber quality QTLs not reported in previous studies were detected in the introgressed region. SSR markers are useful for defining the positions of recombinants and for clarifying the borders of informative introgressions. Substitution mapping enabled us to distinguish between linkage and pleiotropy for several loci involved in the quantitative traits affected by the 7.57-cM introgression from Hai7124 loci affecting fiber length, strength, and micronaire; these traits are clearly controlled by different genes. Fiber strength and fiber length are usually positively correlated, but it is not clear whether this correlation is due to linkage or pleiotropy. Although QTL fine-mapping using a substitution mapping strategy revealed that qFL-chr.7 and qFS-chr.7 are located in the same 0.36 cM interval, the possibility that one pleiotropic QTL or two closely linked QTLs exist in this region should be further tested and verified via higher resolution mapping.
References
Alpert KB, Tanksley SD (1996) High-resolution mapping and isolation of a yeast artificial chromosome contig containing fw2.2: a major fruit-weight quantitative trait locus in tomato. Proc Natl Acad Sci USA 93:15503–15507
An CF, Jenkins JN, Wu JX, Guo YF, McCarty JC (2010) Use of fiber and fuzz mutants to detect QTL for yield components, seed, and fiber traits of upland cotton. Euphytica 172:21–34
Bernacchi D, Beck-Bunn T, Emmatty D, Eshed Y, Inai S, Lopez J, Petiard V, SayamaH UJ, Zamir D, Tanksley SD (1998) Advanced backcross QTL analysis in tomato. II: evaluation of near-isogenic lines carrying single-donor introgressions for desirable wild QTL-alleles derived from Lycopersicon hirsutum and L. pimpinellifolium. Theor Appl Genet 97:170–180
Brouwer DJ, Clair DA (2004) Fine mapping of three quantitative trait loci for late blight resistance in tomato using near isogenic lines (NILs) and sub-NILs. Theor Appl Genet 108:628–638
Cao ZB, Wang P, Chen H, Zhu XF, Zhang TZ (2014) Microsatellite-assisted improvement of fiber qualities in Gossypium hirsutum using G. barbadense introgressed lines. Theor Appl Genet 127:587–594
Chen L, Zhang ZS, Hu MC, Wang W, Zhang J, Liu DJ, Zheng J, Zheng FM, Ma J (2008) Genetic linkage map construction and QTL mapping for yield and fiber quality in upland cotton (Gossypium hirsutum L.). Acta Agron Sin 34:1199–1205
Chen GD, Li HB, Zheng Z, Wei YM, Zheng YL, McIntyre CL, Zhou MX, Liu CJ (2012) Characterization of a QTL affecting spike morphology on the long arm of chromosome 3H in barley (Hordeum vulgare L.) based on near isogenic lines and a NIL-derived population. Theor Appl Genet 125:1385–1392
Ding XP, Li XK, Xiong LZ (2011) Evaluation of near-isogenic lines for drought resistance QTL and fine mapping of a locus affecting flag leaf width, spikelet number, and root volume in rice. Theor Appl Genet 123:815–826
Fray A, Nesbitt TC, Fray A, Grandillo S, Knaap E, Cong B, Liu JP, Meller J, Elber R, Alpert KB, Tanksley SD (2000) fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289:85–88
Fridman E, Pleban T, Zamir D (2000) A recombination hotspot delimits a wild species quantitative trait locus for tomato sugar content to 484 bp within an invertase gene. Proc Natl Acad Sci USA 97:4718–4723
Grandillo S, Ku H-M, Tanksley SD (1996) Characterization of fs8.1 a major QTL influencing fruit shape in tomato. Mol Breed 2:251–260
Guo WZ, Cai CP, Wang CB, Zhao L, Wang L, Zhang TZ (2008) A preliminary analysis of genome structure and composition in Gossypium hirsutum. BMC Genom 9:314
He DH, Lin ZX, Zhang XL, Nie YC, Guo XP, Zhang YX, Li W (2007) QTL mapping for economic traits based on a dense genetic map of cotton with PCR-based markers using the interspecific cross of Gossypium hirsutum × G. barbadense. Euphytica 153:181–197
Jiang CX, Wright RJ, El-Zik KM, Paterson AH (1998) Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proc Natl Acad Sci USA 95:4419–4424
Kohel RJ, Yu J, Park YH, Lazo GR (2001) Molecular mapping and characterization of traits controlling fiber quality in cotton. Euphytica 121:163–172
Kosambi D (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lacape JM, Nguyen TB, Courtois B, Belot JL, Giband M, Gourlot JP, Gawryziak G, Roques S, Hau B (2005) QTL analysis of cotton fiber quality using multiple Gossypium hirsutum × Gossypium barbadense backcross generation. Crop Sci 45:123–140
Lacape JM, Jacobs J, Arioli T, Derijcker R, Forestier-Chiron N, Llewellyn D, Jean J, Thomas E, Viot C (2009) A new interspecific, Gossypium hirsutum × G. barbadense, RIL population: towards a unified consensus linkage map of tetraploid cotton. Theor Appl Genet 119:281–292
Lacape JM, Llewellyn D, Jacobs J, Arioli T, Becker D, Calhoun S, Al-Ghazi Y, Liu SM, Pala O, Georges S, Giband M, Assun HD, Barroso PAV, Claverie M, Gawryziak G, Jean J, Vialle M, Viot C (2010) Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol 10:132
Lin HX, Liang ZW, Sasaki T, Yano M (2003) Fine mapping and characterization of quantitative trait loci, Hd4 and Hd5, controlling heading date in rice. Breed Sci 53:51–59
Lyman RL, Mackay FC (1998) Candidate quantitative trait loci and naturally occurring phenotypic variation for bristle number in Drosophila melanogaster. The Delta-Hairless gene region. Genetics 149:983–998
Mei M, Syed NH, Gao W, Thaxton PM, Smith CW, Stelly DM, Chen ZJ (2004) Genetic mapping and QTL analysis of fiber-related traits in cotton (Gossypium). Theor Appl Genet 108:280–291
Meredith WR (1984) Quantitative genetics. In Kohel RJ, Lewis CF (eds) Cotton, pp 131–150
Ooijen JW, Voorrips RE (2002). JoinMap: version 3.0: software for the calculation of genetic linkage maps. University and Research Center
Park YH, Alabadt MS, Ulloa M, Sicker B, Wilkins TA, Yu J, Stelly DM, Kohel RJ, El-Shihy OM, Cantrell RG (2005) Genetic mapping of new cotton fiber loci using EST-derived microsatellites in an interspecific recombinant inbred line cotton population. Mol Genet Genom 274:428–441
Paterson AH, Deverna JW, Lanini B, Tanksley SD (1990) Fine mapping of quantitative trait loci using selected overlapping recombinant chromosomes in an interspecies cross of tomato. Genetics 124:735–742
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Report 11(2):122–127
Paterson AH, Saranga Y, Menz M, Jiang CX, Wright R (2003) QTL analysis of genotype × environment interaction affecting cotton fiber quality. Theor Appl Genet 106:384–396
Percival AE, Wendel JF, Stemart JM (1999) Taxonomy and germplasm resourses. In: Smith CW, Cothren JT (eds) Cotton: origin, histrory, technology, and production. Wiley, New York, pp 33–63
Qin HD, Guo WZ, Zhang YM, Zhang TZ (2008) QTL mapping of yield and fiber traits based on a four-way cross population in Gossypium hirsutum L. Theor Appl Genet 117:883–894
Shappley ZW, Jenkins JN, Zhu J, McCarty JC (1998) Quantitative trait loci associated with agronomic and fiber traits of upland cotton. J Cotton Sci 2:153–163
Shen XL, Guo WZ, Zhu XF, Yuan YL, Yu JZ, Kohel RJ, Zhang TZ (2005) Molecular mapping of QTL for fiber qualities in three diverse lines in upland cotton using SSR markers. Mol Breed 15:169–181
Shen XL, Zhang TZ, Guo WZ, Zhu XF, Zhang XY (2006) Mapping QTLs with main effects, epistatic effects and QTL × environment interaction for fiber and yield traits in recombinant. Crop Sci 46:61–66
Sun FD, Zhang JH, Wang SF, Gong WK, Shi YZ, Liu AY, Li JW, Gong JW, Shang HH, Yuan YL (2012) QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol Breed 30:569–582
Szalma SJ, Hostert BM, LeDeaux JR, Stuber CW, Holland JB (2007) QTL mapping with near-isogenic lines in maize. Theor Appl Genet 114:1211–1228
Tanksley SD (1993) Mapping polygenes. Annu Rev Genet 27:205–233
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Theor Appl Genet 92:191–203
Tanksley SD, Ganal MW, Martin GB (1995) Chromosome landing: a paradigm for map-based gene cloning in plants with large genomes. Trends Genet 11:63–68
Thomson MJ, Edwards JD, Septiningsih EM, Harrington SE, McCouch SR (2006) Substitution mapping of dth1.1, a flowering-time quantitative trait locus (QTL) associated with transgressive variation in rice, reveals multiple sub-QTL. Genetics 172:2501–2514
Tuinstra MR, Ejeta G, Goldsbrough PB (1998) Evaluation of near-isogenic sorghum lines contrasting for QTL markers associated with drought tolerance. Crop Sci 38:835–842
Ulloa M, Meredith WR (2000) Genetic linkage map and QTL analysis of agronomic and fiber quality traits in an intraspecific population. J Cotton Sci 4:161–170
Voorrips RE (2006) MapChart 2.2: software for the graphical presentation of linkage maps and QTLs. Plant Research International, Wageningen
Wang SC, Basten J, Zeng ZB (2010) Windows QTL Cartographer 2.5. Department of Statistics. North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTL.Cart.html
Wang B, Nie YC, Lin ZX, Zhang XL, Liu JJ, Bai J (2012a) Molecular diversity, genomic constitution, and QTL mapping of fiber quality by mapped SSRs in introgression lines derived from Gossypium hirsutum × G. darwinii Watt. Theor Appl Genet 125:1263–1274
Wang P, Zhu YJ, Song XL, Cao ZB, Ding YZ, Liu BL, Zhu XF, Wang S, Guo WZ, Zhang TZ (2012b) Inheritance of long staple fiber quality traits of Gossypium barbadense in G. hirsutum background using CSILs. Theor Appl Genet 124:1415–1428
Wu JX, Gutierrez OA, Jenkins JN, McCarty JC, Zhu J (2009) Quantitative analysis and QTL mapping for agronomic and fiber traits in an RI population of upland cotton. Euphytica 165:231–245
Xiao J, Wu K, Fang DD, Stelly DM, Yu J, Cantrell RG (2009) New SSR markers for use in cotton (Gossypium spp.) improvement. J Cotton Sci 13:75–157
Xie XB, Song MH, Jin FX, Ahn SN, Suh JP, Hwang HG, McCouch SR (2006) Fine mapping of aa grain weight quantitative trait locus on rice chromosome 8 using near-isogenic lines derived from a cross between Oryza sativa and Oryza rufipogon. Theor Appl Genet 113:885–894
Xu P, Zhu J, Zhang XG, Ni WC, Xu YJ, Shen XL (2012) Molecular mapping and identification of QTLs for fiber micronaire on Chromosome 7 from Gossypium klotzschianum. Acta Agron Sin 38(3):447–453
Yamamoto T, Lin HX, Sasaki T, Yano M (2000) Identification of heading date quantitative trait locus Hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genetics 154:85–891
Young ND, Tanksley SD (1989) RFLP analysis of the size of chromosomal segments retained around the tm-2 locus of tomato during backcross breeding. Theor Appl Genet 77:353–359
Yu Y, Yuan D, Liang S, Li X, Wang X, Lin Z, Zhang XL (2011) Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genom 12:15
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136(4):1457–1468
Zhang JF, Lu Y, Adragna H, Hughs E (2005a) Genetic improvement of New Mexico acala cotton germplasm and their genetic diversity. Crop Sci 45:2363–2373
Zhang ZS, Xiao YH, Luo M, Li XB, Luo XY, Hou L, Li DM, Pei Y (2005b) Construction of a genetic linkage map and QTL analysis of fiber-related traits in upland cotton (Gossypium hirsutum L.). Euphytica 144:91–99
Zhang YS, Luo LJ, Liu TM, Xu CG, Xing YZ (2009a) Four rice QTL controlling number of spikelets per panicle expressed the characteristics of single Mendelian gene in near isogenic backgrounds. Theor Appl Genet 118:1035–1044
Zhang ZS, Hu MC, Zhang J, Liu DJ, Zheng J, Zhang K, Wang W, Wan Q (2009b) Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol Breed 24:49–61
Zhao H, Liu J, Shi L, Xu F, Wang Y (2010) Development of boron-efficient near isogenic lines of Brassica napus and their response to low boron stress at seedling stage. Russ J Genet 46:57–63
Acknowledgments
This study was financially supported in part by Grants from the National Science Foundation of China (31330058), National High Technology Research and Development Program of China (863 Program) (2011AA10A102), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the 111 Project (B08025).
Author contributions
TZ Zhang designed the study; ZB Cao performed the laboratory and field experiments and analyzed the data; XF Zhu and H Chen performed the field experiments in Xinjiang and Jiangpu; and TZ Zhang and ZB Cao wrote the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Cao, Z., Zhu, X., Chen, H. et al. Fine mapping of clustered quantitative trait loci for fiber quality on chromosome 7 using a Gossypium barbadense introgressed line. Mol Breeding 35, 215 (2015). https://doi.org/10.1007/s11032-015-0393-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0393-3