
Abstract
Root-knot nematodes (Meloidogyne spp.) are major pests attacking carrots (Daucus carota) worldwide, causing galling and forking of the storage roots, rendering them unacceptable for market. Genetic resistance could significantly reduce the need for broad-spectrum soil fumigants in carrot production. In this study, genetic resistance to Meloidogyne incognita was mapped. Three diverse sources of resistance, from Syria (HM), Europe (SFF) and South America (Br1091), were identified. Two F2 mapping populations were developed using these parents, (Br1091 × HM1) and (SFF × HM2), as well as a segregating population derived from the self-pollination of a HM plant (HM3). Analysis revealed four QTLs conditioning resistance in Br1091 × HM1, three in SFF × HM2, and three in HM3. A consensus genetic map of the three populations revealed five non-overlapping QTLs for M. incognita resistance, one each on carrot chromosomes 1, 2, 4, 8, and 9. One QTL was present in all three populations, in the same region of chromosome 8 as Mj-1 which imparts resistance to M. javanica.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carrot (Daucus carota) is a major crop worldwide, providing an economically important crop for farmers and important nutritional benefits to consumers. Root-knot nematodes (Meloidogyne spp., RKN) are major pests in many carrot production regions causing direct economic loss of the crop due to galling and forking of the carrot roots, rendering an infected carrot unmarketable (Roberts 1987). M. hapla is the predominant RKN species in cooler production areas, while M. javanica and Meloidogyne incognita are major pests in warmer areas. Limited control of RKN through crop rotations and non-chemical management is possible, but difficult due to wide host ranges, including many weed species (Hunt and Handoo 2010). Current control measures rely on soil-applied nematicides that are effective, but expensive and harmful to the environment. This has led to the conclusion that genetic resistance provides an ideal solution to RKN control (e.g., Nyczepir and Thomas 2010).
RKN resistance has been characterized in many species including alfalfa (Potenza et al. 2001), common bean (Omwega and Roberts 1992), cowpea (Roberts et al. 1996; Ehlers et al. 2000), grape (Cousins et al. 2003), lima bean (Roberts et al. 2008), pepper (Djian-Caporalino et al. 2007), soybean (Li et al. 2001), sugar beet (Yu et al. 1999), sweet potato (Jones and Dukes 1980), tobacco (Yi et al. 1998), and most extensively in tomato (Solanum lycopersicum). The Mi-1 locus in tomato confers resistance to M. incognita, M. javanica, and M. arenaria (Dropkin et al. 1969), as well as conferring resistance to potato aphids (Rossi et al. 1998; Vos et al. 1998) and whiteflies (Nombela et al. 2003). Mi-1 was mapped to the short arm of chromosome 6 and since then several other genomic regions of resistance have also been identified (Williamson 1998). At least one of these (Mi-9) is homologous to Mi-1 and mapped to the same genomic region (Jablonska et al. 2007). The mode of action of Mi-1 allows the second-stage juvenile (J2) nematodes to penetrate the root and migrate through intercellular space, but when the nematode attempts to establish a feeding site, a hypersensitive plant cell response is activated and infection, feeding, and root-galling are prevented (Paulson and Webster 1972). Other modes of action conferring genetic resistance to RKN have been described in cotton (Gossypium hirsutum) where two major QTLs were identified, one on chromosome 11 influencing gall production, and the other on chromosome 14 allowing galling but limiting egg production (Gutierrrez et al. 2010).
The Mj-1 locus of carrot was discovered in a ‘Brasilia’ cultivar, line ‘Br1252,’ and it imparts resistance to M. javanica as a monogenic dominant trait (Boiteux et al. 2000; Simon et al. 2000). Resistance to M. javanica in ‘Brasilia’ conferred by the Mj-1 locus suppresses root penetration by infective J2, development of juvenile and adult stages, egg production, and root-galling symptoms (Huang 1986; Simon et al. 2000; Boiteux et al. 2004). The close correlation between egg production and root-galling reaction in heterozygous and homozygous plant groups for Mj-1 alleles (Simon et al. 2000; Boiteux et al. 2004) has led to the use of root-galling indices as a primary phenotype measure for resistance in breeding selection. Co-dominant STS flanking markers were developed from dominant AFLP and RAPD flanking markers to facilitate selection (Boiteux et al. 2004). Simon et al. (2000) also observed partial resistance to M. incognita in Br1252 and derivatives, in which determination of resistance could not be explained by Mj-1 alone. Recently, a second M. javanica resistance locus, Mj-2, was mapped in PI 652188 from China and found to be distantly linked to Mj-1, on chromosome 8 (Ali et al. 2014).
The widespread occurrence of M. incognita in carrot production regions and partial resistance observed in Br1252 has led to a wider search for genetic resistance to M. incognita. This research was undertaken to determine inheritance patterns and map chromosomal regions responsible for resistance to M. incognita identified in Brazilian, Middle Eastern, and European carrot cultivars, to gain a broader picture of M. incognita resistance in these diverse germplasm resources.
Materials and methods
Plant materials
Preliminary screening of several hundred cultivated and wild carrot populations led to the discovery of M. incognita resistance in “Brasilia” seed lot 1091 (Br1091) (Matthews et al. 1999), the Syrian cultivar “Homs” (HM), and in a population derived from a cross between the European cultivars “Scarlet Fancy” and “Favourite” (SFF). A Br1091 plant was crossed with a HM plant to generate the Br1091 × HM1 F2 population from a single F1 plant. A SFF plant was crossed to a second HM plant to generate the SFF × HM2 F2 population from a single F1 plant. HM1 and HM2 were siblings derived from a self-pollinated HM selection. A third HM plant was self-pollinated to generate the HM3 population. This population had undergone five generations of selfing before the final self-pollination to produce the population HM3, and it was still segregating for resistance. All parent plants (Br1091, SFF, HM1, HM2, and HM3) had been previously evaluated in a greenhouse screen as described below (Br1091) or evaluated in a M. incognita infested field and identified as resistant to M. incognita with limited gall formation on the carrot root (unpublished data). F3 families were derived from 95 Br1091 × HM1 F2 and 34 SFF × HM2 F2 plants.
Nematode screening
All RKN screening was performed under greenhouse conditions at the University of California, Riverside, CA. The Br1091 × HM1 F2, and SFF × HM2 F2 populations were evaluated in the same trial in 2009, the HM3 population was evaluated in 2010, and 11 plants from each Br1091 × HM1 and SFF × HM2 F3 family were evaluated in the same trial in 2012. All evaluations also included susceptible control plants of cv. “Imperator 58”. Individual plants were grown in 10-cm pots filled with fine sand, and the resistance screening was carried out according to the methods described by Simon et al. (2000). Inoculations were made using M. incognita race 1 isolate “Beltran” cultured on susceptible tomato plants. Based on estimates of a 20 % mean hatch of extracted eggs in hatch tests, the egg inoculum density was adjusted to 50,000 eggs per plant to provide 10,000 s-stage juveniles per plant. The inoculum of eggs suspended in water was injected into the root zone using a syringe with three holes along the length of the needle. One-month-old plants were inoculated and evaluated approximately 60 days after inoculation. Fibrous roots of individual plants were evaluated on a 0 (no galls)- to 8 (severely galled)-point scale modified from Bridge and Page (1980), and the resulting scores were used for the QTL analysis and heritability estimation. Screened plants were shipped to Wisconsin for vernalization and planted in the University of Wisconsin, Department of Horticulture greenhouse at Arlington, WI.
DNA extraction and marker evaluations
Leaves were sampled and lyophilized from the greenhouse in Arlington, WI, for the Br1091 × HM1 F2, Br1091 × HM1 F3, and SFF × HM2 F2 populations. Leaves from the HM3 population were harvested in the greenhouse at the University of California, Riverside, CA, and placed in plastic bags with silica gel to desiccate the leaves. DNA for all populations was extracted according to Murray and Thompson (1980) and quantified on 1 % agarose gel electrophoresis.
AFLP reactions were performed according to Vivek and Simon (1999) with slight modifications. DNA was digested with a combination of EcoRI and MseI restriction enzymes, and for amplification, the EcoRI primer had a HEX fluorochrome tag for fluorescent evaluation. Fluorescent SSR primers and PCR procedures were performed according to Cavagnaro et al. (2011) and Iorizzo et al. (2011). AFLP and SSR markers were evaluated at the University of Wisconsin Biotechnology Center using an ABI 3730xl capillary sequencer. Polymorphic SSRs were identified by screening a subset of 10 individuals from each population. GeneMarker version 1.5 was used to score alleles (SoftGenetics, State College, Pennsylvania).
SNPs were evaluated using the KASPar system (http://www.KBioscience.co.uk). The Br1091 × HM1 F2 population (138 individuals), 12 individuals from SFF × HM2 F2 population, and 6 bulk samples of 8 individuals each from HM3 population were evaluated with a panel of 4000 SNPs previously developed by Iorizzo et al. (2013a). Selected polymorphic SNPs in the SFF × HM2 F2 and HM3 populations were evaluated in the full population (Table 1). To validate the QTLs identified in the Br1091 × HM1 F2 population, SSR markers within the QTL support intervals were evaluated in 507 of the Br1091 × HM1 F3 individuals as described above.
Genetic map construction
Linkage maps were constructed with JoinMap 3.0 software (Van Ooijen and Voorrips 2001). Markers and genotypes with more than 10 % missing data and markers that significantly deviated from expected segregation ratios using a Chi-square test (P < 0.01) were removed. For linkage groups with clusters of markers with significant segregation distortion (P < 0.0005), all markers were used to generate the linkage map. Linkage groups were obtained at a LOD threshold >3.0. The regression mapping algorithm was used with Haldane’s mapping function to calculate distances between markers. Haldane’s mapping function was chosen because it provided a more accurate marker placement according to the carrot physical map than the Kosambi’s mapping function (data not shown). Each marker was coded twice, once for each parental phase. The linkage groups were properly phased by using marker scores for individuals related to the parents (Gomez et al. 1996; Vivek and Simon 1999). The marker order was further examined using CheckMatrix (http://www.atgc.org/XLinkage, Truco et al. 2013) for inconsistencies, and markers with more than one inconsistent score were removed. To remove redundant markers in the Br1091 × HM1 population, a genetic bin map was developed. For each linkage group, pair-wise recombination values among all markers were calculated. Adjacent markers with zero recombination among them were assigned to the same genetic bin. In addition, adjacent markers with “false” recombination due to missing data were considered to belong to the same genetic bin. The marker with the least number of missing data points was chosen to represent each genetic bin.
SNPs and SSRs with known chromosome locations were used to anchor the linkage groups (Iorizzo et al. 2013b; Cavagnaro et al. 2011; Iovene et al. 2011; Yildiz et al. 2013). After being assigned to chromosomes, linkage groups were oriented and numbered following the chromosome orientation and classification of Iovene et al. (2011).
Map merging
JoinMap version 3.0 was used to merge the maps. For each pair-wise comparison, common co-linear markers were identified as anchoring markers and used to develop the consensus map. QTL coordinates were transferred from the individual maps to the merged maps according to the location of the nearest flanking markers mapped in each specific linkage map. MapChart version 2.2 was used to draw all chromosome diagrams (Voorrips 2002).
QTL mapping
QTL analysis was performed in all three populations using R/qtl with the multiple imputations method (Broman and Sen 2009). QTL detection for each population included preliminary QTL identification using scanone followed by QTL modeling. The largest LOD peak from the analysis was added to the QTL model, and if the QTL model was significant, it was retained. This process was then repeated using addqtl, instead of scanone, followed by QTL modeling and testing for interactions until adding additional QTL to the model was no longer significant. The final step used addpair to add a pair of interacting QTL or the interaction between a QTL in the model and a newly identified QTL. The support intervals were calculated using a 1.5-LOD drop (Broman and Sen 2009). QTLs were named Mi-population-C_-Q_ where “population” is the population in which the QTL was identified, “C_” is the chromosome on which the QTL was identified and “Q_” is the QTL identifier from the QTL model. For example, Mi-BrHM1-C2-Q1 was mapped in the Br1091 × HM1 population, is on chromosome 2, and is QTL that explains the most variation in the model.
Heritability estimation and QTL validation
Parent offspring regression was used to estimate broad-sense heritability using F3 family averages in the Br1091 × HM1 (95 families) and SFF × HM2 populations (334 families, Nyquist 1991) calculated with the statistical program R. For the Br1091 × HM1 F3 QTL validation, F3 individuals were used. Markers that best approximated the F2 QTL were used to fit a QTL model with the F3 individuals. To better customize the model to the F3 population, refineqtl was used and the refined qtl positions used in the final fitqtl analysis.
Results
Genetic linkage maps
The Br1091 × HM1 linkage map was generated from the evaluation of 138 individuals and included 389 SNP markers (Table 1; Supplemental Figure 1). The total genetic distance covered 563.3 cM with an average marker spacing of 1.5 cM. Markers on chromosome 4 and 9 displayed significant segregation distortion from the 1:2:1 expected ratio (Supplemental Figure 1).
The SFF × HM2 linkage map was generated from the evaluation of 113 individuals and included 138 AFLP, SSR, and SNP markers (Table 1; Supplemental Figure 2). The total genetic distance covered 520.1 cM with an average marker spacing of 4.0 cM.
The HM3 linkage map was generated from the evaluation of 281 individuals and included 70 SSR and SNP markers (Table 1; Supplemental Figure 3). The total genetic distance covered 219.2 cM with an average marker spacing of 3.4 cM. HM3 had no segregating markers on three chromosomes and relatively few segregating markers on the six other chromosomes. Out of 3636 SNPs screened, only 226 were polymorphic and many of those SNPs were clustered together in a few chromosomal regions.
The merged linkage map for all three populations included 445 markers covering a total genetic distance of 556.9 cM and an average marker spacing of 1.3 cM (Table 1; Fig. 1). The order of markers was conserved in the merged map, relative to each individual map (data not shown).

Merged linkage map of carrot chromosomes that incorporates significant QTL for M. incognita nematode resistance from three populations (Br1091 × HM1, SFF × HM2, HM3). The bars represent 1.5-LOD support intervals, and the populations are coded with Br1091 × HM1 as solid bars, SFF × HM2 as open bars, and HM3 as cross hashed bars. Numbers in parentheses indicate the largest LOD score followed by the percent phenotypic variation explained by that QTL
QTL mapping
QTLs for M. incognita resistance in the Br1091 × HM1 population were located on chromosomes 1, 2, 8, and 9, and they accounted for 55.5 % of the phenotypic variation of the resistance reaction (Table 2; Fig. 1, Supplemental Figure 4, Supplemental Figure 5, Supplemental Figure 6, Supplemental Figure 7). QTLs attributable to the Br1091 parent included those on chromosomes 1 and 8. QTLs on chromosomes 2 and 9 were derived from the HM1 parent. All four QTLs displayed additive effects ranging from 0.6 to 1.4. The additive effect is half the difference between the susceptible and resistant homozygous means.
QTLs for M. incognita resistance in the SFF × HM2 population were on chromosomes 2, 4, and 8, and they accounted for 34.0 % of the phenotypic variation in the resistance reaction (Table 2; Fig. 1, Supplemental Figure 5, Supplemental Figure 6, Supplemental Figure 8). QTLs on chromosomes 4 and 8 were derived from the SFF parent, while the QTL on chromosome 2 was derived from the HM2 parent. The QTL displayed additive effects ranging from 0.8 to 1.1.
QTLs for M. incognita resistance in the HM3 population were on chromosomes 1, 8, and 9, and they accounted for 35.7 % of the phenotypic variation (Table 2; Fig. 1, Supplemental Figure 4, Supplemental Figure 5, Supplemental Figure 7). The QTL on chromosomes 1 and 8 had additive effects of 0.4 and 0.9, respectively, while the QTL on chromosome 9 displayed over-dominance with the heterozygous genotype more resistant than either homozygous genotype (Supplemental Figure 7).
Heritability and QTL validation
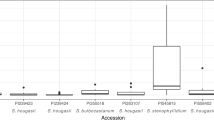
Broad-sense heritability for resistance to M. incognita was 0.33 and 0.25 in Br1091 × HM1 and SFF × HM2, respectively (Supplemental Figure 9). For validating the QTL in Br1091 × HM1, the full model including the QTL on chromosomes 1, 2, 8, and 9 accounted for 23.7 % of the variation with individual QTL effects ranging from 3.0 to 12.5 % (Table 3). All QTL effects were additive (data not shown) as in the Br1091 × HM1 F2 population.
Discussion
A significant QTL for resistance to M. incognita was detected on chromosome 8 in each of the three populations evaluated and those QTL support intervals spanned the same region as Mj-1, which confers resistance to M. javanica and co-segregates with GSSR-044 (Ali et al. 2014). While the QTL on chromosome 8 co-localize to Mj-1 and may be indicating a common resistance determinant, the relationship between the QTL identified in this work and Mj-1 remains to be determined. The additive effects of this QTL in each population were also similar with values of 0.8, 0.9, and 1.0 for SFF × HM2, HM3, and Br1091 × HM1, respectively (Table 2; Supplemental Figure 6). Since M. incognita resistance was noted in M. javanica-resistant segregants derived from a different ‘Brasilia’ cross (Boiteux et al. 2000; Simon et al. 2000), it was not surprising to observe Mi-BrHM1-C8-Q2 in the Br1091 × HM1 cross, but the discovery of similar QTLs in the unrelated SFF × HM2 and HM3 populations was unexpected. It remains to be determined whether the same alleles are responsible for resistance to M. incognita in each population and also how the M. incognita resistance relates to alleles at the Mj-1 locus, but their co-segregation is worth noting. Resistance genes can occur in tandem clusters. In fact, many of the tomato resistance genes are clustered with Mi-1 and Mi-9 on chromosome 6, linkage between Mi-2 and Mi-8 (position not reported), linkage between Mi-6 and Mi-7 (position not reported), and linkage between Mi-3 and Mi-5 on chromosome 12 (Williamson 1998; Jablonska et al. 2007). Clusters of resistance genes can have greater numbers of non-synonymous versus synonymous mutations, and it has been hypothesized that this allows for plasticity in the sequence to adapt to emerging pathogens (Michelmore and Meyers 1998). In fact, variation in copy numbers of tandem repeated nematode resistance genes (Rhg1) has been observed in soybean (Cook et al. 2012). Simon et al. (2000) also presented evidence for the scenario that two fairly closely linked genes in coupling phase explain resistance to M. javanica at the Mj-1 locus, so there is some evidence for more than one gene affecting nematode resistance in the region. Fine mapping and testcrosses between the different sources of resistance, coupled with extensive molecular evaluation of the carrot genome, will be necessary to validate a common allelic basis for the QTL conferring resistance to M. incognita and M. javanica.
In addition to the common QTL on chromosome 8, a QTL in both the Br1091 × HM1 and HM3 populations mapped near the end of the short arm of chromosome 9. It is worth noting that the QTL on Br1091 × HM1 Ch9 is located in a region with distorted markers (DMs). Zhang et al. (2010) concluded that if the distance between distorted and non-DMs is small, the genetic variation around that region can affect the detection of linked QTLs, but will not have a great effect on the position and effect estimations of QTL. In the Br1091 × HM1Ch9 linkage group, all the markers covering the QTL support interval had segregation distortion, which will not be expected to differentially bias the level of genetic variation between closely linked markers. The resistance in Br1091 × HM1 is derived from the HM1 parent, and given the shared ancestry of HM1 and HM3, it is possible that the QTL on chromosome 9 could be identical by descent between the two populations. However, the QTL effects of the two populations differ, with Mi-HM3-C9-Q1 displaying over-dominance and Mi-BrHM1-C9-Q4 being additive. The average score of each homozygous genotype at Mi-HM3-C9-Q1 is 5.4, while the average score of the heterozygous genotype is 4.4 (Supplemental Figure 7). It is possible that the QTLs represent different alleles of the same locus in each population, or two closely linked genomic regions each coming from a different resistant parent.
Br1091 × HM1 and HM3 also share a QTL on chromosome 1, but in this case, the resistance comes from Br1091 and HM3, respectively (Table 2; Supplemental Figure 4). Both of these QTLs have relatively minor effects (6.1 and 4.3 % of the variation explained for Mi-BrHM1-C1-Q3 and Mi-HM3-C1-Q3, respectively), and the QTL support intervals are quite large (23 and 54 cM, respectively, Fig. 1), so it is difficult to draw firm conclusions about the map locations of these two QTLs.
QTL for M. incognita resistance in the SFF × HM2 population had large support intervals (60, 33, and 31 for the QTL on chromosomes 2, 4, and 8, respectively) and small QTL effects (8–13 %). The large support intervals may be due to a relatively small number of individuals (N = 113), small individual QTL effects, low marker density (average spacing of 4 cM but not evenly distributed throughout the linkage map, Supplemental Figure 2), or a combination of these factors. Other researchers have shown each of these constraints can limit the detection of QTL and the refinement of QTL support intervals (e.g., Darvasi et al. 1993; Li et al. 2010; Stange et al. 2013). Mi-SFFH2-C2-Q3 and Mi-BrHM1-C2-Q1 both mapped to chromosome 2, but given the very large support interval of Mi-SFFH2-C2-Q3, they may not be allelic even though the resistance comes from the HM parent in each cross. Furthermore, because Mi-BrHM1-C2-Q1 accounted for 34 % of the variation, compared to only 8 % by Mi-SFFH2-C2-Q3, it might be conjectured that these QTLs either are at different loci or are different alleles of the same locus (Table 2; Supplemental Figure 5).
The full QTL model for each population accounted for 55.5, 34.8, and 35.7 % of the variation in Br1091 × HM1, SFF × HM2, and HM3, respectively (Table 2). There were no significant interaction effects among QTLs for a given cross, and with the exception of over-dominance noted for Mi-HM3-C9-Q1, all QTL effects were additive. Major RKN resistance genes have been identified in other crops, but RKN QTLs are more difficult to characterize. Tamulonis et al. (1997) identified two QTLs in soybean that accounted for 54 % of the variation in M. javanica root-galling, and Gutierrrez et al. (2010) identified two QTLs in cotton accounting for 41 % of the variation for M. Incognita root-gall index for M. incognita. The QTLs detected in this work demonstrate similar values for the percent variation explained.
There were three QTLs identified in the HM3 population, but with six generations of selection for RKN resistance, limited polymorphism was observed. Only 6 chromosomes had segregating markers, and only chromosome 1 had extensive marker coverage, compared to the merged map (Supplemental Figure 3). When screening this population for segregating markers, only 226 SNPs were polymorphic out of 3636 SNPs screened. This reduced polymorphism means only a small part of the genome was actually analyzed in the QTL analysis and other, homozygous regions of the genome might contain nematode resistance genes fixed for resistance or susceptibility. Even so, the HM3 population segregated widely for nematode resistance (Table 1). A cross using resistant individuals from the HM3 population to a genetically unrelated susceptible cultivar would help detect any fixed QTL as well as those QTL segregating in HM3.
The Br1091 × HM1 and SFF × HM2 populations were both derived from intercrosses between unrelated sources of genetic resistance with the presumption that at least one resistance locus was derived from each parent. That expectation was validated in the QTL analysis and the low broad-sense heritability estimates of 0.33 and 0.25 in the Br1091 × HM1 and SFF × HM2 populations, respectively. The validation of the F2 QTL in the F3 generation of Br1091 × HM1 confirms that the QTLs are significant. Because the QTL detected displayed additive effects, the percent of the phenotypic variation explained estimates narrow-sense heritability (Broman and Sen 2009). In the F3 generation, the percent of the phenotypic variation explained was 23.7 % and represents a narrow-sense heritability of 0.237. This compares to the broad-sense heritability from the parent–offspring regression of 0.33 in the same population, indicating resistance in the Br1091 × HM1 population is mostly additive. The finding of quantitative inheritance for M. incognita resistance in these populations makes the development of uniformly resistant lines difficult, but with recurrent selection and molecular markers, progress is being made to develop resistant germplasm.
The nematode screening for this work was carried out on an individual plant basis with each population evaluated at one time. While there was no replication of the individual populations, the combination of the populations and the detection of co-localized QTL support the results. Also, the Br1091 × HM1 QTLs were validated as significant in the F3 population. The evaluations for RKN screening in this work were carried out in different greenhouse trials, but the same nematode isolate and the same susceptible check, ‘Imperator 58’, were used in all trials. The gall-rating of Imperator 58 varied from 5 to 8 with an average rating of 6.5 and a standard deviation of 0.80, so there is variation in the plant response to the RKN attack. While the plant response to RKN attack is difficult to evaluate on an individual plant basis, the results obtained here are encouraging, particularly because individual plants of a single F2 population are more economical to produce, evaluate, and genotype than F3 families or recombinant inbred lines.
The parents in all three populations exhibited unique resistance QTLs that are being introgressed into susceptible germplasm and combined to develop what may be a more durable resistance effective against both M. incognita (from the QTLs detected here) and M. javanica (from the Mj-1 locus). Further research is needed to determine whether all QTLs are required to provide adequate, durable resistance to commercial three-way hybrids under field conditions and whether certain combinations of resistance alleles confer broader resistance. These studies will also provide further insights into the question of whether QTL from different genetic backgrounds are allelic or different, closely linked genes and whether M. javanica resistance imparted by the Mj-1 locus is a pleiotropic function of a M. incognita QTL or a separate gene.
The Br1091 × HM1 linkage map presented here represents the densest collection of sequence-based markers developed for carrot to date. Previously, only 117 markers with known sequence information had been mapped in carrot (Yildiz et al. 2013; Alessandro et al. 2013; Cavagnaro et al. 2011). The Br1091 × HM1 map shares over 300 markers with other linkage maps containing QTL mapping data for traits of interest (Iorizzo et al. 2014). This provides the opportunity to develop the first integrated linkage map for carrot, allowing for anchoring the upcoming sequenced carrot genome (Iorizzo et al. 2014). In depth, fine mapping of M. incognita resistance alleles will allow identifying candidate genes responsible for major QTLs identified in this study and establish a foundation to understand the genetic mode of action in impeding nematode infection. Molecular markers linked to M. incognita resistance QTLs identified in this study provide useful tools for the carrot breeding community to implement marker-assisted selection in breeding programs.
References
Alessandro MS, Galmarini CR, Iorizzo M, Simon PW (2013) Molecular mapping of vernalization requirement and fertility restoration genes in carrot. Theor Appl Genet 126(2):415–423
Ali A, Mathews WC, Cavagnaro PF, Iorizzo M, Roberts PA, Simon PW (2014) Inheritance and mapping of Mj-2, a new source of root-knot nematode (Meloidogyne javanica) resistance in carrot. J Hered 105(2):288–291
Boiteux LS, Belter JG, Roberts PA, Simon PW (2000) RAPD linkage map of the genomic region encompassing the root-knot nematode (Meloidogyne javanica) resistance locus in carrot. Theor Appl Genet 100:439–446. doi:10.1007/s001220050057
Boiteux LS, Hyman JR, Bach IC, Fonseca MEN, Matthews WC, Roberts PA, Simon PW (2004) Employment of flanking codominant STS markers to estimate allelic substitution effects of a nematode resistance locus in carrot. Euphytica 136:37–44. doi:10.1023/B:EUPH.0000019508.78153.dd
Bridge P, Page S (1980) Estimation of root-knot infestation levels on roots using a rating chart. Trop Pest Manag 26:296–298
Broman KW, Sen S (2009) A guide to QTL mapping with R/qtl. Springer, New York. doi:10.1007/978-0-387-92125-9
Cavagnaro PF, Chung S-M, Manin S, Yildiz M, Ali A, Alessandro MS, Iorizzo M, Senalik DA, Simon PW (2011) Microsatellite isolation and marker development in carrot-genomic distribution, linkage mapping, genetic diversity analysis and marker transferability across Apiaceae. BMC Genomics 12:386. doi:10.1186/1471-2164-12-386
Cook DE, Lee TG, Guo X, Melito S, Wang K, Bayless AM, Wang J, Hughes TJ, Willis DK, Clemente TE, Diers BW, Jiang J, Hudson ME, Bent AF (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338:1206–1209. doi:10.1126/science.1228746
Cousins P, Lauver M, Boyden L (2003) Genetic analysis of root-knot nematode resistance derived from Vitis mustangensis. Acta Hortic 603:149–155
Darvasi A, Weinreb A, Minke V, Weller J, Soller M (1993) Detecting marker QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics 134:943–951
Djian-Caporalino C, Fazari A, Arguel MJ, Vernie T, VandeCasteele C, Faure I, Brunoud G, Pijarowski L, Palloix A, Lefebvre V, Abad P (2007) Root-knot nematode (Meloidogyne spp.) Me resistance genes in pepper (Capsicum annuum L.) are clustered on the P9 chromosome. Theor Appl Genet 114:473–486. doi:10.1007/s00122-006-0447-3
Dropkin VH, Helgeson JP, Upper CD (1969) The hypersensitivity reaction of tomatoes resistant to Meloidogyne incognita: reversal by cytokinins. J Nematol 1:55–61
Ehlers JD, Matthews WC, Hall AE, Roberts PA (2000) Inheritance of a broad-based form of root-knot nematode resistance in cowpea. Crop Sci 40:611–618
Gomez R, Angel F, Bonierbale MW, Rodriguez F, Tohme J, Roca WM (1996) Inheritance of random amplified polymorphic DNA markers in cassava (Manihot esculenta Crantz). Genome 39:1039–1043. doi:10.1139/g96-130
Gutierrrez OA, Jenkins JN, McCarty JC, Wubben MJ, Hayes RW, Callahan FE (2010) SSR markers closely associated with genes for resistance to root-knot nematode on chromosomes 11 and 14 of upland cotton. Theor Appl Genet 121:1323–1337. doi:10.1007/s00122-010-1391-9
Huang SP (1986) Penetration, development, reproduction, and sex ratio of Meloidogyne javanica in three carrot cultivars. J Nematol 18:408–412
Hunt DJ, Handoo ZA (2010) Taxonomy, identification and principal species. In: Perry RN, Moens M, Starr JL (eds) Root-knot nematodes. CABI, Wallingford, pp 55–97
Iorizzo M, Senalik DA, Grzebelus D, Bowman M, Cavagnaro PF, Matvienko M, Ashrafi H, Van Deynze A, Simon PW (2011) De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 12:389. doi:10.1186/1471-2164-12-389
Iorizzo M, Senalik DA, Ellison SL, Grzebelus D, Cavagnaro PF, Allender C, Brunet J, Spooner DM, Van Deynze A, Simon PW (2013a) Genetic structure and domestication of carrot (Daucus carota subsp sativus) (Apiaceae). Am J Bot 100:930–938. doi:10.3732/ajb.1300055
Iorizzo M, Ellison S, Grzebelus D, Cavagnaro PF, Spooner D, Van Deynze A, Simon P (2013b) Development of a high-throughput SNP resource to advance genomic, genetic and breeding research in carrot (Daucus carota L.). XXI Plant and Animal Genome Meeting, San Diego, USA, P0762
Iorizzo M, Senalik D, Ellison S, Cavagnaro P, Cheng S, Zheng P, Zheng Z, Van Deynze A, Simon P (2014) The building of the first Apiaceae genome. XXII Plant and Animal Genome Meeting, San Diego, USA, January 12–15
Iovene M, Cavagnaro PF, Senalik D, Buell CR, Jiang J, Simon PW (2011) Comparative FISH mapping of Daucus species (Apiaceae family). Chromosome Res 19:493–506. doi:10.1007/s10577-011-9202-y
Jablonska B, Ammiraju JSS, Bhattarai KK, Mantelin S, Martinez de Ilarduya O, Roberts PA, Kaloshian I (2007) The Mi-9 gene from Solanum arcanum conferring heat-stable resistance to root-knot nematodes is a homolog of Mi-1. Plant Physiol 143:1044–1054. doi:10.1104/pp.106.089615
Jones A, Dukes P (1980) Heritabilities of sweet potato resistances to root-knot caused by Meloidogyne incognita and Meloidogyne javanica. J Am Soc Hortic Sci 105:154–156
Li Z, Jakkula L, Hussey RS, Tamulonis JP, Boerma HR (2001) SSR mapping and confirmation of the QTL from PI96354 conditioning soybean resistance to southern root-knot nematode. Theor Appl Genet 103:1167–1173. doi:10.1007/s001220100672
Li H, Hearne S, Baenziger M, Li Z, Wang J (2010) Statistical properties of QTL linkage mapping in biparental genetic populations. Heredity 105:257–267. doi:10.1038/hdy.2010.56
Matthews W, Simon P, Roberts P (1999) Influence of temperature on expression of resistance in carrot to Meloidogyne javanica. J Nematol 31:553
Michelmore RW, Meyers BC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth and death process. Genome Res 8:1113–1130
Murray M, Thompson W (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325. doi:10.1093/nar/8.19.4321
Nombela G, Williamson VM, Muniz M (2003) The root-knot nematode resistance gene Mi-1.2 of tomato is responsible for resistance against the whitefly Bemisia tabaci. Mol Plant Microbe Interact 16:645–649. doi:10.1094/MPMI.2003.16.7.645
Nyczepir AP, Thomas SH (2010) Current and future management strategies in intensive crop production systems. In: Perry RN, Moens M, Starr JL (eds) Root-knot nematodes. CABI, Wallingford, pp 412–443
Nyquist W (1991) Estimation of heritability and prediction of selection response in plant populations. Crit Rev Plant Sci 10:235–322. doi:10.1080/07352689109382313
Omwega C, Roberts P (1992) Inheritance of resistance to Meloidogyne spp. in common bean and the genetic basis of its sensitivity to temperature. Theor Appl Genet 83:720–726
Paulson RE, Webster JM (1972) Ultrastructure of the hypersensitive reaction in roots of tomato, Lycopersicon esculentum L., to infection by the root-knot nematode, Meloidogyne incognita. Physiol Plant Pathol 2:227–234
Potenza C, Thomas SH, Sengupta-Gopalan C (2001) Genes induced during early response to Meloidogyne incognita in roots of resistant and susceptible alfalfa cultivars. Plant Sci 161:289–299. doi:10.1016/S0168-9452(01)00415-0
Roberts P (1987) The influence of planting date of carrot on Meloidogyne incognita reproduction and injury to roots. Nematologica 33:335–342
Roberts PA, Matthews WC, Ehlers JD (1996) New resistance to virulent root-knot nematodes linked to the Rk locus of cowpea. Crop Sci 36:889–894
Roberts PA, Matthews WC, Ehlers JD, Helms D (2008) Genetic determinants of differential resistance to root-knot nematode reproduction and galling in lima bean. Crop Sci 48:553–561. doi:10.2135/cropsci2007.07.0384
Rossi M, Goggin FL, Milligan SB, Kaloshian I, Ullman DE, Williamson VM (1998) The nematode resistance gene Mi of tomato confers resistance against the potato aphid. Proc Natl Acad Sci USA 95:9750–9754. doi:10.1073/pnas.95.17.9750
Simon PW, Matthews WC, Roberts PA (2000) Evidence for simply inherited dominant resistance to Meloidogyne javanica in carrot. Theor Appl Genet 100:735–742. doi:10.1007/s001220051346
Stange M, Utz HF, Schrag TA, Melchinger AE, Wuerschum T (2013) High-density genotyping: an overkill for QTL mapping? Lessons learned from a case study in maize and simulations. Theor Appl Genet 126:2563–2574. doi:10.1007/s00122-013-2155-0
Tamulonis JP, Luzzi BM, Hussey RS, Parrott WA, Boerma HR (1997) DNA markers associated with resistance to Javanese root-knot nematode in soybean. Crop Sci 37:783–788
Truco MJ, Ashrafi H, Kozik A, van Leeuwen H, Bowers J, Wo SRC, Stoffel K, Xu H, Hill T, Van Deynze A, Michelmore RW (2013) An ultrahigh density, transcript based, genetic map of lettuce. G3-Genes Genomes Genet 3:617–631. doi:10.1534/g3.112.004929
Van Ooijen JW, Voorrips RE (2001) JoinMap® 3.0, software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Vivek BS, Simon PW (1999) Linkage relationships among molecular markers and storage root traits of carrot (Daucus carota L. ssp sativus). Theor Appl Genet 99:58–64. doi:10.1007/s001220051208
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78. doi:10.1093/jhered/93.1.77
Vos P, Simons G, Jesse T, Wijbrandi J, Heinen L, Hogers R, Frijters A, Groenendijk J, Diergaarde P, Reijans M, Fierens-Onstenk J, de Both M, Peleman J, Liharska T, Hontelez J, Zabeau M (1998) The tomato Mi-1 gene confers resistance to both root-knot nematodes and potato aphids. Nat Biotechnol 16:1365–1369. doi:10.1038/4350
Williamson VM (1998) Root-knot nematode resistance genes in tomato and their potential for future use. Annu Rev Phytopathol 36:277–293. doi:10.1146/annurev.phyto.36.1.277
Yi HY, Rufty RC, Wernsman EA, Conkling MC (1998) Mapping the root-knot nematode resistance gene (Rk) in tobacco with RAPD markers. Plant Dis 82:1319–1322. doi:10.1094/PDIS.1998.82.12.1319
Yildiz M, Willis DK, Cavagnaro PF, Iorizzo M, Abak K, Simon PW (2013) Expression and mapping of anthocyanin biosynthesis genes in carrot. Theor Appl Genet 126:1689–1702. doi:10.1007/s00122-013-2084-y
Yu MH, Heijbroek W, Pakish LM (1999) The sea beet source of resistance to multiple species of root-knot nematode. Euphytica 108:151–155. doi:10.1023/A:1003616612201
Zhang L, Wang S, Li H, Deng Q, Zheng A, Li S, Li P, Li Z, Wang J (2010) Effects of missing markers and segregation distortion on QTL mapping in F2 populations. Theor Appl Genet 121:1071–1082
Acknowledgments
This work was funded by the California Fresh Carrot Advisory Board grants (to PWS and PAR); Specialty Crop Research Initiative award 2008-51180-04896 (to PWS and PAR); Organic Agriculture Research and Extension Initiative award 2011-51300-30903 (to PWS and PAR); and a Monsanto Graduate Research Fellowship to J.P. through the University of Wisconsin-Madison Plant Breeding and Plant Genetics Program. The authors appreciate the capable assistance provided by Dr. Doug Senalik.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Parsons, J., Matthews, W., Iorizzo, M. et al. Meloidogyne incognita nematode resistance QTL in carrot. Mol Breeding 35, 114 (2015). https://doi.org/10.1007/s11032-015-0309-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0309-2