
Abstract
With increasing global food demands in the face of challenging biotic and abiotic pressures on crop production, there is a vital need for good crop improvement strategies. Gene editing and gene targeting using designer nucleases are relatively new, sophisticated approaches that can be used for crop improvement. Designer nucleases are molecules that can be engineered to cleave virtually any endogenous DNA target sequence, making this technology inherently more powerful over current, essentially random mutation strategies. These molecules can also be used to promote targeted DNA insertions and homologous recombination. Further modifications of these molecules can convert them into designer transcription factors that can activate or suppress a gene of choice. Four designer nuclease platforms are currently available: meganucleases, zinc finger nucleases, TALENs and the more recently developed CRISPR/Cas9 system. All four of these systems have been shown to function in crop plants and have been used for site-specific gene targeting and gene editing. Herein, we describe the basis of each designer nuclease platform, highlighting the advantages and disadvantages of each, and give examples of their application in crop improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In an era where pressure on food production is rapidly growing in the face of challenging global environmental climate changes, the need to produce hardy crops with higher yields but less input is vital (Tester and Langridge 2010). Unfortunately, a number of tools available for crop improvement suffer from a lack of precision and are reliant upon random events for outcomes. For example, mutagenesis, facilitated by mutagenic chemicals, irradiation or DNA insertion sequences, relies upon the random distribution of mutations throughout the plant genome. While screening technologies for mutations in a specific gene have greatly advanced (e.g. TILLING, genome sequencing, flanking sequence tag libraries) (Parry et al. 2009), the actual site of mutation within a gene sequence remains uncontrolled. RNAi technology allows highly specific, targeted post-transcriptional suppression of a gene (Small 2007); however, it results in gene silencing rather than a complete loss of gene activity and does not allow precise, targeted modification of an endogenous gene sequence.
As another example, a major advance in germplasm improvement of crop species has been the development of transgenic plants which has enabled the introduction of DNA sequences from any source, either biological or synthetic, for agronomic benefit (Gasser and Fraley 1989). The introduction of this additional genetic material, however, has been overwhelmingly achieved by random insertion into the plant genome by nonhomologous recombination. This nontargeted gene insertion precludes the subsequent insertion of additional transgenes at the same locus which would greatly simplify future breeding efforts if it was possible. The ability to subtract specific genes present at a multi-transgene locus would also be of commercial benefit, enabling precise gene combinations to be developed depending upon the intellectual property demands of specific commercial relationships.
A further highly desirable technology is the ability to facilitate in planta homologous recombination. This process enables alteration of endogenous gene sequences to create new alleles with beneficial agronomic traits. Some success in altering endogenous gene sequences has been achieved via the introduction of short oligonucleotide sequences into plant cells which can cause sequence change in target alleles by mismatch repair (Beetham et al. 1999; Zhu et al. 1999; Kochevenko and Willmitzer 2003; Okuzaki and Toriyama 2004; Iida and Terada 2005; Dong et al. 2006). However, published reports of this process in tobacco, maize, rice and wheat have shown very low efficiencies. Nonetheless, companies such as Cibus (San Diego, USA) offer this service as a “Rapid Trait Development System”, and the resultant products are claimed to be considered nontransgenic.
Major technological advances have been achieved in overcoming the current limitations described above by the advent of designer nuclease technologies which include meganucleases, Zn finger nucleases, TALENs and more recently CRISPR\Cas9. This review aims to summarise these technologies and provide examples of their current and potential future application to agricultural crop improvement.
Custom-designed nucleases and nickases
Custom-designed nucleases are all similar in that they each can be engineered to specifically recognise any DNA target sequence usually around 20 nucleotides in length and cleave this target sequence to create a double strand (DS) DNA break. DS DNA breaks within the plant genome are primarily repaired either by nonhomologous end joining (NHEJ) or homologous recombination (Puchta 2005). NHEJ occurs far more commonly and is error prone resulting in insertions or deletions at the break site. These nucleases can therefore create small insertions/deletions (indels) at very precise locations within an endogenous DNA sequence allowing highly targeted mutagenesis to be undertaken. DS DNA breaks can also promote the insertion of foreign DNA sequences at these sites by NHEJ. For example, Tzfira reported that 2.5 % of T-DNA insertions occurred preferentially in an enforced break in the tobacco genome amongst 620 transgenic plants produced (Tzfira et al. 2003).
DNA breaks can also promote homologous recombination. When DNA is introduced into plants by either Agrobacterium or biolistic transformation, homologous recombination has been reported to take place once for every 104–107 illegitimate recombination events (Puchta et al. 1996; Hannin et al. 2001; Puchta 2002; Wright et al. 2005 and reference therein; Tzfira et al. 2012). In spite of its rarity, homologous recombination can be detected in plants by either extensive screening or by homologous recombination-dependent selection strategies such as reconstitution of a selectable marker gene (Tzfira et al. 2012). However, the creation of a targeted DS DNA break combined with the introduction of a sequence flanked with homologous ends to this target can dramatically increase the frequency (i.e. up to 10−2) of homologous recombination at this site (Puchta et al. 1996; Puchta 2002; Wright et al. 2005). Modified versions of nucleases termed “nickases”, described below, have also been engineered that cleave only a single strand of DNA at a target site, which further increases the likelihood of homologous recombination occurring, rather than NHEJ (van Nierop et al. 2009; Chan et al. 2011; Fauser et al. 2014).
The application of designer nucleases therefore exploits endogenous DNA repair mechanisms to create site-specific indels or to promote precise DNA insertions or homologous recombination. Similar exploitation of DNA repair systems has been undertaken using site-specific recombination systems such as Cre/loxP, R/RS and FLP/FRT (Wang et al. 2011). However, the major difference between these site-specific recombination systems and designer nucleases is that the former systems are generally limited to a single, specific target sequence or closely related derivative sequences. In contrast, designer nucleases can be engineered to target any short DNA sequence of choice, making them inherently more flexible.
While all designer nucleases are similar in that they generate targeted DNA cleavage, they differ in their origin and the mechanism by which target sequence specificity is achieved. The designer nuclease platforms that are currently available are summarised as follows.
Meganucleases
Meganucleases, or homing nucleases, are natural restriction endonucleases that are components of mobile genetic elements. These enzymes recognise specific DNA sequences that range from >12 to 40 bp in size, whereupon they produce a DS DNA break (Paques and Duchateau 2007). Several hundred members have been identified that are found in eukaryotes, bacteria and archea and are often encoded on mobile class I introns and inteins (Paques and Duchateau 2007). Given the size of meganuclease recognition sites, an entire plant genome may contain no, or just a few, recognition sites for a given nuclease. These rare cutting nucleases have been successfully used to target DNA insertions in a number of plant species including Arabidopsis, tobacco and maize (D’Halluin et al. 2008; Yang et al. 2009). However, obvious limitations exist in that endogenous target sites are uncommon and are fixed a priori, or alternatively, the target site has to be introduced encoded on a transgene. Re-engineering of meganucleases to recognise new DNA sequences has been achieved but has proven complex (Gao et al. 2010; Tzfira et al. 2012) although it continues to improve (Arnould et al. 2011). Meganucleases with nickase activity have also been developed (McConnell-Smith et al. 2009).
Zn finger nucleases
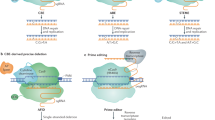
Zinc finger nucleases (ZFNs) are composed of two functional domain types: zinc finger (ZF) DNA recognition domains, common to some transcription factor families, and a non-sequence-specific nuclease domain (Porteus and Carroll 2005). The DNA recognition domain consists of an array of Cys2–His2 ZF domains with each finger binding a specific nucleotide triplet. ZFs have been identified that recognise all GNN and ANN nucleotide triplets and most CNN and TNN triplets. Combining ZFs that have different recognition specificities enables the resultant multimeric protein to bind a specific DNA sequence. The nuclease domain of the ZFN is responsible for DNA cleavage immediately adjacent to the ZFN-binding site. This is usually catalysed by the 196 amino acid C-terminal domain from the nuclease FokI (Kim et al. 1996). This FokI domain functions as a dimer; hence, two ZFNs are required to bind in close proximity to enable dimerisation and production of a DS DNA break at the target site, with each ZFN recognising a different DNA sequence on either side of this site (Fig. 1), (Mani et al. 2005). Typically, each ZFN consists of 3–4 ZF domains with each finger recognising a nucleotide triplet (Klug 2005). A functional pair of ZFNs, each containing 3 ZF domains, would therefore recognise two specific 9 bp sequences that flank an internal 5–7 bp DNA cleavage site (Fig. 1). ZFNs, like TALENs and the CRISPR\Cas9 systems described below, are therefore true designer nucleases in that many DNA sequences can be selectively targeted in the plant genome, making these systems remarkably powerful. In addition, fusion of zinc fingers to transcriptional activation domains can generate synthetic transcription factors that can be potentially designed to target many regulatory sequences of choice (Stege et al. 2002; Sanchez et al. 2006). Some examples of these synthetic transcription factors are described later in this review. ZFN pairs have also been modified by inactivating the FokI cleavage domain in one of the ZFNs to produce a nickase activity (i.e. cleavage of a single DNA strand only) to promote homologous recombination (Gaj et al. 2013).

Three designer nuclease platforms. Schematic diagram of zinc finger nucleases, TALENs and CRISPR/Cas9 nuclease systems. A single Zn finger nuclease (ZFN) consists of three zinc finger (ZF) domains that each recognises a specific nucleotide triplet, coupled to a FokI nuclease domain. A pair of ZFNs is required for activity due to a homo-dimerisation requirement of the FokI nuclease domain. TALENs also function in pairs with a single TALEN molecule consisting of nine individual TALE repeats (rectangles) fused to the FokI nuclease domain. Each TALE repeat recognises a specific nucleotide. The CRISPR/Cas9 system differs in that target sequence recognition is via a small guide RNA (sgRNA, in blue) containing a 20 base sequence (lower case) that recognises a genomic target sequence via complementary base pairing. The target sequence must have a two invariable guanine bases at the 3′ end which form a protospacer-adjacent motif sequence (PAM, underlined in red). Associated with the sgRNA is a Cas9 nuclease protein that subsequently cleaves the target site. All three nuclease systems produce a double-stranded (DS) DNA break unless additional nuclease domain modifications are made. DS DNA breaks are preferentially repaired by nonhomologous end joining (NHEJ) which usually results in insertions (two bases shown in red lower case), deletions or substitutions of a few nucleotides at the target site. Addition of a homologous repair template (green) in the presence of a DS DNA break can facilitate homologous recombination which enables designer alleles to be produced by incorporating sequence modifications (red bases shown in lower case) into the repair template
The requirement for ZFN to act as dimers for DNA cleavage increases targeting specificity as the likelihood of off-target site binding by both ZFNs at the same site is low. However, toxicity of ZFNs has been reported presumably due to some off-target cleavage (Paques and Duchateau 2007; Tzfira et al. 2012). Attempts to ameliorate this problem have been by either engineering ZFNs with increased specificity or by including additional modifications such as a FokI nuclease heterodimerisation requirement or engineering additional cofactor requirements (Szczepek et al. 2007; Miller et al. 2007; Pruett-Miller et al. 2009; Townsend et al. 2009; Ramalingam et al. 2011). Individual ZF domains do not always behave as predicted in a multimeric context; therefore, selective synthesis cycles are required to produce ZFNs with desired specificity outcomes (Joung and Sanders 2013; Straub and LaHaye 2013).
In plants, ZFNs have been successfully used in Arabidopsis (Lloyd et al. 2005; de Pater et al. 2009; Tovkach and Zeevi 2009; Osakabe et al. 2010; Zhang et al. 2010; Qi et al. 2013a; de Pater et al. 2013), tobacco (Bibikova et al. 2003; Wright et al. 2005; Cai et al. 2009; Townsend et al. 2009; Petolino et al. 2010), soybean (Curtin et al. 2011), petunia (Marton et al. 2010) and maize (Shukla et al. 2009; Ainley et al. 2013). Expression of ZFNs in Arabidopsis and tobacco has produced heritable, targeted mutations in transgenes and endogenous genes at frequencies as high as 3–7 %, depending upon the ZFN and target sequence (Townsend et al. 2009; Lloyd et al. 2005; Zhang et al. 2010; Osakabe et al. 2010). In tobacco, targeted transgene integration was as high as 10 % (Cai et al. 2009) and homologous recombination with an endogenous gene to generate herbicide resistance up to 4 % (Townsend et al. 2009). Commercially produced ZFN expression plasmids can be purchased from Sigma-Aldrich as part of a propriety platform (CompaZr) with Sangamo Biosciences (Richmond, CA, USA) which hold ZFN patent rights (Thomas Scott 2005; DeFrancesco 2011). This commercial production alleviates the extensive confirmation of ZFN specificity and activity by the end user (Gaj et al. 2013; Tzfira et al. 2012; Johnson et al. 2013).
TALENs
Transcription activator-like effector nucleases (TALENs) are similar to Zn finger nucleases in that they allow true designer targeting of most DNA sequences. Transcription activator-like effectors (TALEs) are a group of proteins first identified in the bacterial plant pathogen Xanthamonas oryzae (Bogdanove et al. 2010; Schornack et al. 2013). These proteins are directly introduced into plant cells by the bacterium to promote bacterial colonisation. Each TALE binds to a specific DNA sequence in the vicinity of an endogenous plant gene and then transcriptionally activates this host gene to promote bacterial pathogenesis (Bogdanove et al. 2010). Within the TALE protein are 33–35 amino acid repeats that each recognises a specific DNA base, with a hypervariable region at amino acid positions 12 and 13 determining base specificity (Boch et al. 2009; Moscou and Bogdanove 2009). Most engineered TALE repeat arrays published to date use multimers of four domains that contain at hypervariable residues amino acids NN, NI, HD or NG for the recognition of guanine, adenine, cytosine and thymine nucleotides, respectively (Joung and Sander 2013). Having deciphered the DNA-binding code of these proteins, it is now possible to produce synthetic TALEs that transcriptionally activate or repress a gene of interest by targeting a specific sequence in the 5’ region of the chosen gene. This ability is potentially a very powerful tool for altering plant gene expression for desirable traits (Morbitzer et al. 2010; Mahfouz et al. 2012).
Further engineering of TALEs has enabled the development of TALENs by fusion of a FokI nuclease domain to the TALE protein, as described above for ZFNs (Christian et al. 2010; Miller et al. 2011; Li et al. 2011; Mahfouz et al. 2011; Joung and Sanders 2013; Schornack et al. 2013). As for ZFNs, TALENS also function in pairs, again due to the homodimeric requirement for DS DNA cleavage by the FokI nuclease domain, with each TALEN targeting a specific sequence either side of the cleavage site (Fig. 1). TALENS can also be used for nickase activity rather than DS nuclease activity by inactivating one of the FokI domains. TALENs have been suggested to show less target sequence restrictions than ZFNs and equal or better efficiencies at mediating target site cleavage (Cermak et al. 2011). The assembly of tandemly repeated TALE DNA-binding domains, however, is challenging using conventional cloning techniques although improved cloning strategies have been developed (Joung and Sanders 2013; Straub and LaHaye 2013). TALENs have been used in a variety of eukaryotic organisms including Arabidopsis (Cermak et al. 2011), tobacco (Mahfouz et al. 2011; Zhang et al. 2013), rice (Li et al. 2012), wheat (Shan et al. 2013b; Wang et al. 2014); soybean (Haun et al. 2014), maize (Liang et al. 2014) and barley (Wendt et al. 2013; Gurushidze et al. 2014). They are commercially available from companies including Cellectis Bioresearch (Paris, France), Transposagen Biopharmaceuticals (Lexington, KY, USA) and Life Technologies (Grand Island, NY, USA) (Gaj et al. 2013). Two patent positions cover TALEN technology with one being exclusively licensed to the Two Blades Foundation, a USA-based charitable organisation, for commercial use in plants who in turn have licensed these rights to LifeTechnologies while the latter patent has been licensed to Cellectis Research (DeFrancesco 2011).
CRISPR/Cas9 system
The CRISPR/Cas system is a prokaryote defence mechanism found in most archeal (90 %) and bacterial species (40 %) and protects these microbes against invading nucleic acids such as viral genomes and plasmids (Horvath and Barrangou 2010). Clustered regular interspaced short palindromic repeats (CRISPR) are short direct repeats (21–47 bp) separated by spacer sequences (21–72 bp) that are usually segments of captured viral or plasmid DNA. CRISPR repeats are often adjacent to CRISPR-associated (Cas) genes which encode a heterogeneous family of proteins that include nucleases, helicases and polymerases, in addition to noncoding RNAs. CRISPR segments are transcribed and these transcripts are processed to form small RNAs. These small RNAs act as guides by binding to complementary foreign nucleic acid sequences by homologous pairing which targets components of the Cas complex, including an endonuclease called Cas9, to these invading sequences resulting in their degradation (Horvath and Barrangou 2010). Obvious parallels exist between the CRISPR/Cas system and eukaryotic RNAi-mediated gene silencing systems in that target sequence recognition is based upon complimentary nucleic acid pairing; however, apart from this similarity, these two systems are mechanistically distinct.
To aid the utility of this natural system in genome editing applications, the complexity of prokaryotic CRISPR/Cas systems has been substantially reduced by engineering it to consist of just two genes, one encoding the Cas9 nuclease protein and the second to encode a synthetic small guide RNA (sgRNA). This latter molecule is approximately 85 bp in length and negates the RNA processing requirements of the endogenous bacterial system (Jinek et al. 2012; Cong et al. 2013; Mali et al. 2013; Qi et al. 2013b). Located at the 5′ end of the sgRNA are 19–22 bases that recognise the DNA target sequence by complementary nucleotide pairing (Fig. 1). This target sequence requires two invariable guanine bases at the 3′ end of the target site which form a protospacer adjacent motif sequence (PAM) of NGG (Straub and LaHaye 2013). Upon target sequence recognition, the Cas9 nuclease cleaves the complementary and noncomplementary DNA strands three and three to eight nucleotides, respectively, from the PAM site in the region of target sequence and sgRNA complementarity (Lozano-Juste and Cutler 2014).
Similar to Zn finger domain proteins and TAL effector proteins, modification of the Cas9 nuclease can also produce nickase activity rather than DS DNA cleavage (Jinek et al. 2012) to facilitate homologous recombination. Combining a nuclease-deficient Cas9 protein with sgRNAs can also produce a transcriptional repressor when appropriately targeted to regulatory sequences of a gene of interest (Qi et al. 2013b). Similarly, fusing a transcriptional activation domain to an inactive Cas9 protein can generate transcriptional activation of a target gene (Perez-Pinera et al. 2013; Maeder et al. 2013). The CRISPR/Cas9 system is therefore as versatile as Zn finger and TAL technologies in that it can function as a designer nuclease or designer transcription factor. Furthermore, this system is suggested to be significantly simpler in application than ZFN and TALENS as the simple sgRNA defines the cleavage site rather than complex engineered proteins containing multimeric ZF or TALE domains (Straub and LaHaye 2013; Belhaj et al. 2013).
One drawback, however, is that the relatively small number of “programmable” target nucleotides is further constrained by the requirement of the PAM sequence. In spite of these target sequence limitations, over 1.4 million potential target sites have been identified in the Arabidopsis genome with more than 99 % of protein-encoding nuclear genes containing at least one target site (Li et al. 2013) and over 90 % of rice genes predicted to also contain suitable target sites (Xie and Yang 2013). In a similar bioinformatic analysis, suitable sgRNA target sites were identified in at least one exon of 83–98 % of genes present in Arabidopsis, Medicago, tomato, soybean, Brachypodium, sorghum and rice; however, only 30 % of maize genes contained a target site (Xie et al. 2014). Another caveat is that, similar to the other designer nuclease platforms, off-target modifications by CRISPR/Cas9 can occur (Fu et al. 2013; Hsu et al. 2013; Xie and Yang 2013).
The relatively simple CRISPR/Cas9 system has recently been shown to function effectively in Arabidopsis, Nicotiana benthamiana, tobacco, wheat, rice, sweet orange, sorghum and maize cells to generate target site indels and nucleotide substitutions or promote homologous recombination (Shan et al. 2013b; Li et al. 2013; Nekrasov et al. 2013, Upadhyay et al. 2013, Jiang et al. 2013; Feng et al. 2013; Mao et al. 2013; Xie and Yang 2013; Jia and Wang 2014; Liang et al. 2014; Xu et al. 2014; Zhang et al. 2014; Jiang et al. 2014; Fauser et al. 2014). An extensive and insightful summary of many of these experiments is provided by Belhaj et al. (2013). These studies demonstrate the robustness of this technology by its successful application to numerous plants species in such a short span of time. The intellectual property ownership of the CRISPR/Cas9 system remains to be determined; however, the BROAD Institute was recently granted the first patent which covers the components and methodology of this system (Zhang 2014).
Applications
The following examples highlight some of the potential applications for designer nuclease technology in crop plants. These events can be broadly classified as precision gene mutation, in situ engineering of endogenous genes, gene removal, transcriptional reprogramming of endogenous genes and production of large cis transgene stacks.
Precision gene mutation
Unlike conventional mutagens and DNA insertion sequences, designer nucleases offer an unparalleled opportunity to target specific regions in a gene of interest. In two examples, the I-CreI homing endonuclease (meganuclease) from Chlamydomonas reinhardti was engineered to recognise a 21 bp sequence in 5′ juxtaposition to the maize liguless1 gene (Gao et al. 2010) and a 22 bp sequence present in MS26, a maize cytochrome P450 gene required for male fertility (Djukanovic et al. 2013). In the former study, 3 % of T0 plants contained mutations at the target site (Gao et al. 2010), while in the latter study, 6 % of T0 plants contained an indel within this gene, and homozygous progeny produced a male sterile phenotype (Djukanovic et al. 2013).
Targeted gene mutation using CRISPR/Cas9 were also undertaken in rice protoplasts where four rice genes were successfully mutated (Shan et al. 2013b). Mutation frequencies were estimated by PCR amplification of the target site from total protoplast DNA, and a proportion of PCR products were shown to have lost a restriction enzyme site present in the target sequence. Using this method, approximately 25 % of alleles were estimated to have been effectively mutated in each case. Stable rice transgenics were also produced in which the OsPDS and OsBAD genes were targeted and mutations detected in 9 and 7 % of T0 plants, respectively, including biallelic mutations in one-third of OsPDS mutant plants (Shan et al. 2013b). In another study, 11 genes were independently targeted in the rice genome using CRISPR/Cas, and 44 % of T0 plants on average had a mutation at the targeted locus with 4 % of plants containing homozygous mutations (Zhang et al. 2014). These mutations were stably inherited in progeny, and deep sequencing revealed that off-target genome modifications were rare (Zhang et al. 2014).
Wheat protoplasts have also been mutated using the CRIPSR/Cas system at 28 % efficiency (Shan et al. 2013b). In this case, the target gene was the wheat homologue of the barley Mlo gene which is of particular interest given that inactivation of this gene in barley provides broad spectrum resistance to Blumeria graminis (powdery mildew) (Buschges et al. 1997). Subsequently, the simultaneous editing of all three wheat Mlo homoealleles using TALENs was reported resulting in broad spectrum resistance to powdery mildew disease (Wang et al. 2014). Twenty-seven mutant T0 plants were detected amongst 450 transgenics of which 20 were heterozygous for mutations at a single Mlo locus, two plants contained multiple mutations at single loci, four plants had mutations present at two Mlo loci and one line was heterozygous for mutations at all three homologous loci. Progeny from this latter line that was homozygous for mutations at all three homologous Mlo loci were resistant to powdery mildew disease (Wang et al. 2014).
CRISPR/Cas-targeted mutations have also been produced in the wheat inox and PDS genes at around 20 % efficiency in suspension-cultured cells (Upadhyay et al. 2013). A remarkable extrapolation of targeted gene knockout using CRIPSR/Cas9 was recently demonstrated in a human cell line where 64, 751 unique sgRNAs were used to screen 18,080 genes for increased drug resistance upon gene knockout (Shalem et al. 2014). Such high throughput, targeted mutagenesis has yet to be applied to plants, but it is an exciting proposition.
TALENs have also been demonstrated to function effectively in rice and produce highly targeted gene knockouts. In one study, four loci were targeted in the rice genome, and PCR assays confirmed TALEN editing in 3–60 % of callus lines depending upon the TALEN pair used (Shan et al. 2013a). Transgenic plants were regenerated after transformation with two TALEN pairs and mutations detected in 19 and 36 % of T0 plants, respectively. In the same study, similar TALEN efficiencies were observed for Brachypodium distachyon callus transformed with TALEN pairs (Shan et al. 2013a).
TALENS were also used in soybean to produce simultaneous mutations in two fatty acid desaturase genes (FAD2-1A and FAD2-1B) for improved oil quality (Haun et al. 2014). Four out of nineteen transgenic plants contained mutations in both FAD2 genes; however, both mutations were subsequently inherited in T1 progeny from a single plant only. Progeny from this plant were identified that were homozygous for mutations at both genes and that no longer contained TALEN transgenes by segregation. Seed from these plants showed improved oil quality with a dramatic increase in oleic acid and concomitant reduction in linoleic acid (Haun et al. 2014).
All four designer nuclease platforms have therefore been successfully used in crop plants to produce targeted mutations in genes of interest with comparable efficiencies. Unlike conventional mutagenesis, these mutations were targeted to a precise DNA sequence. However, a point to consider is that while designer nucleases may be able to precisely target a short DNA sequence and cause cleavage, there is no control over the subsequent NHEJ process that takes place. Hence, although the site of mutation is highly specific, the resultant structure of the mutated locus is largely random and consists of indels of unspecified size and sequence. Truly precise sequence engineering of an endogenous locus is restricted to homologous recombination.
In situ engineering of endogenous genes
An efficient homologous recombination system is highly desirable in plant improvement as endogenous gene sequences can be altered to encode allelic variants with improved agronomic traits. Unlike NHEJ, this process can provide absolute designer sequence specificity by providing a recombination template of exact sequence choice (Fig. 1). Homologous recombination in plants remains a challenging process; however, several studies have successfully employed designer nucleases to promote sequence replacement and targeted sequence insertion. In rice, homology-directed repair following TALEN cleavage of the PDS locus was achieved by concomitantly providing a 72 bp donor sequence, although the efficiency of this process was undetermined (Shan et al. 2013a). In maize, ZFN-mediated cleavage of the IPK1 gene, which catalyses the last step in phytate production, was coupled with precise insertion of an herbicide-selectable marker gene at this site using homology-dependent repair mechanisms (Shukla et al. 2009). The resultant plants were both herbicide resistant and had reduced levels of phytate, an anti-nutritional component of feed grain that contributes to environmental pollution via animal waste (Shukla et al. 2009). In this study, selection using an herbicide resistance gene with no promoter, but which acquired adjacent regulatory sequences upon correct integration, resulted in a twofold increase in targeted gene insertion when compared with the same gene containing an autonomous promoter.
Gene removal
Removal of specific transgene sequences after the production of transgenic plants, often selectable marker genes, has been undertaken in numerous species including tobacco, Arabidopsis, rice, maize, barley, sorghum, tomato, soybean and wheat using site-specific recombinase systems (reviewed by Ow 2007). Examples include the development of a selectable marker-free corn line, LY038, developed by Monsanto using the Cre-lox system (Ow 2007; Wang et al. 2011). This system was also used to reduce the complexity of a biolistic transgene locus in wheat (Srivastava et al. 1999). However, as pointed out earlier, these site-specific recombination platforms are restricted in that each system is confined to a specific recognition sequence not present in the endogenous plant genome.
The following examples demonstrate the utility of designer nucleases by their ability to produce precise deletions in endogenous sequences at sites of choice. In rice protoplasts and callus tissue, two TALEN pairs were introduced that targeted two endogenous sites separated by 1,322 bp in the genome. Deletion alleles could be identified in both tissues with 5 % of calli containing deletions, and in one callus, an inversion of the intervening sequence was detected (Shan et al. 2013a). In wheat, a duplex sgRNA that recognised two separate regions of the endogenous inox gene resulted in deletion of the intervening 50 bp sequence between each target site in 3 % of sequences amplified (Upadhyay et al. 2013). In Arabidopsis, three different tandemly arrayed gene families were targeted with ZFNs that recognised multiple members within each cluster (Qi et al. 2013a). The resulting double-stranded DNA breaks and NHEJ produced deletions at these loci up to 55 kb in size. A large chromosomal deletion of 9 Mb was also generated using two ZFN pairs that each recognised a locus at either end of the intervening 9 Mb of sequence. The proportion of somatic cells containing deletions was inversely related to the size of the deletion and varied from 1 to less than 0.1 % (Qi et al. 2013a). However, it is noteworthy that no plants in this study were able to be recovered with germline-transmitted deletions, although the diploid nature of Arabidopsis presumably makes it less amenable for transmission of deletions.
Targeted deletions have also been produced in animal cells. Using zebrafish embryos and mRNA injection, TALENs or TALENS in conjunction with ZFNs were used to generate targeted deletions of endogenous sequences (Gupta et al. 2014). Targeted deletion sizes included 39, 69 kb and 5.5 Mb which were achieved at efficiencies of 3.2, 4.9 and 0.7 %, respectively (Gupta et al. 2014). Similarly in human cell lines, ZFN pairs were used to create precise, large deletions that ranged in size from several hundred base pairs to 15 megabases (Lee et al. 2010). The potential application of this approach for plant improvement is obvious. Deleterious genes linked to traits of interest could be removed, introgressed DNA segments from wild relatives could be reduced in size, and groups of candidate genes could be deleted en mass in positional cloning experiments for gene identification, all of which can be carried out with precision.
Transcriptional reprogramming of endogenous genes
A further ingenious application of designer nucleases is the exploitation of their sequence-targeting abilities to reprogram transcriptional regulation of endogenous genes through targeting transcription factor-binding sites or by generating synthetic transcription factors. The following report by Li et al. (2012) is a wonderful example of exploiting a bacterial pathogen’s virulence armoury to create disease-resistant rice using TALEN technology. In rice, an endogenous sucrose transporter gene, Os Sweet14, is targeted by TALE effectors produced by the bacterial pathogen Xanthomomas oryzae pv. oryzicola resulting in upregulation of this gene during bacterial infection. Upregulation of OsSweet14 is essential for successful infection by this pathogen. The Xoo effector-binding sites present within the promoter region of OsSweet14 were identified and then mutagenised using a sequence-specific TALEN. The resultant altered endogenous rice gene was no longer successfully targeted by Xoo effector proteins which resulted in significantly enhanced resistance to this bacterial pathogen.
In an alternative approach, ZF proteins were modified to generate a synthetic transcription factor in Brassica napus to improve oil quality by reducing the level of saturated fat (Gupta et al. 2012). A ZF protein was engineered to recognise a common region located 50 bp 3′ of the transcriptional start site of two B-ketoacyl-ACP synthase II (KASII) genes involved in fatty acid elongation. A transcriptional activation domain (V16) from the herpes simplex virus was fused to the ZF protein domain to generate a synthetic transcription factor which when introduced into canola resulted in a concomitant increase in KASII gene expression. Transgenic lines showed reduced total saturated fatty acid content in seeds due to a reduction in palmitic acid content resulting in improved oil quality.
Producing cis transgene stacks
Transgenic crop plants (cotton, canola, maize) are now being released that contain multiple transgenes (Que et al. 2010) an example being the Dow Agroscience/Monsanto maize line “SmartStax” which contains eight GM traits (Marra et al. 2010). As more useful transgenic traits are developed, the ability to effectively combine and manipulate large numbers of transgenes becomes more imperative. The most advantageous arrangement of multiple transgenes is at a single locus enabling subsequent simple coinheritance of these traits. A true designer multigene locus would offer the flexibility of addition or subtraction of genes at will to tailor the locus to accommodate various regulatory or commercial requirements.
In numerous studies, recombination-mediated integration using Cre/lox, R/RS and FLP/FRT has been used to target a sequence into a pre-existing recombination site and produce single copy insertions. This has been successfully undertaken in Arabidopsis (Vergunst and Hooykaas 1998; Vergunst et al. 1998; Louwerse et al. 2007), tobacco (Albert et al. 1995; Choi et al. 2000; Day et al. 2000; Nanto et al. 2005; Nanto and Ebinuma 2008; Nanto et al. 2009), maize (Baszczynski et al. 2003; Kerbach et al. 2005), rice (Srivastava and Ow 2002; Srivastava et al. 2004; Chawla et al. 2006; Akbudak et al. 2010; Nandy and Srivastava, 2011; Srivastava 2013), soybean (Li et al. 2009) and aspen (Fladung and Becker 2010). However, again a constraint of these approaches is that a target site must be pre-introduced into the genome through transgenesis and that a limited number of target sites are available for each platform. Nonetheless, sequential rounds of targeted gene insertion theoretically make it plausible to generate large multi-gene stacks using these technologies.
Site-specific integration has also been achieved using ZFNs in corn whereby a 4.5 kb sequence that encoded a selectable maker gene (aad1) and flanking sequences with homology to the target site was precisely integrated in juxtaposition to a pre-existing transgene (pat) in 3 % of transgenic events (Ainley et al. 2013). In this study, a ZFN pair was used to cleave a sequence immediately adjacent to the pat transgene. The homologous sequences flanking the incoming aad1 gene enabled potential homologous recombination between the target site and donor sequence.
Perhaps the most advanced demonstration of sequential cis stacking of transgenes in crop plants has been demonstrated in cotton (Dhalluin et al. 2013). A meganuclease was re-engineered to recognise an endogenous target in juxtaposition to a pre-existing transgene sequence that encoded the cry2Ae insecticidal protein and BASTA herbicide tolerance gene (bar). Using meganuclease cleavage to promote homologous recombination, a second 9 kb sequence encoding two herbicide tolerance transgenes, epsp and hppd (5.5 kb in total), and flanked by sequence (3.5 kb) with target locus homology was introduced adjacent to the first transgene locus in 2 % of transformed calli. Analysis of T1 progeny from regenerated plants showed simple inheritance of these four cis stacked transgenic traits. Interestingly, both this study and the maize studies of Shukla et al. (2009) and Ainley et al. (2013) used homology-dependent repair mechanisms to promote precise transgene insertions.
Targeted transgene insertion does not necessarily require homologous recombination-based processes. In tobacco, 2.5 % of T-DNA insertions occurred in an enforced DS DNA break catalysed by the I-SceI meganuclease, and the incoming T-DNA sequence did not contain significant homology to the target integration site (Tzfira et al. 2003). However, in general these insertions lacked the precision of homologous recombination and were frequently associated with small indels at the target site. In a similar set of experiments using ZFNs, a GFP ORF was excised and replaced with a promoterless antibiotic selectable marker gene (hpt) in both Arabidopsis and tobacco in 5 % of regenerated plants (Weinthal et al. 2013). In this latter experiment, both the GFP target and incoming hpt gene were flanked by the same ZFN recognition site. Likewise, a promoterless GFP reporter gene flanked by TALEN sites was inserted in juxtaposition to an endogenous gene promoter (TaMlo) in wheat protoplasts, albeit with small indels again arising from the NHEJ process (Wang et al. 2014).
When considering the current molecular tools available when producing cis transgene stacks, designer nucleases potentially have the advantage in that the number of sequential target sites is not limited. In addition, the initial choice of insertion site within the plant genome can be theoretically predetermined rather than beginning with a random insertion event. This could enable the first transgene insertion to be located next to a desirable endogenous trait, or, as in the cotton example above, a pre-existing transgene that will contribute to the utility of the final transgene stack. Sensible construct design would enable sequential removal of the previous selectable marker during insertion of the next transgene by flanking this selectable marker with appropriate nuclease target sites.
Regulatory considerations: Are they transgenic?
A caveat for the use of designer nucleases is that firstly the species of choice must have a functional transformation system available, and secondly the resulting plants will be considered as transgenic. Or will they? A designer nuclease can be used to precisely cleave a DNA target site which is then repaired by endogenous DNA repair systems. The nuclease transgene can then be segregated away by selecting progeny plants that contain only the targeted mutation and not the transgene. These plants may potentially be considered as nontransgenic. Logically, these plants differ very little to plants with a mutation in the same gene that have arisen by EMS or radioisotope mutagenesis. The only differences being that the designer nuclease-produced plants will contain a mutation in a precisely defined region of choice in the target gene and will also have far less, if any, unknown background mutations when compared with mutagen-derived plants. The regulation and classification of these precision-engineered crops in terms of their GM or nonGM status is yet to be determined (Kuzma and Kokotovich 2011; Waltz 2012; Lusser and Davies 2013; Hartung and Schiemann 2014).
In summary, a number of designer nuclease platforms are available for crop plant improvement. Their applications range from targeted mutations, deletions, homologous recombination, production of cis transgene stacks and transcriptional reprogramming of endogenous genes. These technologies have been demonstrated to function effectively in a number of important crop species, and it is likely that new cultivars will contain improved germplasm derived from these technologies in the very near future. This adoption would be greatly facilitated by a sensible ruling regarding the nonGM status of these plants in simple targeted mutation applications.
References
Ainley WM, Sastry-Dent L, Welter ME, Murray MG, Zeitler B, Amora R, Corbin R, Miles RR, Arnold NL, Strange TL, Simpson MA, Cao Z, Carroll C, Pawelczak KS, Blue R, West K, Rowland LM, Perkins D, Samuel P, Dewes CM, Shen L, Sriram S, Evand SL, Rebar EJ, Zhang L, Gregory PD, Urnov FD, Webb SR, Petolino JF (2013) Trait stacking via targeted genome editing. Plant Biotechnol J 11:1126–1134
Akbudak MA, More AB, Nandy S, Srivastava V (2010) Dosage-dependent gene expression from direct repeat locus in rice developed by site-specific gene integration. Mol Biotechnol 45:15–23
Albert H, Dale EC, Lee E, Ow DW (1995) Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J 7:649–659
Arnould S, Delenda C, Grizot S, Desseaux C, Paques F, Silva GH, Smith J (2011) The I-CreI meganuclease and its engineered derivatives: applications from cell modification to gene therapy. Protein Eng Des Sel 24:27–31
Baszczynski CL, Gordon-Kamm WJ, Lyznik LA, Peterson DJ, Zhao ZY (2003) Site-specific recombinases and their uses for targeted gene manipulation in plant systems. In: Stewart CN Jr (ed) Transgenic plants: current innovations and future trends. Horizon, Wymondham, pp 157–178
Beetham PR, Kipp PB, Sawycky XL, Arntzen CJ, May GD (1999) A tool for functional plant genomics: chimeric RNA/DNA oligonucleotides cause in vivo gene-specific mutations. Proc Natl Acad Sci USA 96:8774–8778
Belhaj K, Chaparro-Garcia A, Kamoun S, Nekrasov V (2013) Plant genome editing made easy: targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods 9:39–49
Bibikova M, Golic M, Golic KG, Carroll D (2003) Enhancing gene targeting with designed zinc finger nucleases. Science 300:764
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, LaHaye T, Nickstadt A, Bonas U (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Bogdanove AJ, Schornack S, Lahaye T (2010) TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol 13:394–401
Buschges R, Hollricher K, Panstruga R, Simons G, Wolter M, Frijters A, van Daelen R, van der Lee T, Diergaarde P, Groenendijk J, Topsch S, Vos P, Salamini F, Schulze-Lefert P (1997) The barley Mlo gene: a novel control element of plant pathogen resistance. Cell 88:695–705
Cai CQ, Doyon Y, Ainley WH et al (2009) Targeted transgene integration in plant cells using designed zinc finger nucleases. Plant Mol Biol 69:699–1709
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39:e82
Chan SH, Stoddard BL, Xu SY (2011) Natural and engineered nicking endonucleases—from cleavage mechanism to engineering of strand-specificity. Nucleic Acids Res 239:1–18
Chawla R, Ariza-Nieto M, Wilson AJ, Moore SK, Srivastava V (2006) Transgene expression produced by biolistic-mediated, site-specific gene integration is consistently inherited by the subsequent generations. Plant Biotechnol J 4:209–218
Choi S, Begum D, Koshinsky H, Ow DW, Wing RA (2000) A new approach for the identification and cloning of genes: the pBACwich system using Cre/lox-site specific recombination. Nucleic Acids Res 28:e19
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genet 186:757–761
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jian W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using the CRISPR/Cas systems. Sci 339:819–823
Curtin SJ, Zhang F, Sander JD, Haun WJ, Starker C, Baltes NJ, Reyon D, Dalborg EJ, Goodwin MJ, Coffman AP, Dobbs D, Joung JK, Voytas DF, Stupar RM (2011) Targeted mutagenesis of duplicated genes in soybean with zinc finger nucleases. Plant Physiol 156:466–473
D’Halluin K, Vandderstraeten C, Stals E, Cornelissen M, Ruiter R (2008) Homologous recombination: a basis for targeted genome optimisation in crop species such as maize. Plant Biotechnol J 6:93–102
Day CD, Lee E, Kobayashi J, Holappa LD, Albert H, Ow DW (2000) Transgene integration into the same chromosomal location can produce alleles that are differentially silenced. Genes Dev 14:2869–2880
de Pater S, Neuteboom LW, Pinas JE, Hooykaas PJ, van der Zaal BJ (2009) ZFN-induced mutagenesis and gene-targeting in Arabidopsis through agrobacterium-mediated floral dip transformation. Plant Biotechnol J 7:821–835
de Pater S, Pinas JE, Hooykaas PJJ, van der Zaal BJ (2013) ZFN-mediated gene targeting of the Arabidopsis protoporhyrinogen oxidase gene through Agrobacterium-mediated floral dip transformation. Plant Biotechnol J 11:510–515
DeFrancesco L (2011) Move over ZFNs. Nat Biotechnol 29:681–684
Dhalluin K, Vanderstraeten C, Van Hulle J, Rosolowska J, Van Den Brande I, Pennewaert A, Dhont K, Bossut M, Jantz D, Ruiter R, Broadvest J (2013) Targeted molecular trait stacking in cotton through targeted double-strand break induction. Plant Biotechnol J 11:933–941
Djukanovic V, Smith J, Lowe K, Yang M, Gao H, Jones S, Nicholson MG, West A, Lape J, Bidney D, Falco SC, Jantz D, Lyznik LA (2013) Male-sterile plants produced by targeted mutagenesis of the cytochrome P450-like gene (MS26) using a re-designed I-CreI homing endonuclease. Plant J 76:888–899
Dong C, Beetham P, Vincent K, Sharp P (2006) Oligonucleotide-directed gene repair in wheat using a transient plasmid gene repair assay system. Plant Cell Rep 25:457–465
Fauser F, Schiml S, Puchta H (2014) Both CRISPR/Cas-based nuclease and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. Plant J 79:348–359
Feng Z, Zhang B, Ding W, Liu X, Yang D-L, Wei P, Cao F, Zhu S, Zhang F, Mao Y, Zhu J-K (2013) Efficient genome editing in plants using a CSRIPR/Cas system. Cell Res 23:1229–1232
Fladung M, Becker D (2010) Targeted integration and removal of transgenes in hybrid aspen (Populus termula L. × P. tremuloides Michx.) using site-specific recombination systems. Plant Biol 12:334–340
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High frequency off-target mutagenesis induced CRISPR-Cas nucleases in human cells. Nat Biotechnol 31:8
Gaj T, Gerbach CA, Barbas CF III (2013) ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Gao H, Smith J, Yang M, Jones S, Djukanovic V, Nicholson MG, West A, Bidney D, Falco SC, Jantz D, Lyznik LA (2010) Heritable targeted mutagenesis in maize using designed endonucleases. Plant J 61:176–187
Gasser CS, Fraley RT (1989) Genetically engineering plants for crop improvement. Science 244:1293–1299
Gupta M, DeKelver RC, Palta A, Clifford C, Gopalan S, Miller JC, Novak S, Desloover D, Gachotte D, Connell J, Flook J, Patterson T, Robbins K, Rebar EJ, Gregory PD, Urnov FD, Petolino JF (2012) Transcriptional activation of Brassica napus B-ketoacyl-ACP synthase II with an engineered zinc finger protein transcription factor. Plant Biotechnol J10:783–791
Gupta A, Hall VL, Kok FO, Shin M, McNulty JC, Lawson ND, Wolfe SA (2014) Targeted chromosomal deletions and inversions in zebrafish. Genome Res 23:1008–1017
Gurushidze M, Henesel G, Hiekel S, Schedel S, Valkov V, Kumlehn J (2014) True-breeding targeted gene knock-out in barley using designer TALE-nuclease in haploid cells. PLoS. doi:10.1371/journal.pone.0092046
Hannin M, Volrath S, Bogucki A, Briker M, Ward E, Paszkowski J (2001) Gene targeting in Arabidopsis. Plant J 28:671–677
Hartung F, Schiemann J (2014) Precise plant breeding using new genome editing techniques: opportunities, safety and regulation in the EU. Plant J 78:742–752
Haun W, Coffman A, Clasen BM, Demorest ZL, Lowy A, Ray E, Retterath A, Stoddard T, Juillerat A, Cedrone F, Mathis L, Voytas DF, Zhang F (2014) Improved soybean oil quality by targeted mutagenesis of the fatty acid desaturase 2 gene family. Plant Biotechnol J. doi:10.1111/pbi.12201
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832
Iida S, Terada R (2005) Modification of endogenous natural genes by gene targeting in rice and other higher plants. Plant Mol Biol 59:205–219
Jia H, Wang N (2014) Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS ONE 9:e93806
Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP (2013) Demonstration of CRIPSR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. doi:10.1093/nar/gkt780
Jiang W, Yang B, Weeks DP (2014) Efficient CRISPR/Ca9-mediated gene editing in Arabidopsis thaliana and inheritance of modified genes in the T2 and T3 generations. PLoS ONE 9:e99225
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Sci 337:816–821
Johnson RA, Gurevich V, Levy AA (2013) A rapid assay to quantify the cleavage efficiency of custom-designed nucleases in planta. Plant Mol Biol 82:207–221
Joung JK, Sander JD (2013) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14:49–55
Kerbach S, Lorz H, Becker D (2005) Site specific recombination in Zea mays. Theor Appl Genet 111:1608–1616
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA 93:1156–1160
Klug A (2005) Towards therapeutic applications of engineered zinc finger proteins. FEBS Lett 579:892–894
Kochevenko A, Willmitzer L (2003) Chimeric RNA/DNA oligonucleotide-based site-specific modification of the tobacco acetolactate synthase gene. Plant Physiol 132:174–184
Kuzma J, Kokotovich A (2011) Renegotiating GM crop regulation. EMBO Reps 12:883–888
Lee HJ, Kim E, Kim J-S (2010) Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res 20:81–89
Li Z, Xing A, Moon BP, McCardell RP, Mills K, Falco SC (2009) Site-specific integration of transgene cassettes in soybean via recombinase-mediated DNA cassette exchange. Plant Physiol 151:1087–1095
Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, Yang B (2011) TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res 39:359–372
Li T, Liu B, Spalding MH, Weeks DP, Yang B (2012) High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat Biotechnol 30:390–392
Li J-F, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31:688–691
Liang Z, Zhang K, Chen K, Gao C (2014) Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system. J Genet Genomics 41:63–68
Lloyd A, Plasier CL, Carroll D, Drews GN (2005) Targeted mutagenesis using zinc-finger nucleases in Arabidopsis. Proc Natl Acad Sci USA 102:2232–2237
Louwerse JD, van Lier MCM, van der Steen DM, de Vlaam CMT, Hooykaas PJJ, Vergunst AC (2007) Stable recombinase-mediated cassette exchange in Arabidopsis using Agrobacterium tumefaciens. Plant Physiol 145:1282–1293
Lozano-Juste J, Cutler SR (2014) Plant genome engineering in full bloom. Trends Plant Sci. doi:10.1016/j.tplants.2014.02.014
Lusser M, Davies HV (2013) Comparative regulatory approaches for groups of new plant breeding techniques. New Biotechnol 30:437–4456
Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung KJ (2013) CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10:977–979
Mahfouz MM, Li L, Shamimuzzaman M, Wibowo A, Fang X, Zhu JK (2011) De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc Natl Acad Sci USA 108:2623–2628
Mahfouz MM, Li L, Piatek M, Fang X, Mansour H, Bangarusamy DK, Zhu JK (2012) Targeted transcriptional repression using chimeric TALE-SRDX repressor protein. Plant Mol Biol 78:311–321
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Sci 339:823–826
Mani M, Smith J, Kandavelou K, Berg JM, Chandrasegaran S (2005) Binding of two zinc finger nuclease monomers to two specific sites is required for effective double-strand DNA cleavage. Biochem Biophys Res Commun 334:1191–1197
Mao Y, Zhang H, Xu N, Zhang B, Gou F, Zhu J-K (2013) Application of the CRIPSR-Cas system for efficient genome engineering in plants. Mol Plant 6:2008–2011
Marra MC, Piggott NE, Goodwin BK (2010) The anticipated value of SmartStax™ for US corn growers. AgBioForum 13:1–12
Marton I, Zuker A, Shklarman E, Zeevi V, Tovkach A, Roffe S, Ovadis M, Tzfira T, Vanstein A (2010) Non-transgenic genome modification in plant cells. Plant Physiol 154:1079–1087
McConnell-Smith A, Takeuchi R, Pellenz S, Davis L, Maizels N, Monnat RJ, Stoddard BL (2009) Generation of a nicking enzyme that stimulates site-specific gene conversion from the I-Anil LAGLIDADG homing endonuclease. Proc Natl Acad Sci 106:5099–5104
Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL et al (2007) An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25:778–785
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–148
Morbitzer R, Romer P, Boch J, Lahaye T (2010) Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc Natl Acad Sci USA 107:21617–21622
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Sci 326:1501
Nandy S, Srivastava V (2011) Site-specific gene integration in rice genome mediated by the FLP–FRT recombination system. Plant Biotechnol J 9:713–721
Nanto K, Ebinuma H (2008) Marker-free site-specific integration plants. Transgenic Res 17:337–344
Nanto K, Yamada-Watanabe K, Ebinuma H (2005) Agrobacterium-mediated RMCE approach for gene replacement. Plant Biotechnol J 3:203–214
Nanto K, Sato K, Katayama Y, Ebinuma H (2009) Expression of a transgene exchanged by the recombinase mediated cassette (RMCE) method in plants. Plant Cell Rep 28:777–785
Nekrasov V, Staskawicz B, Weigel D, Jones JDG, Kamoun S (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31:691–693
Okuzaki A, Toriyama K (2004) Chimeric RNA/DNA oligonucelotide-directed gene targeting in rice. Plant Cell Rep 22:509–512
Osakabe K, Osakabe Y, Toki S (2010) Site-directed mutagenesis in Arabidopsis using custom-designed zinc finger nucleases. Proc Natl Acad Sci USA 107:12034–12039
Ow D (2007) GM maize from site-specific recombination technology, what next? Curr Opin Biotechnol 18:115–120
Paques F, Duchateau P (2007) Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr Gene Therapy 7:49–66
Parry MAJ, Madgwick PJ, Bayon C, Tearall K, Hernandez-Lopez A, Baudo M, Rakszegi M, Hamada W, Al-Yassin A, Ouabbou H, Labhilili M, Phillips AL (2009) Mutation discovery for crop improvement. J Exp Bot 60:2817–2825
Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, Guilak F, Crawford GE, Reddy TE, Gersbach CA (2013) RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10:973–976
Petolino JF, Worden A, Curlee K, Connell J, Strange Moynahan TL, Larsen C, Russell S (2010) Zinc finger nuclease-mediated transgene deletion. Plant Mol Biol 73:617–628
Porteus MH, Carroll D (2005) Gene targeting using zinc finger nucleases. Nat Biotechnol 23:967–973
Pruett-Miller SM, Reading DW, Porter SN, Porteus MH (2009) Attenuation of zinc finger nuclease toxicity by small-molecule regulation of protein levels. PLoS Genet 5(2):e1000376. doi:10.1371/journal.pgen.1000376
Puchta H (2002) Gene replacement by homologous recombination in plants. Plant Mol Biol 48:173–182
Puchta H (2005) The repair of double stranded DNA breaks in plants. J Exp Bot 56:1–14
Puchta H, Dujon B, Hohn B (1996) Two different but related mechanisms are used in plants for the repair of genomic double-strand breaks by homologous recombination. Proc Natl Acad Sci USA 93:5055–5060
Qi Y, Li X, Zhang Y, Starker CG, Baltes NJ, Zhang F, Sander JD, Reyon D, Joung JK, Voytas DF (2013a) Targeted deletion and inversion of tandemly arrayed genes in Arabidopsis thaliana using zinc finger nucleases. G3 3:1707–1715
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weismann JS, Arkin AP, Lim WA (2013b) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183
Que Q, Chilton M-D, de Fontes CM, He C, Nuccio M, Zhu T, Wu Y, Chen JS, Shi L (2010) Trait stacking in transgenic crops. GM Crop 1:220–229
Ramalingam S, Kandavelou K, Rajenderan R, Chandrasegaran S (2011) Creating designed zinc-finger nucleases with minimal cytotoxicity. J Mol Biol 405:630–641
Sanchez JP, Ullman C, Moore M, Choo Y, Chua NH (2006) Regulation of Arabidopsis thaliana 4-coumarate: coenzyme-A ligase-1 expression by artificial zinc finger chimeras. Plant Biotechnol J 4:103–114
Schornack S, Moscou MJ, Ward ER, Horvath DM (2013) Engineering plant disease resistance based on TAL effectors. Ann Rev Phytopathol 51:383–406
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root D, Doench JG, Zhang F (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Sci 343:84–87
Shan Q, Wang Y, Chen K, Liang Z, Li J, Zhang Y, Zhang K, Liu J, Voytas DF, Zheng X, Zhang Y, Gao C (2013a) Rapid and efficient gene modifications in rice and Brachypodium using TALENs. Mol Plant. doi:10.1093/mp/ss162
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu J-L, Gao C (2013b) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31:686–688
Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, Mitchell JC, Arnold NL, Gopalan S, Meng X, Choi VM, Rock JM, Wu YY, Katibah GE, Zhifang G, McCaskill D, Simpson MA, Blakeslee B, Greenwalt SA, Butler HJ, Hinkley SJ, Zhang L, Rebar EJ, Gregory PD, Urnov FD (2009) Precise genome modification in the crop species Zea mays using zinc finger nucleases. Nat 259:442–445
Small I (2007) RNAi for revealing and engineering plant gene functions. Curr Opin Biotechnol 18:148–153
Srivastava V (2013) Site specific gene integration in rice. In: Rice protocols, methods in molecular biology. vol. 958, Humana Press, Pennsylvania, USA, pp 83–93
Srivastava V, Ow DW (2002) Biolistic site-specific integration in rice. Mol Breeding 8:345–350
Srivastava V, Anderson OD, Ow DW (1999) Single-copy transgenic wheat generated through the resolution of complex integration patterns. Proc Natl Acad Sci USA 96:11117–11121
Srivastava V, Ariza-Nieto M, Wilson AJ (2004) Cre-mediated site-specific gene integration for consistent transgene expression in rice. Plant Biotechnol J 2:169–179
Stege JT, Guan X, Ho T, Beachy RN, Barbas CF III (2002) Controlling gene expression in plants using zinc finger transcription factor. Plant J 32:1077–1086
Straub A, LaHaye T (2013) Zinc fingers, TAL effectors or Cas9-based DNA binding proteins: What’s best for targeting desired genome loci? Mol Plant 6:1384–1387
Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ et al (2007) Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25:786–793
Tester M, Langridge P (2010) Breeding Technologies to increase crop production in a changing world. Sci 327:818–822
Thomas Scott C (2005) The zinc finger nuclease monopoly. Nat Biotechnol 23:915–918
Tovkach A, Zeevi V, Tzfira T (2009) A toolbox and procedural notes for characterising novel zinc finger nucleases for genome editing in plant cells. Plant J 57:747–757
Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF (2009) High-frequency modification of plant genes using engineered zinc-finger nucleases. Nat 459:442–445
Tzfira T, Frankmen L, Vaidya M, Citovsky V (2003) Site-specific integration of Agrobacterium tumefacians T-DNA via double-stranded intermediates. Plant Physiol 133:1011–1023
Tzfira T, Weinthal D, Marton I, Zeevi V, Zuker A, Vainstein A (2012) Genome modification in plant cells by custom-made restriction enzymes. Plant Biotechnol J 10:373–389
Upadhyay SK, Kumar J, Alok A, Tuli R (2013) RNA-guided genome editing for target gene mutations in wheat. G3 3:2233–2238
van Nierop GP, de Vries AA, Holkers M, Vrijsen KR, Goncalves MA (2009) Stimulation of homology-directed gene targeting at an endogenous human locus by a nicking endonuclease. Nucleic Acids Res 37:5725–5736
Vergunst AC, Hooykaas PJJ (1998) Cre/lox-mediated site specific integration of Agrobacterium T-DNA in Arabidopsis thaliana by transient expression of Cre. Plant Mol Biol 38:393–406
Vergunst AC, Jansen LET, Hooykaas PJJ (1998) Site-specific integration of Agrobacterium T-DNA in Arabidosis thaliana mediated by Cre recombinase. Nucleic Acids Res 26:2729–2734
Waltz E (2012) Tiptoeing around transgenics. Nat Biotechnol 30:215–217
Wang Y, Yau Y-Y, Perkins-Balding D, Thomson JG (2011) Recombinase technology: applications and possibilities. Plant Cell Rep 30:267–285
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu J-L (2014) Simultaneous editing of three homoealleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol. doi:10.1038/nbt.2969
Weinthal DM, Taylor RA, Tzfira T (2013) Nonhomologous end joining-mediated gene replacement in plant cells. Plant Physiol 162:390–400
Wendt T, Holm PB, Starker CG, Christian M, Voytas DF, Brinch-Pedersen H, Holme IB (2013) TAL effector nuclease induced mutations at a pre-selected location in the genome of primary barley transformants. Plant Mol Biol 83:279–285
Wright DA, Townsend JA, Winfrey RJ, Irwin PA, Rajagopal J, Lonosky PM, Hall BD, Jondle MD, Voytas DF (2005) High-frequency homologous recombination in plants mediated by zinc-finger nucleases. Plant J 44:693–705
Xie K, Yang Y (2013) RNA-guided genome editing plants using a CRISPR-Cas system. Mol Plant 6:1975–1983
Xie K, Zhang J, Yang Y (2014) Genome-wide prediction of highly specific guide RNA spacers for CRISPR-Cas9-mediated genome editing in model plants and major crops. Mol Plant 7:923–926
Xu R, Li H, Qin R, Wang L, Li L, Wei P, Yang J (2014) Gene targeting using the Agrobacterium tumefaciens-mediated CRISPR-Cas system in rice. Rice 7:5
Yang M, Djukanovic V, Stagg J, Lenderts B, Bidney D, Falco SC, Lyznik LA (2009) Targeted mutagenesis in the progeny of maize transgenics. Plant Mol Biol 70:669–779
Zhang F (2014) CRISPR-Cas systems and methods for altering expression of gene products. US Patent No. 8,697,359
Zhang F, Maeder ML, Unger-Wallace E, Hoshaw JP, Reyon D, Christian M, Li X, Pierick CJ, Dobbs D, Peterson T, Joung KJ, Voytas DF (2010) High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc Natl Acad Sci USA 107:12028–12033
Zhang Y, Zhang F, Li X, Baller JA, Qi Y, Starker CG, Bogdanove AJ, Voytas DF (2013) Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol 161:20–27
Zhang H, Zhang J, Wei O, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Zhang H, Xu N, Zhu J-K (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J12:797–807
Zhu T, Peterson DJ, Tagliani L, Clair G, Baszczynski CL, Bowen B (1999) Targeted manipulation of maize genes in vivo using chimeric RNA/DNA oligonucleotides. Proc Natl Acad Sci USA 96:8768–8773
Acknowledgments
We wish to thank the Australian Grains Research and Development Corporation (GRDC) for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rinaldo, A.R., Ayliffe, M. Gene targeting and editing in crop plants: a new era of precision opportunities. Mol Breeding 35, 40 (2015). https://doi.org/10.1007/s11032-015-0210-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0210-z