
Abstract
Oils with high oleic acid content are in great demand because they have optimal properties for food and non-food uses. Two different levels of high oleic acid content (>75 and >84%) have been reported in safflower (Carthamus tinctorius L.). The trait is mainly controlled by partially recessive alleles at a major gene Ol, but the highest levels have been attributed to modifying genes. The objectives of this research were to map the Ol locus and modifying genes involved in oleic acid content of safflower seeds and to determine the nature of Ol through a candidate gene approach. Two F2 mapping populations from the nuclear male-sterile line CL-1 and the high oleic acid lines CR-6 (>75% oleic acid) and CR-9 (>84%) were developed and phenotyped for oleic acid content at the F2 and F3 seed level. A genetic linkage map comprising 15 linkage groups and 116 random amplified polymorphic DNA, simple sequence repeat (SSR), and sequence-characterized amplified regions marker loci was constructed for the CL-1 × CR-9 population. The Ol gene was mapped to linkage group (LG) T3 tightly linked to the SSR marker ct365, which was confirmed in the CL-1 × CR-6 population. Additionally, a quantitative trait locus with a minor effect on increasing oleic acid content was identified on LG T2. The candidate gene approach indicated that an oleoyl-phosphatidylcholine desaturase FAD2-1 locus underlies the Ol gene. Both the genetic information and the markers developed in this research will contribute to marker-assisted selection for high oleic acid content in safflower.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Safflower (Carthamus tinctorius L.) has been cultivated since antiquity for its flowers, used for flavouring and colouring foods and for making dyes, and for its seed oil (Dajue and Mündel 1996). Nowadays safflower is an underutilized minor oilseed crop with great potential in many areas of the world because of its agronomic performance and seed oil quality (Knowles 1989). Conventional safflower oil has a high linoleic acid content (>70%), which is a unique trait amongst oilseed crops. Additionally, safflower germplasm producing high oleic acid oil has been identified (Knowles 1989). High oleic acid oils are highly appreciated for food and non-food applications because they combine a hypocholesterolemic effect (Mensink and Katan 1989) with a much greater oxidative stability than oils with greater polyunsaturation levels (Yodice 1990).
High oleic acid safflower was initially reported as having oleic acid content between 64 and 83% of the total fatty acids, which was produced by partially recessive alleles ol at a single locus Ol (Knowles and Hill 1964). Another different allele ol 1 at the same locus produced, in homozygous condition, between 35 and 50% oleic acid, compared to 10–15% in safflower plants carrying the wild-type alleles Ol (Knowles and Hill 1964). Fernández-Martínez et al. (1993) identified safflower germplasm with higher oleic acid content (86–91%) than that previously reported by Knowles and Hill (1964). Hamdan et al. (2009a) demonstrated that these very high oleic acid levels were produced by the recessive alleles ol in combination with modifying genes with a positive effect on oleic acid content. The role of modifying genes in increasing oleic acid content beyond the limits produced by the ol alleles had been previously suggested by Knowles (1972). Hamdan et al. (2009a) also identified an effect of modifying genes on reducing oleic acid content below the expected levels in germplasm carrying the ol alleles. A similar negative effect of modifying genes on oleic acid content has been reported in sunflower (Urie 1985; Fernández-Martínez et al. 1989).
In the fatty acid biosynthetic pathway, the microsomal enzyme oleoyl-phosphatidylcholine desaturase (FAD2) catalyses the desaturation step from oleic acid (18:1) to linoleic acid (18:2) (Okuley et al. 1994). Whereas only a single copy of the FAD2 gene has been identified in plants such as Arabidopsis and maize (Okuley et al. 1994; Beló et al. 2008), the genome of the oil crops sunflower, soybean, and canola contains multiple copies of the gene (Scheffler et al. 1997; Martínez-Rivas et al. 2001; Schlueter et al. 2007). From these multiple copies, a highly expressed or seed-specific microsomal FAD2 (FAD2-1) has been demonstrated to underlie the elevated oleic acid levels in the seeds of the oil crops mentioned (Pérez-Vich et al. 2002; Schuppert et al. 2006; Hu et al. 2006; Falentin et al. 2007; Lacombe et al. 2009; Pham et al. 2010, 2011). Recently, three different FAD2 genes (FAD2-1, FAD2-2, and FAD2-3) have been identified in safflower (Guan et al. 2011a, b). From them, FAD2-1 is strongly expressed in developing seeds and has a high similarity to sunflower FAD2-1. Additionally, Guan et al. (2011a) determined that the FAD2-1 transcript levels in a high oleic acid safflower genotype were significantly lower than in standard (high linoleic) genotypes during seed development. Accordingly, an alteration of the function of the enzyme encoded by safflower FAD2-1 is a priori hypothesized as the factor underlying the high oleic acid trait in this crop.
Unlike most oilseed crops, research on the development of molecular markers for trait selection has been scarce in safflower. The use of molecular markers in this crop has mainly focused on the evaluation of genetic diversity (Yazdi-Samadi et al. 2001; Ravikumar et al. 2005; Sehgal and Raina 2005; Johnson et al. 2007; Yang et al. 2007; Amini et al. 2008) and the taxonomy of the genus Carthamus (Vilatersana et al. 2005; Chapman and Burke 2007). Nevertheless, SCAR (sequence-characterized amplified regions) markers have been developed linked to the Li, Ms, HSya, and Tph2 genes determining very high linoleic acid content, nuclear male sterility, accumulation of hydroxysafflor yellow A, and high gamma-tocopherol content, respectively (Hamdan et al. 2008; Zhang et al. 2009; García-Moreno et al. 2011). Moreover, the recent development of safflower SSR (simple sequence repeats) and RFLP (restriction fragment length polymorphism) markers (Chapman et al. 2009; Mayerhofer et al. 2010; Hamdan et al. 2011) and a first genetic linkage map (Mayerhofer et al. 2010) has established the foundation for genomic studies in this species and supplied a critical mass of sequence-based DNA markers for safflower molecular trait breeding.
The objectives of this research were to map the Ol locus and modifying genes controlling high oleic acid content in safflower seeds and to determine the nature of Ol through a candidate gene approach.
Materials and methods
Plant material, phenotypic analyses, and DNA extraction
CR-6 is a safflower line with high oleic acid content (75–83%) isolated from the US Department of Agriculture (USDA) germplasm accession PI 560177 (www.ars-grin.gov/npgs/). CR-9 is a safflower line with very high oleic acid content (84–88%) developed from the USDA germplasm accession PI 401479. CL-1 is a nuclear male-sterile (NMS) safflower line with conventional fatty acid profile (oleic acid content from 14 to 22%) isolated from the USDA-ARS germplasm accession PI 560161. The lines CR-6 and CR-9 carry the recessive alleles ol for high oleic acid content at the Ol gene. The different levels of oleic acid in both lines are determined by modifying genes with a small positive effect on the trait (Hamdan et al. 2009a). Male sterility in CL-1 is controlled by recessive alleles at a single gene Ms (Heaton and Knowles 1982).
Heads of male sterile plants of CL-1 were bagged before flowering and pollinated with fresh pollen of CR-6 and CR-9 plants. F1 plants from the CL-1 × CR-6 and the CL-1 × CR-9 crosses were grown and self-fertilized by bagging the heads before the beginning of flowering. One population of 119 F2 seeds from the cross CL-1 × CR-6 and another population of 116 F2 seeds from the cross CL-1 × CR-9 were used for the molecular analyses. These populations were analysed for fatty acid composition by the half-seed technique, as described below. The F2 half-seeds were germinated and the corresponding plants were grown in the field in 2005 and self-fertilized as described above to produce the F3 seed. Since ms alleles determining NMS were segregating in these populations, male fertility/sterility was recorded for each F2 plant. Additionally, male-sterile plants were testcrossed with plants of CR-6 (for the CL-1 × CR-6 cross) or CR-9 (for the CL-1 × CR-9 cross) in order to determine their genotype for oleic acid content. From 24 to 48 F3 seeds from each male-fertile F2 plant and from 24 to 48 testcross seeds from each male-sterile F2 plant were analysed to determine presence or absence of segregation for oleic acid content. F2 plants were classified as OlOl if they had a low oleic acid content in their respective F2 half-seed and also showed uniformly low oleic acid levels in F3 or testcross seeds, Olol if their F3 or testcross seeds segregated for low and high oleic acid content, and olol if both the F2 half-seed and the F3 or testcross seeds showed high oleic acid content. Male-fertile F2 plants not producing a minimum of 24 F3 seeds for fatty acid analyses (46 out of 97 in the CL-1 × CR-6 population, and 13 out of 79 in the CL-1 × CR-9 population) were classified as Ol_ if oleic acid content in the F2 half-seed was <30% and olol if oleic acid content in the F2 half-seed was >75%. For the Ms gene, F2 plants were classified as Ms_ if they were male-fertile, and msms if they were male-sterile.
The fatty acid composition of the oil in half-seeds was analyzed by simultaneous extraction and methylation of the fatty acids (Garcés and Mancha 1993) followed by gas–liquid chromatography (GLC) using a Perkin-Elmer Autosystem gas–liquid chromatograph (Perkin-Elmer Corporation, Norwalk, CT, USA). A 2-m-long column packed with 3% SP-2310/2% SP- 2300 on Chromosorb WAW (Supelco Inc., Bellefonte, PA, USA) was used. The oven, injector, and flame ionization detector were held at 198, 275, and 250°C, respectively.
For DNA extraction, ten fully expanded leaves were cut from individual F2 plants from the CL-1 × CR-6 and CL-1 × CR-9 populations and frozen at −80°C. The leaf tissue was lyophilised and ground to a fine powder in a laboratory mill. DNA was isolated from ground leaf tissue from each F2 plant using a modified version of the protocol described by Rogers and Bendich (1985). DNA was also isolated from five plants of the CL-1, CR-6, and CR-9 parental lines.
Molecular analysis of the CL-1 × CR-9 population
The CL-1 × CR-9 population was used to map the Ol gene and to identify molecular markers linked to modifying genes affecting its expression. A genetic linkage map from this population was constructed using random amplified polymorphic DNA (RAPD), SCAR, and SSR markers. For RAPD analysis, 122 RAPD primers (Operon Technologies, Alameda, CA, USA; complete kits G, H, L, M, and S, and two to six primers from kits A, B, F, J, and AA) and 313 pairwise combinations of these RAPD primers were surveyed in two replicate samples of the parental lines CL-1 and CR-9 and four F2 individuals. PCR mixture composition and reaction conditions were as described by Hamdan et al. (2008). Eighty primers revealing intense and clearly scorable polymorphic bands were chosen for the analysis of the CL-1 × CR-9 F2 population. Additionally, five SCAR markers developed in a previous study by Hamdan et al. (2008) linked to the Li gene determining very high linoleic acid content were screened in the parental lines CL-1 and CR-9, according to the protocols reported by Hamdan et al. (2008). Three of them (IASCA-39, IASCA-42, and IASCA-45) were polymorphic and mapped in the CL-1 × CR-9 population.
For SSR analysis, 109 genomic safflower SSR primers developed by Hamdan et al. (2011) (prefix CAT), 104 EST (expressed sequence tag)-based SSR primers reported by Chapman et al. (2009) (prefixes CT1 and EL), 112 EST-based and genomic safflower SSR primers developed and mapped by Mayerhofer et al. (2010) (prefixes ct, gd, and VL), and two EST-based Carthamus maculosa SSR primers (prefix cm) were screened in two replicate samples of the parental lines and four F2 individuals. The PCR reaction mixture (30 μl) contained 1 × PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTPs (Invitrogen/Life Technologies, Carlsbad, CA, USA), 0.3 μM of primers, 0.7 U of Taq DNA polymerase (BioTaq™ DNA Polymerase, Bioline, London, UK), and 50 ng of template DNA. To reduce non-specific amplification, touchdown PCR was used with an initial denaturation at 94°C for 2 min, followed by one cycle of 94°C for 30 s, final annealing temperature (T A) +10°C for 30 s, and 72°C for 30 s. The annealing temperature was decreased by 1°C per cycle during each of the nine following cycles, at which time the products were amplified for 32 cycles at 94°C for 30 s, T A for 30 s, and 72°C for 30 s with a final extension of 20 min at 72°C. Final annealing temperatures varied from 46 to 62°C. Amplified products were separated on 3% Metaphor® (BMA, Rockland, ME, USA) agarose gels in 1 × TBE buffer with ethidium bromide incorporated in the gel. A total of 60 SSR primers revealing clear polymorphisms were genotyped in the CL-1 × CR-9 F2 population.
For all RAPD, SSR, and SCAR loci, Chi-squared analyses were carried out on each locus to detect deviations from the expected Mendelian ratios for co-dominant (1:2:1) or dominant (3:1) markers. A RAPD–SCAR–SSR linkage map was constructed using the software MAPMAKER/EXP version 3.0b (Whitehead Institute, Cambridge, MA, USA) (Lander et al. 1987). Two-point analysis was used to identify linkage groups (LGs) at a LOD score of 4 and a maximum recombination frequency of 0.40. Three-point and multi-point analyses were used to determine the order and interval distances between the markers in each LG. The Haldane mapping function was used to compute the map distances in centiMorgans (cM) from the recombination fractions. Multiple loci detected by a single probe were coded with the probe name plus the suffix “a”, “b” or “c”. Linkage group maps were drawn using the MapChart software (Voorrips 2002). Nomenclature for the linkage groups followed, when possible, that of Mayerhofer et al. (2010). The Ol and Ms genes were mapped in the genetic linkage map using their segregation data. The genotypes for the Ol and Ms genes were inferred from oleic acid and male-sterility phenotypes, respectively, as described above. Two-point analysis was used to group the marker loci, Ol, and Ms with a LOD threshold of 6 and a maximum recombination fraction of 0.4 as linkage criteria. The significance of each marker’s association with the phenotypic trait was determined by one-way analysis of variance (ANOVA) using the statistical package IBM SPSS Statistics version 19.0 (IBM Corp., Armonk, NY, USA), with marker genotypes being classes.
Composite interval mapping (CIM) (Zeng 1994) was used to scan the genome for quantitative trait loci (QTL) affecting oleic acid content, in order to strengthen and corroborate the Ol mapping results, evaluate the existence of additional QTL, and estimate the interaction and global effect of all the detected QTL. Phenotypic data consisted of the oleic acid content of each F2 half-seed and also the oleic acid content of the corresponding F2:3 family in the case of male-fertile F2 plants (average oleic acid content from 24–48 F3 seeds from self-fertilized F2 plants). Since minor or modifying genes are not detectable when the major gen Ol is segregating in the population, QTL analyses were also performed on a high oleic F3 subpopulation (F3 seeds classified as olol) to scan the genome for minor QTL without the highly significant effect of the Ol gene. This subpopulation contained F2:3 families with uniformly high oleic acid content coming only from olol F2 genotypes, and F2:3 families segregating for low and high oleic acid coming only from Olol genotypes. For the latter, only the phenotypic data of the high oleic acid class were used, since the analysis of F3 seeds from these Olol F2 genotypes revealed clear bimodal distributions in which the high oleic acid class could be clearly identified. The analyses were carried out by using the average oleic acid content, the minimum oleic acid content, and the maximum oleic acid content within the high oleic acid class (olol F3 genotypes) in each Olol and olol F2:3 family.
CIM was performed first with the marker closest to a highly significant oleic acid QTL, detected by initial simple interval mapping analyses, as a co-factor. If new QTL were found, the whole genome was re-scanned including as co-factor the marker locus closest to the new QTL. Genome-wide threshold values (α = 0.05) for declaring the presence of QTL were estimated from 1,000 permutations of each phenotypic trait (Churchill and Doerge 1994). The thresholds of the LOD score were 2.66 for the F2 oleic acid content, 2.63 for the average F3 oleic acid content (whole population), 2.52 for the average F3 oleic acid content in the high oleic acid class (Olol + olol subpopulation), 2.64 for the minimum F3 oleic acid content in the high oleic acid class (Olol + olol subpopulation), and 2.72 for the maximum F3 oleic acid content in the high oleic acid class (Olol + olol subpopulation). Estimates of QTL positions were obtained at the point where the LOD score assumes its maximum in the region under consideration. One-LOD support limits for the position of each QTL were also calculated (Bohn et al. 1996). The proportion of phenotypic variance explained by each individual QTL was calculated as the square of the partial correlation coefficient (R 2). Estimates of the additive (a i) and dominance (d i) effects were computed following Falconer (1989). The software package used to perform CIM was PLABQTL 1.2 (Utz and Melchinger 1996).
Finally, those RAPD fragments identified by CIM as associated with oleic acid QTL were transformed into more consistent SCAR markers. RAPD fragments amplified from the wild-type (CL-1) or the very high oleic acid parental line (CR-9) were separated on a 1.5% agarose gel before being excised and purified by means of the QIAquick gel extraction kit (Qiagen GmbH, Hilden, Germany). The purified products were ligated in a T/A vector (pCR2.1) and cloned with the TOPO-TA cloning kit (Invitrogen/Life Technologies) as described by the manufacturer. Transformed E. coli were plated on selective media containing ampicillin and X-gal. Five white colonies per marker were picked from the plate and cultured overnight. The QIAprep Spin Miniprep Kit (Qiagen) was used for plasmid DNA extraction. PCR with M13 forward and reverse vector primers was performed to confirm the presence and size of the insert. Sequencing in both forward and reverse orientations of the cloned fragments (three clones per marker) was carried out at GATC Biotechnology (Konstanz, Germany) using the M13 forward and reverse sequencing primers. Sequence analysis was conducted with the aid of the software Vector NTI Advance 10.3.0 (Invitrogen/Life Technologies). The consensus sequence for the three clones sequenced for each RAPD fragment was used to design the SCAR markers. A first set of primer pairs (F1 and R1) was designed containing the original 10 bases of the RAPD primer plus the next 8–15 internal bases. A second set of primer pairs (F2 and R2) located more internally was designed with the software Vector NTI Advance 10.3.0. SCAR-PCR amplifications were carried out as described by Hamdan et al. (2008), adjusting annealing temperatures according to the primer-specific T m. SCAR markers were genotyped in the F2 CL-1 × CR-9 individuals, mapped, and CIM analyses were re-run as described above. This final QTL analysis with the new SCAR markers included in the map is the one described in the Results section.
Molecular analysis of the CL-1 × CR-6 population
The CL-1 × CR-6 population was used to confirm the effect of the Ol gene and its map position. All SCAR and SSR markers linked to Ol in the CL-1 × CR-9 population were screened for polymorphisms in duplicated samples of the parental lines and four F2 individuals of the CL-1 × CR-6 population. PCR mixture composition and reaction conditions were as described above. Since two SCAR markers flanking the Ol gene were not polymorphic, their corresponding RAPD markers were tested for polymorphisms. RAPD reactions were also as described above. Three SCAR, three SSR, and one RAPD primers revealing polymorphisms were genotyped in the CL-1 × CR-6 F2 population. Linkage analysis was run with MAPMAKER using segregation data for RAPD, SCAR, and SSR marker loci, as well as for the Ol and Ms loci. The genotypes for the Ol and Ms genes were inferred from oleic acid and male-sterility phenotypes, respectively, as described above. Two-point analysis was used to group the SCAR and SSR marker loci, Ol, and Ms with a LOD threshold of 3 and a maximum recombination fraction of 0.40. Multi-point analysis was used to determine the order and interval distances between the markers. Recombination fractions were converted to cM using the Haldane mapping function. The significance of each marker’s association with the phenotypic trait was determined by one-way ANOVA as described above.
Candidate gene analysis
The FAD2-1 gene has been shown to be the candidate gene for high oleic acid content in other oilseed crops, since mutations in this gene disrupt the synthesis of linoleic acid and cause a build-up of oleic acid in the seeds (Pérez-Vich et al. 2002; Schuppert et al. 2006; Lacombe et al. 2009; Hu et al. 2006; Falentin et al. 2007). Twenty primers (10 forward and 10 reverse; Table 1) based on the safflower FAD2-1 sequence (GenBank accesion no. HM165274) were designed and screened for polymorphisms in the parental lines CL-1, CR-6, and CR-9. Since no polymorphisms were found, other parental lines from mapping populations other than CL-1 × CR-9 and CL-1 × CR-6 from our safflower breeding program were screened for polymorphisms. Parental lines from an F2 population segregating for palmitic acid content (CL-1 × CR-50; Hamdan et al. 2009b) showed a robust one-band co-dominant polymorphism for HM165274-based primers and accordingly this population (145 F2 individuals) was used to map the FAD2-1 gene. All polymorphic SSR and SCAR markers from the LG in which the Ol gene was mapped were also genotyped in the CL-1 × CR-50 population.
Since the three mapping populations CL-1 × CR-9, CL-1 × CR-6, and CL-1 × CR-50 shared CL-1 as a parent, identical alleles among the three populations could be identified. A consensus map for the three populations was made using the common markers mapped in these populations as anchor loci, i.e. a common data file for the three populations was constructed in which markers segregating in only one population were coded as missing data in the others. Two-point analysis was used to group all marker loci with a LOD threshold of 6 and a maximum recombination fraction of 0.40. Three-point and multi-point analyses were used to determine the order and interval distances between the markers. Recombination fractions were converted to cM using the Haldane mapping function.
Results
Phenotypic segregations
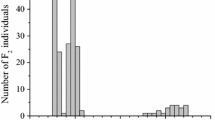
Oleic acid content in F2 seeds from the cross CL-1 × CR-6 followed a bimodal distribution, with a low–intermediate oleic acid class ranging from 12.9 to 33.3% (n = 94) and a high oleic acid class ranging from 77.7 to 81.5% (n = 25), which fitted the expected 3:1 (low–intermediate:high oleic acid content) ratio (χ2 = 1.01, p = 0.31) corresponding to the segregation of the recessive ol alleles. Ninety-seven F2 plants were male-fertile and 22 F2 plants were male-sterile, which fitted the expected 3:1 (male-fertile:male-sterile) ratio (χ2 = 2.69, p = 0.10) for the segregation of the recessive ms alleles. The analysis of the F3 seed generation (Fig. 1) uncovered the three genotypic classes OlOl, with uniformly low oleic acid content (n = 10), Olol, segregating for oleic acid content (n = 27), and olol, with uniformly high oleic acid content (n = 14), which did not differ significantly from an 1:2:1 (OlOl:Olol:olol) ratio (χ2 = 0.80, p = 0.67).

Oleic acid content (% of total fatty acids) in individual F2 half-seeds and their corresponding F2 plants (F3 seeds averaged) in a population of 51 male-fertile F2 plants from the cross between the nuclear male-sterile safflower line CL-1, with wild-type oleic acid content, and the line CR-6, with high oleic acid content (>75%), and in a population of 66 male-fertile F2 plants from the cross CL-1 × CR-9, with very high oleic acid content (>84%)
The analysis of F2 seeds from the cross CL-1 × CR-9 also revealed a bimodal distribution with 88 individuals with low–intermediate oleic acid content (10.4–30.9%) and 28 individuals with high oleic acid content (82.5–87.2%), which fitted a 3:1 ratio (χ2 = 0.05, p = 0.83). Seventy-nine F2 plants were male-fertile and 37 F2 plants were male-sterile, which fitted the expected 3:1 ratio (χ2 = 2.94, p = 0.09). The analysis of the F3 seed generation (Fig. 1) allowed the classification of F2 individuals within genotypic classes OlOl, with uniformly low oleic acid content (n = 15), Olol, segregating for oleic acid content (n = 35), and olol, with uniformly high oleic acid content (n = 16), which fitted a 1:2:1 (OlOl:Olol:olol) ratio (χ2 = 0.27, p = 0.87).
Oleic acid content in F3 seeds classified as olol, including F3 seeds from both olol and Olol F2 genotypes, ranged from 58.1 to 83.8% in the cross CL-1 × CR-6 and from 59.4 to 88.9% in the cross CL-1 × CR-9. Oleic acid content in the parents grown in the same environment ranged from 78.7 to 83.4% in CR-6 and from 84.0 to 88.9% in CR-9.
CL-1 × CR-9 linkage map construction
A preliminary RAPD–SCAR–SSR map was constructed with 172 RAPD, SCAR, and SSR polymorphic markers. After removal of closely linked RAPD loci, the final CL-1 × CR-9 linkage map comprised 116 marker loci. Of these, 47 were RAPD, 60 were SSR, four were SCAR developed by Hamdan et al. (2008) linked to the Li gene (IASCA-39, IASCA-42, IASCA-44, and IASCA-45), three were SCAR newly developed in this study presumably linked to oleic acid QTL (IASCA-73, IASCA-75, and IASCA-76), one was a sunflower InDel marker (prefix IN), and one was a phenotypic marker corresponding to the Ms gene determining nuclear male sterility mapped as a Mendelian trait. Of the 60 SSR included in the map, 26 of them had been mapped previously by Mayerhofer et al. (2010) whereas 34 of them were mapped for the first time in this study. The linkage map spanned a distance of 816.4 cM with an average marker interval of 7.8 cM (Fig. 2). The loci coalesced into 15 linkage groups, of which eight could be matched to those described by Mayerhofer et al. (2010) based on at least two shared marker loci (Fig. 2), and four were tentatively matched to those of Mayerhofer et al. (2010) based on one marker locus (Fig. 2). The remaining three LGs which did not show any shared marker loci were named as LGs Tx, Ty, and Tz. Linkage group T4a of Mayerhofer et al. (2010) was split into two LGs in our map (LG T4a_I and LG T4a_II) and LGs T10a and T10b of Mayerhofer et al. (2010) were also maintained as two independent LGs in our map (Fig. 2). The linkage groups ranged in genetic length from 5.1 to 192.5 cM and comprised 2–23 marker loci (Fig. 2). 99.7% of the mapped genome was within 20 cM of the nearest marker. None of the RAPD, SSR, or SCAR loci deviated significantly from the expected segregation ratios (p < 0.001).

Genetic linkage map of the CL-1 × CR-9 population. Linkage groups (LG) T1–T10 follow the nomenclature of Mayerhofer et al. (2010). Genetic distances are given in centiMorgans (Haldane) on the left of each LG. QTL locations for F2 and F3 seed oleic acid content (complete population), and for the mean and maximum oleic acid content of the high oleic acid class in the F3 (subpopulation Olol + olol) are indicated by arrows at the point where the LOD score reaches its maximum and by boxes corresponding to 1-LOD support intervals. The QTL names and their associated traits, as indicated in Table 3, are shown at the left of the QTL boxes. The position of the Ms gene determining nuclear male sterility mapped as Mendelian trait is also indicated in LG T3. Marker loci are labeled as follows: prefixes OP and F are RAPD marker loci, prefixes CAT, CT1, EL, ct, gd, VL, and cm are SSR marker loci, prefix IASCA are SCAR marker loci, and prefix IN are InDel marker loci
Ol mapping in the CL-1 × CR-9 and the CL-1 × CR-6 populations
The Ol gene was mapped to LG T3 in the CL-1 × CR-9 population (Fig. 3a); the Ms gene was also mapped to the same LG (Fig. 3a). Initially, the Ol gene was flanked by the SCAR marker IASCA-45 developed by Hamdan et al. (2008) and the RAPD fragments OPG2 and OPL4-OPB5 of approximately 1,730 and 150 bp, respectively. The OPG21730 and OPL4-OPB5150 RAPD fragments were transformed into the SCAR markers IASCA-73 and IASCA-74, respectively (Table 1). IASCA-74 (F1/R1 and F2/R2) was not polymorphic in the CL-1 × CR-9 population. IASCA-73 gave a co-dominant amplification pattern. The final LG T3 containing the Ol gene spanned 192.5 cM and contained 24 loci, including the phenotypic loci Ol and Ms, five SCAR, one sunflower InDel, eight RAPD, and eight SSR marker loci (Fig. 3a). From these SSR, three of them were previously mapped to LG T3 by Mayerhofer et al. (2010), one of them (ct309) was mapped by the same authors to LG T2, and the other four SSR loci were mapped for the first time in the present study (Fig. 3a). The Ol locus mapped between the SSR marker ct365 and the SCAR marker IASCA-73, at genetic distances of 0.4 and 39.1 cM, respectively. The Ms locus mapped between the SSR markers CAT83 (8.3 cM) and EL375341 (12.1 cM), and the genetic distance between Ms and Ol was 65.3 cM (Fig. 3a). ANOVA analyses with genotypes at the ct365 marker locus used as classes revealed significant (p < 0.0001) differences between the marker class means for oleic acid content at the F2 and the F3 generations (Table 2).

Genetic mapping of the Ol gene for high oleic acid content, the Ms gene for nuclear male sterility, and the FAD2-1 (microsomal oleoyl phosphatidylcholine desaturase) gene. a Genetic mapping of Ol and Ms in CL-1 × CR-9; b genetic mapping of Ol and Ms in CL-1 × CR-6; c genetic mapping of FAD2-1 in CL-1 × CR-50; d consensus map for the three CL-1 × CR-9, CL-1 × CR-6, and CL-1 × CR-50 LG T3 maps. The cumulative distances in centiMorgans (Haldane) are shown at the left of each map. SSR and SCAR marker loci shared by at least two of the populations are highlighted in bold
Once the Ol gene was mapped in the CL-1 × CR-9 population, all SCAR and SSR markers from the Ol linkage group (LG T3) were tested for polymorphisms in the CL-1 × CR-6 population. The SCAR markers IASCA-39, IASCA-42, and IASCA-45, and the SSR markers CAT26, ct365, and ct599 were polymorphic and genotyped in the CL-1 × CR-6 population. Since the SCAR markers IASCA-73 and IASCA-74 linked to the Ol gene were not polymorphic, their corresponding RAPD markers OPG2 and OPL4-OPB5 were tested for polymorphisms. A polymorphic 150-bp band from the OPL4-OPB5 RAPD marker was genotyped in CL-1 × CR-6. The Ol locus mapped between the SSR marker loci ct365 and ct599 at genetic distances of 2.2 and 57 cM (Fig. 3b). The Ms locus was also mapped to this LG and was flanked by the SCAR marker locus IASCA-39 and the SSR marker locus CAT26. The Ol locus mapped 63.8 cM from the Ms locus (Fig. 3b). ANOVA analyses based on F2 and F3 data also revealed a significant association (p < 0.0001) of the markers flanking the Ol gene with oleic acid content. Results for marker ct365 are shown in Table 2.
QTL analyses in the CL-1 × CR-9 population
One major QTL (Ol3.1) affecting F2 and F3 oleic acid content was found on LG T3, explaining 99.4 and 96.3% of the phenotypic variation in the F2 and F3 oleic acid concentrations, respectively (Table 3). Its position corresponded to that of the Ol gene (Table 3; Figs. 2, 3a). The CR-9 allele at this QTL increased oleic acid content (Table 3). The Ol3.1 QTL also showed a significant effect on palmitic acid, stearic acid, and linoleic acid, which was directly related to the correlation found between oleic acid and these other three fatty acids (Table 4).
As expected, no effect of the Ol major QTL on LG T3 was detected when the subpopulation of Olol and olol F2 plants was analysed using the phenotypic data of mean and maximum oleic acid content in F3 seeds of the high oleic acid class of each Olol and olol F2:3 family. However, a QTL with a minor effect (Ol2.1) on oleic acid content was identified on LG T2 (peak LOD 2.54 for the mean oleic acid content of the high oleic acid class and 3.49 for the maximum oleic acid content of the high oleic acid class) (Table 3; Fig. 2). This QTL explained 20.9% of the variation for the mean oleic acid content of the high oleic acid class, and 27.5% of the variation for maximum oleic acid content of the high oleic acid class (Table 3). The CR-9 allele at this QTL increased oleic acid content (Table 3). The mean oleic acid content in the high oleic acid class from plants with the CL-1 allele on the Ol2.1 closest marker (CAT23) was 81.0%, whereas the mean oleic acid content from plants with the CR-9 allele was 83.6% (Table 2). No QTL with significant effect on oleic acid content were found when the QTL analyses were carried out using the minimum oleic acid content within the high oleic acid class.
Candidate gene mapping
Markers based on the safflower FAD2-1 sequence (GenBank accesion no. HM165274) were not polymorphic among the parental lines CL-1, CR-6, and CR-9. A primer combination (CAR_FAD2-1_F104 and CAR_FAD2-1_R106, Table 1) showed a one-locus co-dominant polymorphism between the parental lines CL-1 and CR-50 from an F2 population of 145 individuals segregating for palmitic acid content (Hamdan et al. 2009b). This primer combination, together with all SSR and SCAR markers from LG T3 showing polymorphism, was genotyped in the CL-1 × CR-50 population. Using a LOD threshold of 4.8 and a maximum recombination frequency of 0.45, a linkage map comprising six SSR marker loci and the CAR_FAD2-1_F104/R106 marker locus was constructed (Fig. 3c). The CAR_FAD2-1_F104/R106 marker locus mapped between markers IASCA-42 and IASCA-73 (Fig. 3c).
The CL-1 × CR-9, CL-1 × CR-6, and CL-1 × CR-50 populations shared four SSR markers and four SCAR markers on LG T3 (Fig. 3), which allowed the construction of a composite map (Fig. 3d). This map was constructed including all markers in the region between the SSR marker loci CAT26 and ct309 (Fig. 3d). The consensus map contained 17 marker loci and spanned 142.6 cM (Fig. 3d). Ol mapped to a similar position in consensus and individual maps (Fig. 3). The CAR_FAD2-1_F104/R106 marker locus was located 0.4 cM from the Ol gene (Fig. 3d).
Discussion
Previous genetic studies indicated that high oleic acid content in safflower is mainly controlled by partially recessive alleles at the Ol gene (Knowles and Hill 1964; Hamdan et al. 2009a). This gene is present in a homozygous recessive condition in the safflower lines CR-6, with high oleic acid content (>75%), and CR-9, with very high oleic acid content (>84%), and has been mapped to LG T3 of the safflower genetic map. QTL analyses confirmed the position of the Ol gene on LG T3 and showed no evidence of additional major factors controlling the high oleic acid trait.
The Ol gene has been mapped tightly linked to an oleoyl-phosphatidyl choline desaturase FAD2-1 locus. Three FAD2 family members (FAD2-1, FAD2-2, and FAD2-3) have been reported in safflower (GeneBank accessions nos. HM165274, HQ179940, and HQ179941). From the existing FAD2 family members in other oilseed crops, FAD2-1 is strongly expressed in developing seeds, whereas FAD2-2, and FAD2-3 are weakly expressed in developing seeds and exhibit more a “housekeeping” pattern of expression (Hongtrakul et al. 1998; Martínez-Rivas et al. 2001). This pattern of expression has been recently confirmed for safflower FAD2-1 and FAD2-2, but not for FAD2-3, which also shows a strong seed expression and does not belong to any of the “seed-type” or “housekeeping-type” plant FAD2 desaturases (Guan et al. 2011b). FAD2-1 plays an essential role in linoleic acid synthesis in developing seeds, and, when mutated, disrupts the synthesis of linoleic acid, leading to a build-up of oleic acid in the seeds, as has been shown in different oilseed crops such as sunflower, canola, and soybean (Pérez-Vich et al. 2002; Schuppert et al. 2006; Hu et al. 2006; Falentin et al. 2007; Lacombe et al. 2009; Pham et al. 2010). Additionally, Guan et al. (2011a) have determined that the expression of FAD2-1 in high oleic acid safflower genotypes is significantly lower than in standard (high linoleic) genotypes during seed development, and, when comparing the FAD2-1 sequence between high oleic and standard genotypes, these authors found differences that might be related to an altered protein function. Results from this study and recent evidence on the role of FAD2-1 in safflower (Guan et al., 2011a) strongly support that the FAD2-1 gene is the locus underlying the major Ol gene in safflower.
Since alleles determining high oleic acid content are partially recessive, the heterozygote is not clearly distinguishable from the wild-type homozygote. This fact makes it difficult to select plants carrying the ol allele in backcross programs focused on the high oleic acid trait. The use of marker-assisted selection (MAS) can contribute to overcoming such a limitation. Molecular markers developed in this research will greatly support introgression of ol alleles into elite lines. FAD2-1-based markers are highly predictive for the phenotype and optimal for selection. Additionally, the linkage map in which the Ol gene has been integrated, with robust SCAR and SSR markers flanking the gene, is an efficient tool for selecting against donor parent alleles around this gene.
In this study, the Ms gene controlling nuclear male sterility was also mapped to LG T3 at a genetic distance to Ol of 63.8 cM in the CL-1 × CR-6 population and 65.3 cM in the CL-1 × CR-9 population. Linkage between the Ms and Ol genes was not detected previously in classical genetic studies (Hamdan et al. 2009a) because the genetic distance between both loci was beyond the 50 cM expected from independent assortment. Previous studies by Hamdan et al. (2008) mapped the Ms gene 31.9–27.4 cM downstream of the SCAR markers IASCA-45, IASCA-42, and IASCA-39. In this study, the map position of the Ms gene in relation to these SCAR markers has been confirmed in two new mapping populations, and four new SSR markers (CAT26, CAT83, EL375341, and ct002) linked to the Ms gene have been identified. The major use of the Ms gene controlling nuclear male sterility in safflower breeding is the production of F1 hybrid seed, the development of testers for inbred line evaluation, and as a parent for genetic studies because of the tedious and time-consuming manual emasculation of safflower flowers (Singh and Nimbkar 2007). The use of MAS for the identification of lines carrying the male-sterile or male-fertile alleles would allow early removal of fertile types in crossing blocks and also avoid progeny testing in backcrossing programs, which will increase the efficiency of breeding procedures using the Ms gene for nuclear male sterility.
Besides the Ms gene, Hamdan et al. (2008) also reported the map position of the Li gene, determining very high linoleic acid content in safflower. This gene was linked to Ms, and was located in the IASCA-39-Ms interval. According to the results of the present study, the Ol and Li genes are expected to be located in the same linkage group at an estimated genetic distance of around 45–50 cM. In a classical genetic study, Futehally and Knowles (1981) found that the major recessive allele li controlling very high linoleic acid content was at a different locus from the ol allele governing high oleic acid levels, which has been demonstrated in this study using a molecular approach. However, Futehally and Knowles (1981) reported that the Ol and the Li genes were segregating independently. Similarly to the above discussion on the the Ms–Ol linkage, the genetic distance between Ol and Li on LG T3 is sufficiently large for the Ol–Li linkage to be detected using only phenotypic data on oleic acid and linoleic acid content.
In addition to the major locus Ol, Knowles (1972) postulated the possible existence of modifying genes affecting high oleic acid content in safflower. Modifying genes are defined as genes having no known effect except to intensify or diminish the expression of a major gene (Briggs and Knowles 1967). Hamdan et al. (2009a) determined that the very high oleic acid content in the safflower line CR-9 was produced by the combination of the ol alleles and modifying genes with a small positive effect on oleic acid content. These genes with a minor effect are difficult to characterize, since they are masked by the large effect of major genes. Pérez-Vich et al. (2004) demonstrated that the highly significant effect of macromutations significantly reduces the power of QTL analysis to identify QTL with smaller effects. To avoid this masking effect, these authors proposed the study of populations segregating only for genes with a minor effect, independently of the macromutation or major gene controlling the trait. Another approach is the one followed in the present research, i.e. to analyze the variation produced by modifying genes in a subpopulation that includes only those phenotypes that are fixed for the major gene and accordingly are expected to segregate only for modifying genes. This approach has allowed the identification and mapping of a modifying gene on LG T2 of the CL-1 × CR-9 population with an effect of further increasing oleic acid content in individuals homozygous for the ol allele. Despite Hamdan et al. (2009a) also describing modifying genes having an effect on lowering oleic acid content, we have not identified modifying genes affecting negatively oleic acid levels. The complex role of modifying genes has also been described in sunflower, where their number and mode of action on oleic acid expression is still a matter of controversy (Fernández-Martínez et al. 2004).
The key components required for an efficient system for molecular breeding are the identification and characterization of suitable genetic markers and the development of a reference genetic map using both an internationally accepted linkage group nomenclature system and publicly available markers, useful for cross-referencing maps and mapped gene locations. Although safflower is lagging behind other oilseed crops in terms of development of these necessary molecular breeding tools, the recent development of sequence-based markers (Chapman et al. 2009; Mayerhofer et al. 2010; Hamdan et al. 2011) and a first genetic linkage map for this species (Mayerhofer et al. 2010) has laid the basis for future genomic studies in this species. Since the safflower map from Mayerhofer et al. (2010) comprised 116 marker loci and was not saturated due to a limited polymorphism found within their mapping populations, the next short-term strategy for improving molecular breeding tools in safflower might be to add new mapped loci to this reference map. In this study, a total of 67 sequence-based markers (60 SSR and seven SCAR) have been mapped, 41 of them (34 SSR and 7 SCAR) being mapped for the first time, which increases the marker density of the safflower genetic map and improves its coverage.
Oils rich in oleic acid have important market niches for both food and non-food uses. Safflower cultivars with high oleic acid content are already available in the market and most breeding companies include high oleic acid as a major target trait in their breeding programs. The present research has shed light on the genetic basis underlying high oleic acid phenotypes, including the dominant role of the FAD2-1 gene and the involvement of at least one modifying gene that contributes to a further increase of oleic acid content. Additionally, molecular markers for selecting for high oleic acid content have been developed, which will contribute to facilitating selection for this trait within breeding programs in which MAS is routinely used.
References
Amini F, Saeidi G, Arzani A (2008) Study of genetic diversity in safflower genotypes using agro-morphological traits and RAPD markers. Euphytica 163:21–30
Beló A, Zheng P, Luck S, Shen B, Meyer DJ, Li B, Tingey S, Rafalski A (2008) Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol Genet Genom 279:1–10
Bohn M, Khairallah MM, González-de-León D, Hoisington DA, Utz HF, Deutsch JA, Jewell DC, Mihm JA, Melchinger AE (1996) QTL mapping in tropical maize: I. Genomic regions affecting leaf feeding resistance to sugarcane borer and other traits. Crop Sci 36:1352–1361
Briggs FD, Knowles PF (1967) Introduction to plant breeding. Reinhold Publishing Corporation, New York
Chapman MA, Burke JM (2007) DNA sequence diversity and the origin of cultivated safflower (Carthamus tinctorius L.; Asteraceae). BMC Plant Biol 7:60. doi:10.1186/1471-2229-7-60
Chapman MA, Hvala J, Strever J, Matvienko M, Kozik A, Michelmore RW, Tang S, Knapp SJ, Burke JM (2009) Development, polymorphism, and cross-taxon utility of EST-SSR markers from safflower (Carthamus tinctorius L.). Theor Appl Genet 120:85–91
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Dajue L, Mündel H-H (1996) Safflower (Carthamus tinctorius L.). Monograph n. 7, Promoting the Conservation and Use of Underutilized and Neglected Crops. Institut für Pflanzengenetik und Kulturpflanzenzüchtung (IPK), Gatersleben, Germany and International Plant Genetic Resources Institute (IPGRI), Rome, Italy
Falconer DS (1989) Introduction to quantitative genetics, 3rd edn. Longman Group, London
Falentin C, Brégeon M, Lucas MO, Deschamps M, Leprince F, Fournier MT, Delourne R, Renard M (2007) Identification of fad2 mutations and development of allele-specific markers for high oleic acid content in rapeseed (Brassica napus L.). In: Proceedings of 12th international rapeseed conference, Wuhan, vol II, pp 117–119
Fernández-Martínez JM, Jiménez A, Domínguez J, García JM, Garcés R, Mancha M (1989) Genetic analysis of the high oleic content in cultivated sunflower (Helianthus annuus L.). Euphytica 41:39–51
Fernández-Martínez JM, del Rio M, de Haro A (1993) Survey of safflower (Carthamus tinctorius L.) germplasm for variants in fatty acid composition and other seed characters. Euphytica 69:115–122
Fernández-Martínez JM, Velasco L, Pérez-Vich B (2004) Progress in the genetic modification of sunflower oil quality. In: Proceedings of 16th international sunflower conference, Fargo, ND, vol I, pp 1–14
Futehally S, Knowles PF (1981) Inheritance of very high levels of linoleic acid in an introduction of safflower (Carthamus tinctorius L.) from Portugal. In: Knowles PF (ed) Proceedings of 1st international safflower conference, Davis, CA
Garcés R, Mancha M (1993) One-step lipid extraction and fatty acid methyl esters preparation from fresh plant tissues. Anal Biochem 211:139–143
García-Moreno MJ, Fernández-Martínez JM, Velasco L, Pérez-Vich B (2011) Molecular tagging and candidate gene analysis of the high gamma-tocopherol trait in safflower (Carthamus tinctorius L.). Mol Breed 28:367–379
Guan L-L, Xu Y-W, Wang Y-B, Chen L, Shao J-F, Wu W (2011a) Isolation and characterization of a temperature-regulated microsomal oleate desaturase gene (CtFAD2-1) from safflower (Carthamus tinctorius L.). Plant Mol Biol Rep. doi:10.1007/s11105-011-0349-7
Guan L-L, Wang Y-B, Shen H, Hou K, Xu Y-W, Wu W (2011b) Molecular cloning and expression analysis of genes encoding two microsomal oleate desaturases (FAD2) from safflower (Carthamus tinctorius L.). Plant Mol Biol Rep. doi:10.1007/s11105-011-0322-5
Hamdan YAS, Velasco L, Pérez-Vich B (2008) Development of SCAR markers linked to male sterility and very high linoleic acid content in safflower. Mol Breed 22:385–393
Hamdan YAS, Pérez-Vich B, Velasco L, Fernández-Martínez JM (2009a) Inheritance of high oleic acid content in safflower. Euphytica 168:61–69
Hamdan YAS, Pérez-Vich B, Fernández-Martínez JM, Velasco L (2009b) Novel safflower germplasm with increased saturated fatty acid content. Crop Sci 49:127–132
Hamdan YAS, García-Moreno MJ, Redondo-Nevado J, Velasco L, Pérez-Vich B (2011) Development and characterization of genomic microsatellite markers in safflower (Carthamus tinctorius L.). Plant Breed 130:237–241
Heaton TC, Knowles PF (1982) Inheritance of male sterility in safflower. Crop Sci 22:520–522
Hongtrakul V, Slabaugh MB, Knapp SJ (1998) A seed specific Δ-12 oleate desaturase gene is duplicated, rearranged and weakly expressed in high oleic acid sunflower lines. Crop Sci 38:1245–1249
Hu X, Sullivan-Gilbert M, Gupta M, Thompson SA (2006) Mapping of the loci controlling oleic and linolenic acid contents and development of fad2 and fad3 allele-specific markers in canola (Brassica napus L.). Theor Appl Genet 113:497–507
Johnson RC, Kisha TJ, Evans MA (2007) Characterizing safflower germplasm with AFLP molecular markers. Crop Sci 47:1728–1736
Knowles PF (1972) The plant geneticist’s contribution towards changing lipid and amino acid composition of safflower. J Am Oil Chem Soc 49:27–29
Knowles PF (1989) Safflower. In: Downey RK, Röbbelen G, Ashri A (eds) Oil crops of the world. McGraw-Hill, New York, pp 363–374
Knowles PF, Hill AB (1964) Inheritance of fatty acid content in the seed oil of a safflower introduction from Iran. Crop Sci 4:406–409
Lacombe S, Souyris I, Bervillé AJ (2009) An insertion of oleate desaturase homologous sequence silences via siRNA the functional gene leading to high oleic acid content in sunflower seed oil. Mol Genet Genom 281:43–54
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Martínez-Rivas JM, Sperling P, Lühs W, Heinz E (2001) Spatial and temporal regulation of three different microsomal oleate desaturase genes (FAD2) from normal-type and high-oleic varieties of sunflower (Helianthus annuus L.). Mol Breed 8:159–168
Mayerhofer R, Archibald C, Bowles V, Good AG (2010) Development of molecular markers and linkage maps for the Carthamus species C. tinctorius and C. oxyacanthus. Genome 53:266–276
Mensink RP, Katan MB (1989) Effect of a diet enriched with monounsaturated or polyunsaturated fatty acids on levels of low-density and high-density lipoprotein cholesterol in healthy women and men. N Engl J Med 321:436–441
Okuley J, Lightner J, Feldmann K, Yadav N, Lark E, Browse J (1994) Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell 6:147–158
Pérez-Vich B, Fernández-Martinez JM, Grondona M, Knapp SJ, Berry ST (2002) Stearoyl-ACP and oleoyl-PC desaturase genes cosegregate with quantitative trait loci underlying stearic and oleic acid mutant phenotypes in sunflower. Theor Appl Genet 104:338–349
Pérez-Vich B, Fernández-Martinez JM, Leon A, Knapp SJ, Berry ST (2004) Mapping minor QTL for increased stearic acid content in sunflower seed oil. Mol Breed 13:313–322
Pham A-T, Lee J-D, Shannon JG, Bilyeu K (2010) Mutant alleles of FAD2-1A and FAD2-1B combine to produce soybeans with the high oleic acid seed oil trait. BMC Plant Biol 10:195. doi:10.1186/1471-2229-10-195
Pham A-T, Lee J-D, Shannon JG, Bilyeu K (2011) A novel FAD2-1 A allele in a soybean plant introduction offers an alternate means to produce soybean seed oil with 85% oleic acid content. Theor Appl Genet 123:793–802
Ravikumar RL, Shiva Priya M, Patil BS, Satish D (2005) DNA profiling and fingerprinting of selected mutants for marker analysis in safflower (Carthamus tinctorius L.) In: Essendal E (ed) Proceedings of 6th international safflower conference, Istanbul
Rogers SO, Bendich AJ (1985) Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol Biol 5:69–76
Scheffler JA, Sharpe AG, Schmidt H, Sperling P, Parkin IAP, Lühs W, Lydiate DJ, Heinz E (1997) Dasaturase multigene families of Brassica napus arose through genome duplication. Theor Appl Genet 94:583–591
Schlueter JA, Vasylenko-Sanders IF, Deshpande S, Yi J, Siegfried M, Roe BA, Schlueter SD, Scheffler BE, Shoemaker RC (2007) The FAD2 gene family of soybean: Insights into the structural and functional divergence of a paleopolyploid genome. Crop Sci 47(Supplement_1):S-14-26
Schuppert G, Tang S, Slabaugh M, Knapp SJ (2006) The sunflower high-oleic mutant Ol carries variable tandem repeats of FAD2-1, a seed-specific oleoyl-phosphatidyl choline desaturase. Mol Breed 17:241–256
Sehgal D, Raina SN (2005) Genotyping safflower (Carthamus tinctorius) cultivars by DNA fingerprints. Euphytica 146:67–76
Singh V, Nimbkar N (2007) Safflower. In: Singh RJ (ed) Genetic resources, chromosome engineering and crop improvement : oilseed crops, vol 4. CRC Press, Boca Raton, pp 167–194
Urie AL (1985) Inheritance of high oleic acid in sunflower. Crop Sci 25:986–989
Utz HF, Melchinger AE (1996) PLABQTL: a program for composite interval mapping of QTL. J Quant Trait Loci 2:1
Vilatersana R, Garnatje T, Susanna A, Garciajacas N (2005) Taxonomic problems in Carthamus: RAPD markers and sectional classification. Bot J Linn Soc 147:375–383
Voorrips RE (2002) Mapchart: software for the graphical presentation of linkage map and QTL. J Hered 93:77–78
Yang Y-X, Wu W, Zheng Y-L, Chen L, Liu R-J, Huang C-Y (2007) Genetic diversity and relationships among safflower (Carthamus tinctorius L.) analyzed by inter-simple sequence repeats (ISSRs). Genet Resour Crop Evol 54:1043–1051
Yazdi-Samadi B, Maali-Amiri R, Ghannadha MR, Abd-Mishani C (2001) Detection of DNA polymorphism in landrace populations of safflower in Iran using RAPD-PCR technique. In: Bergman JW, Mündel HH (eds) Proceedings of 5th international safflower conference, Williston, ND and Sidney
Yodice R (1990) Nutritional and stability characteristics of high oleic sunflower seed oil. Fat Sci Technol 92:121–126
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang Z, Guo M, Zhang J (2009) Identification of AFLP fragments linked to hydroxysafflor yellow A in Flos Carthami and conversion to a SCAR marker for rapid selection. Mol Breed 23:229–237
Acknowledgments
Y.A.S. Hamdan was the recipient of a grant from the Agencia Española de Cooperación Internacional (AECI). The research was funded by Junta de Andalucía support program to research groups (PAI-AGR-118).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yamen A. S. Hamdan and María J. García-Moreno contributed equally to this work.
Rights and permissions
About this article
Cite this article
Hamdan, Y.A.S., García-Moreno, M.J., Fernández-Martínez, J.M. et al. Mapping of major and modifying genes for high oleic acid content in safflower. Mol Breeding 30, 1279–1293 (2012). https://doi.org/10.1007/s11032-012-9714-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-012-9714-y