
Abstract
Grain weight is one of the three direct yield components, being developed through a dynamic process of grain filling in maize. In this study, 258 recombinant inbred lines derived from a cross between a dent corn and a popcorn inbred were evaluated for grain fresh and dry weight at 10, 20, 30, and 40 days after pollination (DAP) and the activities of ADP-Glc pyrophosphorylase (AGPP), granule-bound starch synthase (GBSS), and soluble starch synthase (SSS) at 30 DAP. Grain-filling rate (GFR) and increasing rate of fresh weight (FWIR) were calculated during all periods. Quantitative trait locus (QTL) mapping was conducted for all traits. Meta-QTL (mQTL) was revealed by meta-analysis using BioMercator. Totally, 161 QTL were detected for six traits. QTL on chromosomes 1, 7, and 10 were detected in most cases, with 43, 54, and 28 QTL, respectively. For each trait, 1–4 QTL were detected but no QTL for GBSS. Three mQTL at bins 7.02–7.03, 1.03–1.04, and 10.05–10.06 included 47, 24, and 23 QTL detected in this study. Together with 28 QTL for grain weight detected in our previous research, they included 53, 28, and 25 QTL, respectively. Five identified expressed sequence tags (EST), five candidate genes with related functions, and QTL for grain weight in other research were co-located in these regions. It is worth concentrating further research on these regions to develop near-isogenic lines (NILs) of common QTL and their chromosome segment substitution lines (CSSL). Also, cloning and function validation for co-located EST and candidate genes could facilitate identification of genes for grain development and final weight.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Grain weight is one of the three direct yield components, being developed through a dynamic process of grain filling in maize (Zea mays L.). Its genetic basis needs to be extensively revealed through all developmental stages. After cell division, differentiation, and expansion, grain weight at mature harvest time is determined by grain-filling rate and duration (Takai et al. 2005). Final grain weight is largely a genetically determined trait (Reddy and Daynard 1983; Seka and Cross 1995; Seka et al. 1995; Borrás et al. 2009), though grain development is affected by many environmental and physiological factors, such as drought (Ouattar et al. 1987), temperature (Wilhelm et al. 1999), nitrogen content (Ta and Weiland 1992), assimilate availability (Borrás et al. 2004), related enzyme activity (Jones and Brenner 1987; Thévenot et al. 2005), kernel water content (Ouattar et al. 1987; Borrás et al. 2003), and kernel number (Hartung et al. 1989; Borrás and Otegui 2001). Ample variations in grain weight, and grain-filling rate and duration have been observed among commercial hybrids (Gambín et al. 2007) and diverse inbreds (Borrás et al. 2009). Different genotypes exhibit a wide range of grain growth patterns (Borrás et al. 2009). Both rate and duration of grain filling are important in grain development (Ercole and Alessandro 1981; Wang et al. 1999; Takai et al. 2005). However, improvement in grain-filling rate should receive more attention than long grain-filling duration in areas such as the Huanghuihai Corn Belt in China, where the growth season for maize is limited (Liu et al. 2011).
In previous research, QTL mapping for grain weight at final harvest time has been extensively conducted using three kinds of maize germplasm: normal maize (Stuber et al. 1987, 1992; Veldboom and Lee 1994; Austin and Lee 1996, 1998; Austin et al. 2000; Mihaljevic et al. 2004; Blanc et al. 2006; Yan et al. 2006; Ma et al. 2007), high-oil maize (Song 2003), and popcorn (Li et al. 2007, 2011). According to public data in Gramene (http://www.gramene.org/), 186 QTL for grain weight had been reported until 31 May 2010, distributed on all ten maize chromosomes. However, very few studies have been carried out on dynamic QTL of grain weight, and grain-filling rate and duration across different developmental stages. Recently, Capelle et al. (2010) detected QTL for fresh grain weight (GFW) and dry grain weight (GDW) at 30, 40, 60, and 80 DAP using an intermated RIL population derived from two normal maize inbreds F2 and F252. Liu et al. (2011) detected QTL for GDW at 16, 23, 32, 40, and 45 DAP, and QTL for grain-filling rate (GFR) during four periods using 203 RIL from a cross between two normal maize inbreds, Huang-C and Xu178.
Starch constitutes 70–75% of grain dry weight in maize (Boyer and Hannah 2001). Therefore, starch concentration plays an important role in grain weight potential, and the starch synthesis pathway could be an important site of regulation. ADP-Glc pyrophosphorylase (AGPP), starch synthase (granule-bound starch synthase, GBSS; soluble starch synthases, SSS), starch branching enzyme (SBE), and starch debranching enzyme (DBE) are four kinds of important enzymes in starch synthesis and accumulation (Prioul et al. 1990, 1994; Slattery et al. 2000). An effective way to determine main QTL for objective traits is to use genetic variability in enzyme activity and carbohydrate concentration. QTL co-located with genes encoding for related functions could be considered as candidate genes (Prioul et al. 1997; Thévenot et al. 2005). Such an approach has been successfully used for carbon metabolism in leaves (Causse et al. 1995; Pelleschi et al. 1999). Thévenot et al. (2005) detected QTL for enzyme activities and soluble carbohydrates involved in starch accumulation at 15, 25, and 35 DAP using 100 RIL from a cross between a late dent line MBS and an early flint line F-2.
Since the results of QTL detection were greatly influenced by different populations, generations, and environments (Austin and Lee 1996; Li et al. 2007), direct comparisons of QTL data across different studies were difficult. Integration of QTL detected across independent studies could be realized through meta-QTL analysis, and true QTL with more accurate confidence intervals and small target regions for candidate genes could be provided (Goffinet and Gerber 2000; Arcade et al. 2004). This method has been used in QTL integration for grain yield components (Wang et al. 2009) and flowering time (Chardon et al. 2004) in maize, and in other crops (Shi et al. 2009).
In this study, a cross between two contrasting genotypes, an early popcorn inbred N04 with small grain size and a late dent inbred Dan232 with large grain size, was used to develop 258 RIL (Li et al. 2011). QTL mapping was conducted for grain dry weight and fresh weight at four stages (10, 20, 30, and 40 DAP), grain-filling rate in dry and fresh weight during six periods, and activities of three enzymes (AGPP, GBSS, and SSS) at 30 DAP. In addition, meta-QTL was analyzed for QTL of all traits detected herein, and together with QTL for grain dry weight at mature harvest time obtained in our previous studies using the same population (Li et al. 2011) and its F2:3 and BC2F2 generations (Li et al. 2007). Our main objectives were to (1) reveal the characteristics of dynamic QTL for grain weight and grain-filling rate, (2) relate QTL for grain weight and grain-filling rate with the activities of starch synthesis-related enzymes, (3) identify consistent QTL across different generations, environments, and related traits, and (4) determine main genetic regions for grain development worthy of elucidation in further research in maize.
Materials and methods
Plant materials and field experiment
The population used in this study consisted of 258 F9 RIL derived by the single-seed descent method from a cross between two contrasting inbred lines, Dan232 and N04. Dan232 is a late dent corn inbred with large-size grain, derived from Lu 9 Kuan × Dan340. N04 is an early popcorn inbred with small-size grain, derived from a Chinese popcorn variety BL03 (Li et al. 2007, 2011).
The 258 F9 RIL and both parents were evaluated using α-designs with three-row plots and two replications at Zhengzhou, Henan, China in 2008 and 2009. The rows were 4 m long with 0.67 m spacing between rows. Plots were planted by hand at density of 60,000 plants per ha. Standard cultivation management practices were used at each environment. All plants were self-pollinated within each plot by hand when more than 80% silks appeared.
Trait evaluation
Three to five ears with uniform grain set were harvested at 10, 20, 30, and 40 DAP. To increase the representativeness and uniformity of the sampled kernels, only kernels on the middle two-thirds of each ear were shelled manually and bulked within plot. Some of the samples collected at 30 DAP were immediately frozen in liquid nitrogen and stored at −80°C for measurement of enzyme activity. This date was chosen because previous studies have shown that the activities of AGPP, GBSS, and SSS peaked at 25–35 DAP (Prioul et al. 1990, 1994; Thévenot et al. 2005; Yang 2010).
One-hundred-grain fresh weight (GFW, g) was tested with two replications at each time point for each plot. After being naturally dried, 100-grain dry weight (GDW, g) was tested accordingly. Grain-filling rate (GFR, g/day) during six periods was calculated, including 10–20 DAP (12 DAP), 10–30 DAP (13 DAP), 10–40 DAP (14 DAP), 20–30 DAP (23 DAP), 20–40 DAP (24 DAP), and 30–40 DAP (34 DAP). Since the GFW for some lines at 40 DAP was lower than at 20 DAP and/or 30 DAP, increasing rate of fresh weight (FWIR, g/day) was calculated for five periods, 10 DAP, 12 DAP, 13 DAP, 14 DAP, and 23 DAP. Trait measurements averaged over two replications were used as the preliminary data in further analysis.
The activities of three enzymes AGPP, GBSS, and SSS at 30 DAP were measured as described by Douglas et al. (1988) and Nakamura and Yuk (1989).
Phenotypic data analysis
Analysis of variance for each trait and correlation coefficients among traits were calculated using the statistical software package SPSS12.0 with random statistical model. Heritability and the confidence intervals of the measured traits were computed according to Knapp et al. (1985). Heritability (H 2B ) was calculated as H 2B = 1 − 1/F, where F = MSg/MSge. MSg = nrσ 2g + rσ 2ge + σ 2e , MSge = rσ 2ge + σ 2e , where σ 2g is the variance of family, σ 2ge is the variance of interaction between family and environment, σ 2e is the error variance, r is the number of replications, and n is the number of locations.
Simple sequence repeat (SSR) analysis and QTL analysis
A total of 207 simple sequence repeat (SSR) markers were used to genotype the 258 RIL using the same method described by Li et al. (2007). The linkage maps covered 10 maize chromosomes with total length of 2,408.8 cM and average interval of 11.6 cM (Li et al. 2011).
Composite interval mapping (CIM) was used to map QTL and estimate their effects for each trait (Zeng 1993, 1994). Model 6 of the Zmapqtl procedure in QTL Cartographer version 2.5 (Wang et al. 2011) was employed, specifying the five markers identified by stepwise regression that explained most of the variation for a given trait as genetic background parameters and a window size of 10 cM on either side of the markers flanking the test site. To identify an accurate significance threshold for each trait according to the traditionally used alpha of 5%, an empirical threshold was determined for CIM using 1,000 permutations (Churchill and Doerge 1994). QTL position was assigned to relevant regions at the point of a maximum likelihood odds ratio (LOD) score. QTL confidence interval was calculated by subtracting one LOD unit on each side from the maximum LOD position. Based on the results of QTL mapping, interactions among detected QTL were analyzed using MIM in WinQTLCart (Wang et al. 2011).
Meta-QTL analysis with BioMercator
In our previous research, three generations (F2:3, BC2F2, and RIL) were derived from the same cross between Dan232 and N04. Using two genetic linkage maps constructed using F2:3 and RIL populations, QTL for GDW at mature harvest time have been detected in three populations (Li et al. 2007, 2011). The field experiments in previous research were conducted under two or four environments in Henan, China.
Since QTL for the same or related traits detected in different experiments and mapped to the same or similar chromosome regions might be several estimates of the position of a single QTL, algorithms for meta-analysis were used to estimate the numbers and positions of their meta-QTL (mQTL) (Goffinet and Gerber 2000) using BioMercator 2.1 software (Arcade et al. 2004). The merged genetic linkage map was obtained by projecting F2:3 map onto RIL map consisting of 237 SSR markers, and was 2,452.2 cM long with average marker interval of 10.35 cM (Li et al. 2011). According to data for multiple individual QTL, a modified Akaike’s information criterion (AIC) was calculated to select among models with varying numbers of mQTL. The model with the lowest test statistic was the most probable model. In each model, a confidence interval was calculated for each mQTL.
Results
Trait analysis of variance, performance, heritability, and correlation in RIL population
Variances for genotype (σ 2g ) and genotype × year interactions (σ 2ge ) were significant for all traits, and those for year (σ 2e ) were significant for most traits (Table 1). Except GFR at 12 DAP and 34 DAP, heritability estimates for other traits were middle to high, with ranges from 0.60 to 0.88.
All traits differed greatly between the two parents. The popcorn inbred N04 had lower values than the dent corn inbred Dan232 for all traits (Table 1). According to the values of skewness and kurtosis, all traits showed normal distributions and transgressive segregations in the RIL population. The coefficients of variation (CV) were high, ranging from 15.3% for GDW at 20 DAP to 46.3% for FWIR at 23 DAP.
For correlations among traits, almost the same tendency was observed for data in 2008 and 2009 and in combined analysis. According to the results in 2008 (Table 2), significant positive correlations were shown in all cases except 11, where the correlations were insignificant, being GFR at 34 DAP with GDW at 30 DAP, GFR at 24 DAP and 34 DAP, FWIR at 13 DAP, 14 DAP, and 23 DAP, and the activity of AGPP with GDW at 10 DAP, FWIR at 23 DAP, and the activities of AGPP and GBSS with GFR at 34 DAP, and the activity of GBSS with GFR at 24 DAP.
QTL identification for GDW and GFW at four stages after pollination
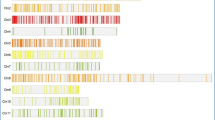
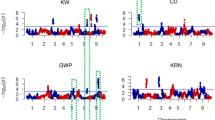
Since the variance components for genotype × year interactions (σ 2ge ) were significant for all traits, QTL mapping was conducted for data for each year. For comparison, combined analyses using means across 2 years were also conducted. A total of 34 QTL for GDW were detected at four stages (Table 3, Supplementary Table 1), being located on chromosomes 1, 3, 4, 5, 7, 9, and 10. Individual QTL explained phenotypic variances from 4.8% to 23.5%, with 23 QTL over 10% and 10 QTL over 15%. The positive alleles of all QTL came from the dent corn parent Dan232 except QTL on chromosomes 3 and 4. QTL on chromosome 7 were consistently detected at all stages under each environment and in combined analysis. They were located in the same marker interval umc2057–umc1567 (bin 7.02–7.03), except at umc1068–umc1066 (bin 7.01–7.02) at 10 DAP in 2008. QTL on chromosome 10 were all detected at four stages in 2009 and in combined analysis, and at 20 DAP in 2008, being located in the same marker interval umc1677–umc2122 (bin 10.05–10.06). On chromosome 1 in the same marker interval phi001–umc2227 (bin 10.05–10.06), QTL were detected at 20, 30, and 40 DAP in 2009, and in combined analysis. On chromosome 5, QTL were detected in umc1478–bnlg565 (bin 5.01–5.02) at 30 DAP in 2008 and at 10 DAP in 2009, and in umc1502–umc1941 (bin 5.05–5.06) at 40 DAP in 2008, and in combined analysis. QTL on chromosomes 3, 4, and 9 were only detected in one case.
For GFW, a total of 37 QTL were detected at four stages (Table 3, Supplementary Table 1), being located on chromosomes 1, 5, 7, 9, and 10. Individual QTL explained phenotypic variances from 4.8 to 21.3%, with 20 QTL over 10% and 5 QTL over 15%. The positive alleles of all QTL came from the dent corn parent Dan232 except the QTL at bins 1.02, 1.02–1.03, and 3.03–3.04. QTL on chromosome 7 were consistently detected at all stages under both environments and in combined analysis. They were located in the same marker interval umc2057–umc1567 (bin 7.02–7.03), except at bnlg2233–umc1068 (bin 7.02) at 10 DAP in 2008. QTL at bin 1.05–1.06 and QTL on chromosome 10 were detected at 10 DAP and 20 DAP, while those QTL at bin 1.03–1.04 and on chromosome 5 were detected at 30 DAP and 40 DAP in most cases. QTL on chromosome 9 was only detected at 10 DAP in 2009.
QTL identification for GFR and FWIR during six periods
Totally, 44 QTL for GFR were detected, located on chromosomes 1, 3, 4, 5, 7, and 10 (Table 4, Supplementary Table 2). Individual QTL explained phenotypic variances from 4.4 to 25.4%, with 26 QTL over 10% and 14 QTL over 15%. Except two QTL on chromosome 3, the positive alleles of all other QTL came from the dent corn parent Dan232. The distributions and locations of GFR QTL were very similar to those for GDW. On chromosome 7, 14 GFR QTL were detected, with 13 in the same marker interval umc2057–umc1567 (bin 7.02–7.03) and one in bnlg2233–umc1068 (bin 7.02). Twelve QTL were detected on chromosome 10. They were located in four marker intervals, with eight QTL in umc1677–umc2122 (bin 10.05–10.06), two in umc1319–umc1567 (bin 10.01–10.02), one in umc1576–umc2034 (bin 10.02), and one in phi059–umc2067 (bin 10.02–10.03). Nine QTL detected on chromosome 1 were all located in the same marker interval phi001–umc2227 (bin 1.03–1.04). Six QTL were detected on chromosome 5, being located in four marker intervals (bnlg656–phi109188, umc1389–umc1162, umc1221–umc1502, and umc1502–umc1941) related with bin 5.02–5.06 (5.02–5.03, 5.03–5.04, 5.04–5.05, and 5.05–5.06). In addition, two and one QTL were detected on chromosomes 3 and 4, respectively.
For FWIR, 44 QTL located on chromosomes 1, 2, 5, 7, 9, and 10 were detected (Table 4, Supplementary Table 2). Individual QTL explained phenotypic variances from 5.2 to 21.0%, with 16 QTL over 10% and 4 QTL over 15%. Except six QTL at bins 1.02, 1.02–1.03, 9.03–9.04, and 10.07, the positive alleles of other QTL came from the dent corn parent Dan232. QTL for FWIR on chromosome 7 were detected under both environments and in combined analysis. They were located in three marker intervals at bins 7.02, 7.02–7.03, and 7.03. The QTL on chromosome 1 were detected in 11 cases, being located in four marker intervals at bins 1.02, 1.02–1.03, 1.03–1.04, and 1.05–1.06. In nine cases, QTL on chromosome 5 were detected in four marker intervals at bins 5.01–5.06 (5.01–5.025, 5.02–5.03, 5.04–5.05, and 5.05–5.06). In addition, QTL on chromosomes 2, 9, and 10 were detected in one, three, and three cases, respectively.
QTL identification for the activities of AGPP, GBSS, and SSS
Only two QTL were detected for the activities of three enzymes, one each for AGPP and SSS. No QTL were detected for GBSS (Table 3, Supplementary Table 1). They were located in the same marker interval umc2057–umc1567 (bin 7.02–7.03). Phenotypic variances explained by individual QTL were 6.34 and 4.48%. All their positive alleles came from the popcorn parent N04.
Digenic epistasis among QTL for GFW, GDW, GFR, and FWIR
For the four traits, 59 pairs of digenic interactions were identified, being 14, 13, 17, and 15 pairs for GFW, GDW, GFR, and FWIR, respectively (data not shown). Phenotypic variance explained by the interaction between umc1307 and umc1867 at bin 9.0–9.01 and umc2163–umc1677 at bin 10.04–10.05 was 11.2% for GFR in 2009. However, the contribution values of other digenic interactions were all low, ranging from 0.1 to 4.8%. These results suggested that the contributions of digenic interactions to grain weight and grain-filling rate were minimal.
Meta-QTL analysis for all traits in this study and GDW at maturity in our previous research
In this study, 161 QTL were detected for GDW, GFW, GFR, and FWIR and the activities of two enzymes. In our previous studies using the same two parents, 28 QTL for GDW at maturity were detected, 22 QTL using the same RIL population under four environments, and three QTL using its F2:3 and BC2F2 populations under two environments. Altogether, 189 QTL were detected. For both 161 QTL and the total 189 QTL, 13 distinct QTL clusters (mQTL) were found (Table 5). These mQTL were located on six chromosomes, three on chromosomes 1 and 5, two on chromosomes 3, 7, and 10, and one on chromosome 9.
For the 161 QTL detected in this study, 155 QTL were located in those cluster regions, accounting for 96.3%. One mQTL included 11.9 QTL on average, with a variation of 2–47 QTL for one to six traits. For the totally detected 189 QTL, 178 QTL were located in those cluster regions, accounting for 94.2%. One mQTL included 13.7 QTL on average, with a variation of 2–53 QTL for one to six traits. In both cases, many more QTL were included in mQTL7-2 (47/53 QTL for six traits), mQTL1-2 (24/28 QTL for four traits), and mQTL10-2 (23/25 QTL for four traits). mQTL1-3, mQTL5-1, and mQTL5-3 included 12/14 QTL for three traits, 10/4 QTL for three traits, and 16/17 QTL for four traits, respectively. Other mQTL included 2–7 QTL for one to four traits each.
Discussion
Three main genetic regions for grain weight and QTL co-location with EST identified from endosperms of the same two parents
Although three populations (F2:3, BC2F2, and RIL) used in our present and previous research were derived from the same parents, the field experiments were not all conducted at the same locations and in the same years, and two linkage maps were constructed and used for QTL mapping. Therefore, the QTL experiments were considered as independent when using the BioMercator software. Through 189 QTL totally detected for GDW, GFW, GFR, FWIR, AGPP, and SSS in this study, and for GDW at maturity in our previous studies using the same RIL population (Li et al. 2011), and its F2:3 and BC2F2 generations (Li et al. 2007) derived from the same two parents, three main genetic regions on chromosomes 1, 7, and 10 were clearly found in determining grain weight and development. The same two parents (Dan232 and N04) were also used to identify differentially expressed EST at 10 and 20 DAP in our previous study (Liu et al. 2010b). For the totally identified 160 unique EST, 70 were assigned to 39 chromosome bins distributed over all ten maize chromosomes. Eleven EST were found to co-localize with previously detected QTL for grain weight. Eight EST were co-localized with six mQTL in this study: mQTL1-1, mQTL1-2, mQTL3-2, mQTL5-2, mQTL7-1, and mQTL7-2. Two EST (PE12C5 and PE15C3) were located in the same marker interval phi001–umc2227 as mQTL1-2. Another two EST (PM44C3 and DM27D9) were co-located with mQTL7-2 in the marker interval umc2057–umc1567.
The most important one was the marker interval umc2057–umc1567 at bin 7.02–7.03, in which 54 QTL were integrated. mQTL7-2 included 47 QTL detected in this study (11 QTL for both GDW and GFW, 13 for GFR, and 10 for FWIR) and seven QTL for GDW detected using the same RIL population, and its F2:3 and BC2F2 generations. Notably, both one QTL for the activities of AGPP and SSS and two EST (PM44C3 and DM27D9) were located in this marker interval. Thévenot et al. (2005) considered that QTL co-located with genes encoding for related functions might be considered as candidate genes. PM44C3 and DM27D9 were deduced to encode opaque-2 modifier and alanine aminotransferase (AlaAT), respectively. Opaque-2 modifiers increase zein synthesis and alter its spatial distribution in maize endosperm. AlaAT could increase the biomass and grain yield significantly through enhancing nitrogen uptake efficiency in rice (Shrawat et al. 2008). According to information from MaizeSequence (http://www.maizesequence.org/index.html), one candidate gene, elongation initiation factor 2 (eIF2), is located in silico in this region. eIF2 is a phosphoprotein, and eIF2a regulates phosphorylation through several intermediate isoforms that correlated with the increase and subsequent reduction in protein synthetic activity during seed development (Le et al. 1998).
In the marker interval phi001–umc2227 at bin 1.03–1.04, 24 QTL were detected in this study, including 6 QTL for GDW, 4 for GFW, 9 for GFR, and 5 for FWIR. In our previous study, four QTL for GDW at maturity were also commonly detected using the same RIL population (Li et al. 2011). In addition, one QTL for GDW was detected near bin 1.04–1.05 (umc2025–bnlg1811 and bnlg1811–bnlg1884) in both F2:3 and BC2F2 generations. Two EST (PE12C5 and PE15C3) and three candidate genes (Glycine rich protein, Grp; PhytochromeB1, phyB1; and Pdc3 pyruvate decarboxylase3, pdc3) were also located in this marker interval. PM12C5 encodes zinc finger (C3HC4-type RING finger) family protein. Zinc finger proteins are transcription factors, most of which regulate important biological processes, such as flower development, light-regulated morphogenesis, pathogen responses, and kernel growth and development in maize (Colasanti et al. 1998; Kozaki et al. 2004; Bluhm et al. 2008; Royo et al. 2009). PE15C3 encodes guanosine triphosphate (GTP) binding protein, which participates in a wide range of biological process, including signal transduction, protein synthesis and secretion, and cellular proliferation (Takatsuji 1998; Flaherty and Woloshuk 2004). Grp encodes an important structural protein in plant cell wall, and the growth of cell wall influences cell elongation. phyB1 encodes a kind of light receptor in plants. Sawers et al. (2005) suggest that phytochrome signaling pathways are attractive targets for manipulating grain yield in cereal grasses.
Now, the full-length complementary DNA (cDNA) sequences for three co-located EST (PM44C3, PE12C5, and PE15C3) have been obtained in our library, and their same putative functions have been reflected (Liu et al. 2010a; unpublished data). For the full-length cDNA sequences of PE12C5 and PE15C3, the GenBank accession numbers are GQ131520.1 and EU816421, being designated ZmC4HC3 and ZmArf2, respectively. Preliminary expression analyses have shown their different expression levels during endosperm development and among different tissues. Based on the comparisons for nucleotide and amino acid sequences of ZmArf2 between the popcorn inbred N04 and the dent corn inbred Dan232, eight nucleotides differed and five amino acids changed between the two inbreds (Liu et al. 2010a).
Totally, 25 QTL were detected in the marker interval umc1677–umc2122 at bin 10.05–10.06, including 23 detected in this study (9 for GDW, 4 for GFW, 8 for GFR, and 2 for FWIR) and 2 QTL for GDW at maturity detected in the same RIL population (Li et al. 2011). mQTL10-3 was co-located with a candidate gene ribosomal protein S11 (Rps11). Rps11 is a major component of ribosome and involved in the process of protein biosynthesis (Lebrun and Freyssinet 1991).
Clearly, the three marker intervals for mQTL7-2, mQTL1-2, and mQTL10-2 are worthy of greater attention in further research. Wang et al. (2009) also obtained meta-QTL for grain weight in these regions. Construction of near-isogenic lines (NIL) for common QTL for GDW located at these regions, and chromosome segment substitution lines (CSSL) for these regions are in progress in our present research. Since QTL for grain-weight-related traits in these regions were consistently detected across different generations, environments, and developmental stages/periods, marker-assisted selection (MAS) could be used to improve grain weight in maize. Moreover, full-length cDNA cloning of co-located EST (DM27D9, PE12C5, PE15C3, PM44C3, and PM44C3), and homologous cloning of co-located candidate genes (eIF2, Grp, phyB1, pdc3, and Rps11), and their function validations in further research might help to reveal grain-weight-related genes in these regions.
QTL detected at different stages and coincidence with related traits
Grain weight at harvest is controlled by a series of genes expressed systemically during different developmental stages. To reveal its dynamic genetic mechanism, QTL for grain weight at four stages and grain-filling rate during all periods in dry and fresh content were detected in this study. Among all the 159 QTL for GDW, GFW, GFR, and FWIR, QTL on chromosomes 1, 5, 7, and 10 were detected almost for all traits and across almost all stages/periods. They could play important roles during the whole process of grain development. For these QTL, MAS could be done for any trait at any stage. Considering measurement convenience, GDW at harvest was the best choice.
However, QTL on chromosomes 2, 3, 4, and 9 were shown to be specific for traits and developmental stages/periods. On chromosome 2, QTL was only detected for FWIR during 20–30 DAP. On chromosome 3, QTL for GDW and GFW at 20 DAP, for GFR during 10–20 DAP and 20–40 DAP were found. On chromosome 4, QTL were detected for GDW at 10 DAP and GFR during 20–40 DAP. On chromosome 9, QTL for GDW and GFW at 10 DAP and for FWIR during 0–10 DAP and 10–40 DAP were found. Clearly, most specific QTL were related with early developmental stages/periods, especially 10–20 DAP, consistent with some kinds of genes functioning only at early developmental stages in endosperm, such as cell division. According to previous research on endosperm development, cell numbers reached maximum at 12–21 DAP (Crane 1964; Linn 1977; Tollenaar 1978; Kiesselbach 1980; Jones 1985; Olsen 2004). For the two parent inbred lines in this study, endosperm microstructures at 3, 5, 7, 10, 15, 20, and 25 DAP showed that 10 DAP endosperms were in the stage of cell division (Liu et al. 2010b). Therefore, QTL on chromosomes 3, 4, and 9 might influence grain weight through controlling cell division. However, this should be proved in further research.
Through analyzing starch synthase genes in the databases of whole genome and full-length cDNA during grain filling in rice, Hirose and Terao (2004) divided the ten starch synthase genes into three groups of early, late and steady expressers. In our present study, QTL on chromosome 7 at bin 7.02–7.03 were detected across all stages and for all traits, which could be considered as steady expressers for both grain weight and grain-filling rate. Accordingly, QTL on chromosomes 3, 4, and 9 were only detected for related traits at 10 and 20 DAP, which were similar to early expressers of the starch synthesis-related genes. QTL for GFW on chromosome 5 only detected at 20, 30, and 40 DAP might be late expressers. In the study of QTL detection for grain weight and grain-filling rate by Liu et al. (2011), no QTL were consistently detected across four stages (16, 23, 32, and 40/45 DAP). Thévenot et al. (2005) found one common QTL at bin 7.04 for grain fresh weight across three stages (15, 25, and 35 DAP). Interestingly, this common QTL was located near bin loci 7.02–7.03 as in this study, in which QTL was detected in all cases (for grain weight at all stages and grain-filling rate during all periods in fresh/dry content). This further reflects the importance of QTL detected on chromosome 7 in controlling grain development and final grain weight. QTL on chromosomes 2, 3, 4, 5, and 9 shown to be specific for various stages/periods were consistent with the fact that different sets of regulatory genes were encountered during different stages/periods (Thévenot et al. 2005). Ohdan et al. (2005) found that the expression of genes encoding for starch biosynthetic enzymes in developing rice endosperm was largely developmental specific. Specific QTL for both stages/periods and related traits demonstrated that QTL detection for developmental traits should be conducted simultaneously in the view of time course.
In conclusion, three main chromosome regions, bins 1.03–1.04, 7.02–7.03, and 10.05–10.06, were consistently found to be related with QTL for grain weight and grain-filling rate in this study. Some identified EST and candidate genes with related functions were co-located with these common QTL. Further research should be concentrated on revealing QTL/genes for grain development and final weight both through development of near-isogenic lines of common QTL and their chromosome segment substitution lines, and through cloning and function validation of co-located EST and candidate genes.
References
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20:2324–2326
Austin DF, Lee M (1996) Comparative mapping in F2:3 and F6:7 generations of quantitative trait loci for grain yield and yield components in maize. Theor Appl Genet 92:817–826
Austin DF, Lee M (1998) Detection of quantitative trait for grain yield and yield components in maize across generations in stress and nonstress environments. Crop Sci 38:1296–1308
Austin DF, Lee M, Veldboom LR, Hallauer AR (2000) Genetic mapping in maize with hybrid progeny across testers and generations: grain yield and grain moisture. Crop Sci 40:30–39
Blanc G, Charcosset A, Mangin B, Gallais A, Moreau L (2006) Connected populations for detecting quantitative trait loci and testing for epistasis: an application in maize. Theor Appl Genet 113:206–224
Bluhm BH, Kim H, Butchko RA, Woloshuk CP (2008) Involvement of ZFR1 of Fusarium verticillioides in kernel colonization and the regulation of FST1, a putative sugar transporter gene required for fumonisin biosynthesis on maize kernels. Mol Plant Pathol 9:203–211
Borrás L, Otegui ME (2001) Maize kernel weight response to postflowering source–sink ratio. Crop Sci 41:1816–1822
Borrás L, Westgate ME, Otegui ME (2003) Control of kernel weight and kernel water relations by post-flowering source–sink ratio in maize. Ann Bot 91:857–867
Borrás L, Slafer GA, Otegui ME (2004) Seed dry weight response to source–sink manipulations in wheat, maize and soybean: a quantitative reappraisal. Field Crops Res 86:131–146
Borrás L, Zinselmeier C, Senior ML, Westgate ME, Muszynski MG (2009) Characterization of grain-filling patterns in diverse maize germplasm. Crop Sci 49:999–1009
Boyer CD, Hannah LC (2001) Kernel mutants of corn. In: Hallauer AR (ed) Specialty corns. CRC, Boca Raton, pp 1–31
Capelle V, Remoué C, Moreau L, Reyss A, Mahé A, Massonneau A, Falque M, Charcosset A, Thévenot C, Rogowsky P, Coursol S, Prioul JL (2010) QTLs and candidate genes for desiccation and abscisic acid content in maize kernels. BMC Plant Biol 10:2
Causse M, Rocher JP, Henry AM, Charcosset A, Prioul JL, de Vienne D (1995) Genetic dissection of the relationship between carbon metabolism and early growth in maize, with emphasis on key-enzyme loci. Mol Breed 1:259–272
Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A (2004) Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 168:2169–2185
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Colasanti J, Yuan Z, Sundaresan V (1998) The indeterminate gene encodes a zinc finger protein and regulates a leaf-generated signal required for the transition to flowering in maize. Cell 93:593–603
Crane JC (1964) Growth substances in fruit setting and development. Ann Rev Plant Physiol 15:303–326
Douglas CD, Tsung MK, Frederick CE (1988) Enzymes of sucrose and hexose metabolism in developing kernels of two inbreds of maize. Plant Physiol 86:1013–1019
Ercole O, Alessandro C (1981) Phenotypic and genetic relationships between yield components in maize. Euphytica 30:601–609
Flaherty JE, Woloshuk CP (2004) Regulation of fumonisin fiosynthesis in fusarium verticillioides by a zinc binuclear cluster-type gene, ZFR1. Appl Environ Microbiol 70:2653–2659
Gambín BL, Borras L, Otegui ME (2007) Kernel water relations and duration of grain filling in maize temperate hybrids. Field Crops Res 101:1–9
Goffinet B, Gerber S (2000) Quantitative trait loci: a meta-analysis. Genetics 155:463–473
Hartung RC, Poneleit CG, Cornelius EL (1989) Direct and correlated responses to selection for rate and duration of grain fill in maize. Crop Sci 29:740–745
Hirose T, Terao T (2004) A comprehensive expression analysis of the starch synthase gene family in rice (Oryza sativa L.). Planta 220:9–16
Jones RJ (1985) Thermal environment during endosperm cell division in maize: Effects on number of endosperm cells and starch granules. Crop Sci 25:830–834
Jones R, Brenner M (1987) Distribution of abscisic acid in maize kernel during grain filling. Plant Physiol 83:905–909
Kiesselbach TA (1980) The structure and reproduction of corn. Nebraka Agric Exp Stn Res Bull 161:85–86
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Kozaki A, Hake S, Colasanti J (2004) The maize ID1 flowering time regulator is a zinc finger protein with novel DNA binding properties. Nucleic Acids Res 32:1710–1720
Le H, Browning KS, Gallie DR (1998) The phosphorylation state of the wheat translation initiation factors eIF4B, eIF4A, and eIF2 is differentially regulated during seed development and germination. J Biol Chem 273:20084–20089
Lebrun M, Freyssinet G (1991) Nucleotide sequence and characterization of a maize cytoplasmic ribosomal protein Sll cDNA. Plant Mol Biol 17:265–268
Li YL, Niu SZ, Dong YB, Cui DQ, Wang YZ, Liu YY, Wei MM (2007) Identification of trait-improving quantitative trait loci for grain yield components from a dent corn inbred line in an advanced backcross BC2F2 population and comparison with its F2:3 population in popcorn. Theor Appl Genet 115:129–140
Li JZ, Zhang ZW, Li YL, Wang QL, Zhou YG (2011) QTL consistency and meta-analysis for grain yield traits in three generations in maize. Theor Appl Genet 122:771–782
Linn BY (1977) Ploidy variation in maize endosperm. J Hered 68:143–149
Liu YY, Li JZ, Li YL, Wei MG, Cui QX, Wang QL (2010a) Molecular cloning, sequence and expression analysis of ZmArf2, a maize ADP-ribosylation factor. Mol Biol Rep 37:755–761
Liu YY, Li JZ, Li YL, Wei MG, Cui QX, Wang QL (2010b) Identification of differentially expressed genes at two key endosperm development stages using two maize inbreds with large and small grain and integration with detected QTL for grain weight. Theor Appl Genet 121:433–447
Liu ZH, Ji HQ, Cui ZT, Wu X, Duan LJ, Feng XX, Tang JH (2011) QTL detected for grain-filling rate in maize using a RIL population. Mol Breed 27:25–36
Ma XQ, Tang JH, Teng WT, Yan JB, Meng YJ, Li JS (2007) Epistatic interaction is an important genetic basis of grain yield and its components in maize. Mol Breed 20:41–51
Mihaljevic R, Utz HF, Melchinger AE (2004) Congruency of quantitative trait loci detected for agronomic traits in testcrosses of five populations of European maize. Crop Sci 44:114–124
Nakamura Y, Yuk K (1989) Carbohydrate metabolism in the developing endosperm of rice grains. Plant Cell Physiol 30:833–939
Ohdan T, Francisco JPB, Sawada T, Hirose T, Terao T, Satoh H, Nakamura Y (2005) Expression profiling of genes involved in starch synthesis in sink and source organs of rice. J Exp Bot 56:3229–3244
Olsen OA (2004) Nuclear endosperm development in cereals and Arabidopsis thaliana. Plant Cell 16:S214–S227
Ouattar S, Jones RJ, Crookston RK et al (1987) Effect of drought on water relations of developing maize kernels. Crop Sci 27:730–735
Pelleschi S, Guy S, Kim JY, Pointe C, Mahe A, Barthes L, Leonardi A, Prioul JL (1999) Ivr2, a candidate gene for a QTL of vacuolar invertase activity in maize leaves. Gene-specific expression under water stress. Plant Mol Biol 39:373–380
Prioul JL, Reyss A, Schwebel-Duguè N (1990) Relationships between carbohydrate metabolism in ear and in the adjacent leaf during grain filling in maize genotypes. Plant Physiol Bioch 28:489–493
Prioul JL, Jeannette E, Reyss A, Grégory N, Giroux M, Hannah LC, Causse M (1994) Expression of ADP-glucose pyrophosphorylase in maize (Zea mays L.) grain and source leaf during grain filling. Plant Physiol 104:179–187
Prioul JL, Quarrie S, Causse M, de Vienne D (1997) Dissecting complex physiological functions through the use of molecular quantitative genetics. J Exp Bot 48:1151–1163
Reddy VM, Daynard TB (1983) Endosperm characteristics associated with rate of grain filling and kernel size in corn. Maydica 28:339–355
Royo J, Gómez E, Barrero C, Muñiz LM, Sanz Y, Hueros G (2009) Transcriptional activation of the maize endosperm transfer cell-specific gene BETL1 by ZmMRP-1 is enhanced by two C2H2 zinc finger-containing proteins. Planta 230:807–818
Sawers RJ, Sheehan MJ, Brutnell TP (2005) Cereal phytochromes: targets of selection, targets for manipulation. Trends Plant Sci 10:138–143
Seka D, Cross HZ (1995) Xenia and maternal effects on maize kernel development. Crop Sci 35:80–85
Seka D, Cross HZ, McClean PE (1995) Maize kernel development in vitro: sucrose concentration, xenia, and maternal effects. Crop Sci 35:74–79
Shi JQ, Li RY, Qiu D, Jiang CC, Long Y, Morgan C, Bancroft I, Zhao JY, Meng JL (2009) Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182:851–861
Shrawat AK, Caroll RT, De Pawn M, Taylor GJ, Good AG (2008) Genetic engineering of improved nitrogen use efficiency in rice by the tissue-specific expression 1 of alanine aminotransferase. Plant Biotech J 6:722–732
Slattery CJ, Kavakli IH, Okita TW (2000) Engineering starch for increased quantity. Trends Plant Sci 5:291–298
Song XF (2003) Identification of QTL for kernel oil content and analysis of related traits in maize. PhD thesis, China Agricultural University, Beijing, China
Stuber CW, Edwards MD, Wendel JF (1987) Molecular marker-facilitated investigations of in maize. II. Factors influencing yield and its component traits. Crop Sci 27:639–648
Stuber CW, Lincoln SE, Wolff DW, Helentjaris T, Lander ES (1992) Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics 132:823–839
Ta CT, Weiland RT (1992) Nitrogen partitioning in maize during ear development. Crop Sci 32:443–451
Takai T, Fukuta Y, Shiraiwa T, Horie T (2005) Time-related mapping of quantitative trait loci controlling grain-filling in rice (Oryza sativa L.). J Exp Bot 56:2107–2118
Takatsuji H (1998) Zinc-finger transcription factors in plants. Cell Mol Life Sci 54:582–596
Thévenot C, Simond-Côte E, Reyss A, Manicacci D, Trouverie J, Guilloux ML, Ginhoux V, Sidicina F, Prioul JL (2005) QTLs for enzyme activities and soluble carbohydrates involved in starch accumulation during grain filling in maize. J Exp Bot 56:945–958
Tollenaar M (1978) Relationship between assimilation source and reproductive sink in maize grown in a short season environment. Agron J 70:219–223
Veldboom L, Lee M (1994) Molecular-marker-facilitated studies of morphological traits in maize. II: Determination of QTLs for grain yield and yield components. Theor Appl Genet 89:451–458
Wang GL, Kang MS, Moreno O (1999) Genetic analyses of grain-filling rate and duration in maize. Field Crops Res 61:211–222
Wang S, Basten CJ, Zeng ZB (2011) Windows QTL cartographer 2.5. Department of statistics, North Carolina State University, Raleigh, NC (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm)
Wang BT, Wu JY, Ding JQ, Xi ZY (2009) Map integration of QTLs for grain yield and its related traits in maize. Acta Agro Sin 35:1836–1843
Wilhelm EP, Mullen RE, Keeling PL, Singletary GW (1999) Heat stress during grain filling in maize: effects on kernel growth and metabolism. Crop Sci 39:1733–1749
Yan JB, Tang H, Huang YQ, Zheng YL, Li JS (2006) Quantitative trait loci mapping and epistatic analysis for grain yield and yield components using molecular markers with an elite maize hybrid. Euphytica 149:121–131
Yang ML (2010) QTL Analysis for maize grain filling related and kernel nutritive traits at different stages use RIL population. Ms D Thesis, Henan Agricultural University, Zhengzhou, China
Zeng ZB (1993) Theoretical basis of separation of multiple linked gene eVects on mapping quantitative trait loci. PNAS 90:10972–10976
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
This work was funded by the Henan Innovation Project for University Prominent Research Talents (2005HANCET-12), the Henan Natural Science Foundation (0511032900), and Henan Development and Reform Commission.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Li, Y., Yang, M., Dong, Y. et al. Three main genetic regions for grain development revealed through QTL detection and meta-analysis in maize. Mol Breeding 30, 195–211 (2012). https://doi.org/10.1007/s11032-011-9610-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-011-9610-x