
Abstract
The current study was conducted to elaborate a novel pharmacophore model to accurately map selective glycogen synthase kinase-3 (GSK-3) inhibitors, and perform virtual screening and drug repurposing. Pharmacophore modeling was developed using PHASE on a data set of 203 maleimides. Two benchmarking validation data sets with focus on selectivity were assembled using ChEMBL and PubChem GSK-3 confirmatory assays. A drug repurposing experiment linking pharmacophore matching with drug information originating from multiple data sources was performed. A five-point pharmacophore model was built consisting of a hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic (H), and two rings (RR). An atom-based 3D quantitative structure–activity relationship (QSAR) model showed good correlative and satisfactory predictive abilities (training set \({R}^{2}= 0.904\); test set: \({Q}^{2}= 0.676\); whole data set: stability \(s = 0.803\)). Virtual screening experiments revealed that selective GSK-3 inhibitors are ranked preferentially by Hypo-1, but fail to retrieve nonselective compounds. The pharmacophore and 3D QSAR models can provide assistance to design novel, potential GSK-3 inhibitors with high potency and selectivity pattern, with potential application for the treatment of GSK-3-driven diseases. A class of purine nucleoside antileukemic drugs was identified as potential inhibitor of GSK-3, suggesting the reassessment of the target range of these drugs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Glycogen synthase kinase-3 (GSK-3) is a member of the CMGC family of serine/threonine protein kinases, which catalyze the transfer of the \(\gamma \)-phosphate group of adenosine triphosphate (ATP) to the target substrate, i.e., a serine/threonine residue [1]. GSK-3 represents one of the most attractive therapeutic targets due to its crucial role in a multitude of biological processes including microtubule stability [2], protein translation [3], and \(\beta \)-catenin degradation [4]. Therefore, potent and selective GSK-3 inhibitors are scrutinized for promising new leads to be launched in clinical trials for diabetes, Alzheimer’s disease, inflammation, and cancer [5,6,7,8]. GSK-3 may act as a “tumor suppressor” for particular types of cancers by decreasing cell survival and proliferation, inducing cell senescence and apoptosis, sensitizing tumor cells to chemotherapeutic agents [9] and ionizing radiation [10], as well as for different types of tumors, such as skin, breast, oral cavity, salivary glands, larynx, and esophagus [11]. Nevertheless, while several structurally diverse ATP-competitive GSK-3 inhibitors [12,13,14,15,16] displaying different degrees of selectivity, such as maleimides [12], indirubins [13], paullones [14], aloisines [15], and hymenialdisine [16], none has reached commercial status. To date, the most advanced inhibitor is tideglusib (NP-12, NP031112), a non-ATP-competitive GSK-3 inhibitor [17], which has undergone Phase IIa [18] and IIb clinical trials for Alzheimer’s disease and paralysis supranuclear palsy [19].
Maleimides show picomolar inhibitory activity and significant selectivity for GSK-3 [20], prevent neuronal death in Alzheimer’s disease [21, 22], and demonstrate therapeutic potential for insulin-resistant type-2 diabetes [23, 24]. Computer-aided development of bioactive small molecules by virtual screening (VS) has become an attractive technique in the early drug discovery stages due to cost savings and time efficiency [25]. Based on substantial biological information about GSK-3 inhibition, computational strategies such as ligand–protein docking [26,27,28,29,30], three-dimensional pharmacophore identification [31,32,33], and quantitative structure–activity relationships (QSAR) [28, 30, 34] have been used to improve two critical aspects: potency and selectivity. Heretofore, 2D QSAR [34] and Hansch analysis [27] revealed that the \(\hbox {GSK-}3\alpha \) inhibitory activity of maleimides is improved by electron-withdrawing and hydrophobic substituents.
A pharmacophore model based on four potent and selective inhibitors of GSK-3 over cyclin-dependent kinase-2 (CDK-2) including 3-anilino-4-arylmaleimide, oxadiazole, triazole, and macrocyclic polyoxygenated bis-7-azaindolylmaleimide derivatives [32] was developed using distance comparison (DISCO) and validated by virtual screening. Agarwal et al. [31] built a pharmacophore model based on 31 GSK-3 ligands, which subsequently identified 50 novel potential GSK-3 ligands from ZincPharmer [35].

Template of maleimide derivatives (class 1. R1 \(=\) H, COMe, \(\hbox {CONMe}_{2}\) and R2 \(=\) aza-annulatedindole, pyridinyl, pyrazolo-pyridinyl [43]; class 2. R3 \(=\) H, Cl, OMe, \(\hbox {NO}_{2}\) and R4 \(=\) H, Cl, SMe, \(\hbox {CO}_{2}\hbox {H}\), Cl–OH [12]; class 3. Ar \(=\) 3-(7-azaindolyl); 1-naphthyl; 2-thienyl; 2-pyridyl; R5 \(=\) H, NEt, Cl, F, \(\hbox {CF}_{3}\), OH, OMe; \(\hbox {R}6 = \hbox {HO(CH}_{2})_{3}\); \(\hbox {MeO(CH}_{2})_{3}\); \(\hbox {MeN(CH}_{2})_{3}\); \(\hbox {Boc-N(CH}_{2})_{3}\); \(\hbox {HC(O)NH(CH}_{2})_{3}\) [41]; class 4. A \(=\) C, N; B \(=\) C, N; \(n=1\)–4 [44]; class 5. \(\hbox {R}7 = \hbox {CH}_{2}\hbox {OH}\), OH, \(\hbox {OCH}_{3}\), F, cyclopropylmethoxy; R8 \(=\) F, Cl, Br, I, CN, OH, \(\hbox {CH}_{2}\hbox {OH}\), \(\hbox {CH}_{2}\hbox {Me}\) [20]; class 6. \(\hbox {R}9 = (\hbox {CH}_{2})\)n-imidazol-1yl, (\(\hbox {CH}_{2})\)n-dimethylamino-1yl, (\(\hbox {CH}_{2})\)n-piperidin-1yl, morpholino; R10 \(=\) H, \(\hbox {OCH}_{3}\), F, Br [42]
Benchmarking data sets are valuable tools for computational chemists to assess their theoretical methods and innovative concepts before implementing to real-world problems [36, 37]. Thus, we assembled novel benchmarking data sets including selective/nonselective GSK-3 inhibitors and experimentally validated GSK-3 inactive compounds, to certificate the accuracy of pharmacophore hypothesis used in this work [38]. Picomolar inhibitors are of interest in drug design because they reveal key interactions with protein [39, 40]. Hence, this study focuses on: (i) the development of a pharmacophore model based on low nanomolar/picomolar selective inhibitors (maleimide derivatives); (ii) validate the pharmacophore hypothesis by means of 3D atom-based QSAR to obtain comprehensive QSAR models that explain the quantitative differences in inhibitory activities; (iii) assemble discriminative benchmarking validation data sets for GSK-3 adjusted for selectivity; (iv) perform pharmacophore-based virtual screening on benchmarking data sets; (v) propose a reliable, integrative drug repositioning experiment by associating pharmacophore search, chemical and biological data.
Materials and methods
Data set for analysis
A broad inspection of the literature concerning GSK-3 inhibition identified the maleimide class as the most potent class of GSK-3 inhibitors. A total of 203 maleimide derivatives (excluding metal complexes) (Fig. 1) were identified with GSK-3 \(\hbox {IC}_{50}\) inhibitory activities [12, 20, 41,42,43,44] (see supplementary information). The ten most active and selective maleimide compounds, which display low nanomolar and picomolar inhibitory activity (class 1 and 3, Fig. 1), were used to build a pharmacophore model.
The experimental \(\hbox {IC}_{50}\) values were converted to a logarithmic scale (\(\hbox {pIC}_{50} = -\hbox {logIC}_{50}\)) before atom-based 3D QSAR modeling. The biological activities are normally distributed, spanning over 5.124 logarithmic units (see supplementary information). The molecular structures of the 203 maleimide derivatives were sketched, saved as isomeric smiles strings using ChemAxon’s MarvinSketch software [45], and checked for duplicates prior to processing.

Distribution of similarity values between actives and inactives (a); the distribution of similarity values between selectives and nonselectives (b)
Validation data set for virtual screening
In the absence of a standardized validation data set for GSK-3 [37], we assembled validation sets focused on selectivity, benchmarked them, and adjusted them correspondingly in size to test the accuracy of our pharmacophore model.
Artificial enrichment was circumvented by providing ligand–decoy similarity for physical properties [36], which most of the existing benchmarking sets comply with [46]. In order to select decoys, the authors of Directory of useful decoys (DUD) enforced a topological dissimilarity filter [36] to select the actives. This favors artificial enrichment since a number of decoys actually bind to targets they were not initially designed for [46, 47]. One of the most challenging issues in the validation of data sets is the false negative problem which lowers ligand enrichment [47].
Maximum unbiased validation (MUV) sets prepared by G. Rohrer and K. Baumann [37] used chemical and biological data from high-throughput experiments (HTS), actives from confirmatory screenings, and inactives from primary screenings. Although MUV sets rely on a robust theory, i.e., spatial statistics, the chemical sphere of MUV is restricted to 14 targets (i.e., e-DUD define 102 targets). Zefirov et al. elaborated a validation data set for GSK-3 based on 1226 actives, 209 inactives, 2000 decoys (compounds from NCI diversity set), and CDK-2 DUD decoys [48, 49]. Thus, we assembled a validation data set for GSK-3 by selecting confirmed actives deposited in ChEMBL [50], Binding DB [51], and from reference [43] showing inhibitory activities below 20 \(\upmu \hbox {M}\) while also displaying inhibition data against other phylogenetically related kinases, such as CDK-2, cyclin-dependent kinase-4 (CDK-4), and protein kinase C (PKC).
After removing duplicates using Instant JChem [52], 237 unique compounds remained which were then divided as function of selectivity ratio (\(\hbox {SR} = \hbox {IC}_{50}\hbox {CDK}\)-2/CDK-4/PKC: \(\hbox {IC}_{50}\hbox {GSK-}3\)). Thus, the data set was divided as follows: 149 compounds are selective (\(11{,}800 < \hbox {SR} > 1\)) and 88 are nonselective (selectivity ratio in the range \(0.00023 < \hbox {SR} \le 1\)). The decoys were chosen from true inactives attested in confirmatory PubChem [38] assays against GSK-3 AID434954 and AID 463203 [38], which matched low-dimensional physicochemical properties of actives. After applying a drug-like filter with the FILTER software [53,54,55,56,57,58,59] (HBA = 1–7, HBD = 1–7, MW = 200–600, RBN = 0–13, XLogP = −1.26–6.2, 2dPSA = 45–155), only 2106 compounds out of 3170 inactive compounds remained displaying 751 unique Bemis–Murko chemotypes. Since the negative validation set (inactives) has a crucial relevance on VS results, we removed the uncertainties related to artificial decoys by using experimentally confirmed inactives [60, 61].
Before proceeding to evaluate the pairwise 2D Tanimoto similarity between the 237 actives and 2106 inactives, the values of continuous descriptors (physicochemical and functional) [53,54,55,56,57,58,59] were mean-centered and scaled such as their values lies between 0 and 1. The actives–inactives pairwise similarity values display a distribution shifted toward high values (0.8–1) (see Fig. 2a), as well as selective–nonselective inhibitors (Fig. 2b, templates shown in Figs. 3, 4) which makes our experiments more challenging. This situation is encountered in combinatorial libraries where a large number of close analogues are enclosed. The selective and nonselective data sets were subjected to scaffold extraction using ChemAxon software [52] resulting in 59 and 52 Bemis–Murko chemotypes [52, 62] from which 14 scaffolds are shared.
According to Nicholls [63], the selectivity directed benchmarking data sets display sufficient sampling, selectives/inactives and nonselectives/inactives ratios of 1/14.14 and 1/23.94, respectively. To the best of our knowledge, this is the first attempt at building a validation data set for GSK-3 using confirmed inactives.

Representative scaffolds of selective GSK-3 inhibitors

Representative scaffolds of nonselective GSK-3 inhibitors
Pharmacophore modeling
The pharmacophore hypotheses were generated using the “Develop Pharmacophore Model” module of the Phase software implemented in the Schrödinger suite [64]. LigPrep [65] was used to convert the 2D structures to 3D and to enumerate the ionization states and tautomers in a pH range of \(7\pm 2\). The conformational space around each ligand was expanded using ConfGen [66] to generate a maximum number of 100 conformers per rotatable bond and 1000 conformers per structure, which were then further subjected to energy minimization based on the optimized potentials for liquid simulations (OPLS)-2005 force field engaging the distance-dependent dielectric algorithm for solvation treatment [67]. The minimized conformers were filtered within an energy window of 50 kJ/mol [68], whereas redundant conformations were discarded if their heavy atoms root mean squared deviation (RMSD) exceed 2 Å. The pharmacophore hypotheses were generated using the 10 most active (\(\hbox {pIC}_{50}> 9\)) (selectivity ratio \(> 10\)) compounds (class 1, 3, see Table 1; Fig. 1), whereas the threshold for inactivity was set to 6 \(\hbox {pIC}_{50}\) units [69] (16 compounds display \(\hbox {pIC}_{50} < 6\), see supplementary information) using Phase [64], with default settings.
The ranking of the generated hypotheses to identify the best alignment of the actives was performed using a maximal root mean squared deviation (RMSD) of 1.2 Å. Each alignment is assessed by three parameters: (i) the root mean squared deviation in the site point locations—denoting the alignment score; (ii) the vector score designated by the average cosine of the angles between the corresponding pairs of vector pharmacophores (aromatic rings, acceptors, and donors) in the aligned molecules; and (iii) the volume score evaluated as function of van der Waals superimposing models of heavy atoms within each pair of ligands. Hydrogen bond acceptor/donor, hydrophobic, and aromatic ring sites were detected in the pharmacophore models generated, and subsequently ranked based on survival score (Eq. 1), calculated as follows [64]:
where W designates the weights and S the scores; \({S}_{\mathrm{site}}\) stands for the alignment score and is calculated as the root mean square deviation in the site point position; \({S}_{\mathrm{vec}}\) denotes the vector score and accounts for the cosine of the angles created by the analogous pairs of vector features in superimposed structures; \({S}_{\mathrm{vol}}\) is the volume score estimated by the overlapping of van der Waals volumes of heavy atoms in each pair of structures; and \({S}_{\mathrm{sel}}\) represents the selectivity score, which describes the fraction of compounds which are expected to match the hypothesis in addition to their biological activity toward the receptor. \({W}_{\mathrm{site}}\), \({W}_{\mathrm{vec}}\), \({W}_{\mathrm{vol}}\), \({W}_{\mathrm{rew}}\) are set up to 1.0 value by default, but \({W}_{\mathrm{sel}}\) has a reference value of 0.0. A penalty for high-energy structures \({W}_{\mathrm{E}}\Delta {E}\) is included as well as an activity penalty term denoted \({W}_{\mathrm{act}}{A}\) where A stands for biological activity. The hypotheses were ranked by survival score [64] which eliminated those hypotheses that do not discriminate actives from inactive compounds. We started generating the pharmacophore hypotheses with 5, 6, and 7 features, but amidst the generated hypotheses, only 5 feature hypotheses were acceptable in accord with the highest survival score and atom-based 3D QSAR validation. Hypotheses that do not discriminate the pharmacophoric pattern of active versus inactive maleimides were discarded.
Effective pharmacophore models have to comply with statistical criteria and to be accurate in identifying active or even selective compounds. The stability of the pharmacophore hypotheses generated was internally validated by means of statistical parameters: the squared correlation coefficient (\({R}^{2})\), correlation coefficient for the test set (\({Q}^{2})\), the standard deviation of regression (SD), Pearson’s correlation coefficient (Pearson’s R), statistical significance (P), and variance ratio (F). The evaluation and validation of a pharmacophore model are crucial before its utilization for virtual activity profiling. Furthermore, the best pharmacophore hypothesis Hypo-1 was validated by checking the overly of ligand conformations identified by pharmacophore Hypo-1 with the experimental structure of 3-(5-fluoro-6-iodo-1-methyl-1H-indol-3-yl)-4-(7-methoxy-1-benzofuran-3-yl)-1H-pyrrole-2,5-dione (PDB ID: TSK) extracted from co-crystallized complexes of GSK-3 with ligand (PDB ID: 3SD0) from the Protein Data Bank (PDB) [70, 71] and evaluation of all heavy atom RMSD.
Atom-based 3D-QSAR
Atom-based 3D-QSAR is more advantageous over pharmacophore-based 3D-QSAR since the former ignores the structural aspects of the ligand beyond the pharmacophore model, i.e., steric clashes with the receptor [72]. The best pharmacophore hypothesis was used to align all data set molecules in order to develop an atom-based 3D-QSAR. In 3D atom-based QSAR, a molecule is depicted as overlapping van der Waals spheres which are assigned to the following categories: hydrogen bond donor (D), electron withdrawing (i.e., including hydrogen bond acceptors, W), hydrophobic (H), negative ionic (N), positive ionic (P), and miscellaneous (X) [64].
The training set includes 70% randomly selected molecules (143 compounds), whereas the remaining 30% (60 compounds) was used to validate the model (test set). To develop a 3D atom-based QSAR model, the van der Waals spheres of the aligned training set compounds were positioned into a standardized grid of cubes, where each cube was ascribed zero or more “bits” to register the distinct types of atoms belonging to the training set which fill the cube. This chart leads to binary-valued patterns that were employed as independent variables to generate partial least squares (PLS) atom-based QSAR models.
The atom-based 3D-QSAR regression model was developed using maximum five PLS factors, since a supplementary increase in the PLS factors did not lead to an improvement in statistics or predictive abilities. Atom-based QSAR models were built for all pharmacophore hypotheses developed using the randomly chosen training set of compounds, with a grid spacing of 1.0 Å. Internal and external validation parameters for atom-based 3D QSAR were squared correlation coefficient (\(R^{2}\) training set), squared correlation coefficient for test set (\(q^{2}\)), standard deviation (SD), Pearson’s correlation coefficient (Pearson’s R for test set), statistical significance (P), and Fisher’s test (F). The Spearman’s (rho) rank correlation coefficient was calculated using the R software to evaluate statistical associations based on the ranks of the data [73].
Virtual screening
Virtual screening experiments were performed to validate the existence of distinct chemical spaces that describe the unique pharmacophore arrangement Hypo-1 which corresponds to GSK-3 high-affinity inhibition. A retrospective virtual screening experiment based on pharmacophore Hypo-1 was carried out using Advanced Pharmacophore Screening implemented in PHASE, against validation data sets for GSK-3 according to the protocol described. The matching of pharmacophore was carried out by applying default tolerance for each inter-feature distance in the pharmacophore hypothesis; hence, all data set molecules whose pharmacophore features miss the distance cutoff are discarded. When a molecule from the data set generates a match, its pharmacophore site points are aligned to the hypothesis by means of standard least squares algorithm. Since not all molecules from the validation data sets showed all five features disposed geometrically to fit Hypo-1, we chose to loose the criteria based on some set of features common to all of them to minimum 2 features. Thus, all molecules including inactives displaying 2–5 features can be evaluated. Only one match per molecule was retained, the best ranked conformer by fitness score. The resulting enrichment lists were evaluated using an in-house developed program called Evaluation Tool In ChemInformatics (ETICI). The impact of pharmacophore model on validation data sets in terms of efficacy of rankings provided in various conditions (actives, selectives, nonselectives) was evaluated comparatively using several evaluation metrics focused on overall enrichment (AUC), overall plus early enrichment (AROCE and eROCE), and early enrichment (TP at 2, 5, 10% FP).
Drug bank repurposing
The old reductionist concept ‘compound-single-target’ is nowadays translated to a more realistic approach named polypharmacology, according to which drugs exert their clinical effects by interacting with multiple (desired) targets [74]. Complex diseases, such as cancer and neurodegenerative diseases, have a genetic background involving a large number of genes [75], i.e., in cancer cells almost 1000 tagged proteins displayed an effective response to a cytotoxic agent [76]. HTS experiments and in silico approaches such as pharmacophore and docking identified several Food and Drug Administration (FDA) approved drugs as actives against novel targets, which facilitate the repositioning for other diseases [77]. Being independent on 3D structure of the target, pharmacophore methods are more general and hold scaffold hopping potential. The pharmacophore Hypo-1 identified on-target selective inhibitors, and it was used for a drug repurposing experiment. In the absence of thoroughly scrutinized chemical and target spaces [78, 79], this experiment may provide intriguing outcomes, i.e., to identify either new ligands for GSK-3 or putative targets for known drug. Moreover, our approach correlates drug information originating from multiple data sources to support a more predictable drug repositioning attempt [74]. Therefore, we considered the biological pathways common to GSK-3 and the hit drug targets on one side and on the other side the main signaling pathways involved in the disease treated by these drugs relevant for the therapeutic mechanism [74]. Hence, we correlate signaling pathways where the identified drugs and GSK-3 take part and are involved in the same therapeutic problem, as reported in the literature. The information about targets associated with drug therapeutic functions was extracted from Drug Bank [80], whereas additional biological activities of the drugs were scrutinized in the PubChem data base [38, 81].
Evaluation techniques
Each individual virtual screening experiment was validated according to widely recognized virtual screening assessment parameter receiver operating characteristic (ROC) [60, 61], which uses sensitivity (Eq. 2) to account for the fraction of actives predicted as actives, and specificity (Eq. 3) which records the fraction of inactives predicted as inactives. The area under the curve (AUC) is the area below the ROC curve, ranging of [0, 1] where the value 1 designates the perfect separation (all the actives are recovered at the top of the hit list) and 0 denotes the complete antiselection (all the actives are found at the end of the hit list) [60, 61].
where TP represents the true positives, TN designates the true negatives, FP denotes the false positives, and FN stands for false negatives, and each of the above quantities was calculated at any threshold.
The AUC metric captures the necessary criteria of a good measure as stated by Jain and Nicholls [82] including robustness, independence to extensive variables, the absence of free parameters, readily interpretable and intuitive, but is not sufficiently sensitive to early enrichment. The enrichment factor (EF) is defined on the early part of the hit list as suggested by Jain and Nicholls [82] to record the performance of virtual screening methods in terms of ROC enrichment, e.g., register the ratio of true positive rates (TPR) at 2, 5, and 10% of false positive rates (FPR). In addition, we calculated the values of two early enrichment evaluation parameters developed by one of us, namely addition of ROC enrichment (AROCE) [83] and exponential ROC enrichment (eROCE) [84]. AROCE consists of the addition of early, discontinued TPR/FPR ratios multigrade relevance intervals for the TPs [83]. The exponential parameter eROCE (Eq. 4) seeks to smooth the effect of hard cutoffs by attributing decreasing weights to each active detected along the hierarchical list [84].
The parameter \(\alpha \) (in Eq. 5) adapts the exponential weights of the TPs. The exponential function \(\varepsilon _{{i}}\) (Eq. 4) fits the weight assigned to the ith TP identified at the respective false positive rate (FPRi). If a TP is detected before all false positives, the FPR is zero results \(\varepsilon _{i}\) of one, and vice versa, if the TPs are situated at the bottom of the ranking list, the FPR is close to one, and the value is close to zero [84]. NA denotes the total number of actives. Similarly to AUC, the eROCE range is between zero and one.
All evaluation parameters were calculated using an in-house developed program called ETICI.
Results and discussions
Pharmacophore modeling
Among all pharmacophore models developed, only five-point pharmacophores displayed the highest values of survival score. A total of 355 five-point pharmacophore hypotheses were generated, from which 238 hypotheses were externally validated by atom-based 3D QSAR (R-Pearson \(> 0.770\)). In Fig. 5, it is shown the pharmacophore displaying the highest adjusted survival score mapped to the most active compound 180. The best pharmacophore model is represented by the Hypo-1 pattern. The distribution of pharmacophore features into the model includes one hydrogen bond donor, one hydrogen bond acceptor, one hydrophobic, and two rings. The maleimide ring is represented by one hydrogen bond acceptor (=O) and one hydrogen bond donor (N) sites, whereas at least one substituent of the most active maleimides is a fused ring, i.e., indole (see Fig. 5). Whereas the interaction with Asp133 and Val135 is crucial to improve the affinity for GSK-3, it does not contribute to selectivity over other kinases [85]. The acceptor and donor group of maleimide ring interacts effectively with GSK-3 binding site residues; thus, the spatial arrangement of acceptor and donor sites inside Hypo-1 hypothesis is supported by experimental evidence [86]. The hydrophobe site, which is placed on 1-methyl substituent of 1H-indol-3-yl (Fig. 6), makes hydrophobic interactions with Pro136. Interactions with Pro136, which strengthen the interaction of the ligand with the backbone, are being encountered in a number of \(\hbox {GSK-}3\beta \) complexes (Fig. 6) [85]. Structural differences between Pro136 and His84 in CDK-2 may afford the formation of the salt bridge in the \(\hbox {GSK-}3\beta \) [87]. A hydrophobic interaction with Asp200 in the 3SD0 co-crystal occurs in the selectivity areas defined by Cys199, Asp200, Gln185, and Arg141. A ruthenium-coordinated GSK-3 maleimide ligand networks with the flexible glycine-rich loop forming a small hydrophobic pocket Ile62, Gly63, Phe67, and Val70 which appears to be essential for activity and selectivity [88]. The benzofuran ring of the ligand TSK in the 3SD0 co-crystal makes hydrophobic interactions with Phe67, Lys85, and Asp200, whereas the indolyl ring interacts with Val70, Pro136, and Leu188 [88].
In \(\hbox {GSK-}3\beta \), Lys85 forms a salt bridge with Glu97 and simultaneously with Asp200 [89]. The interactions of inhibitor with Lys85, Glu97, and Asp200 can increase the activity and selectivity toward GSK-3 [85].

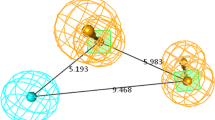
Pharmacophore hypothesis Hypo-1 inter-feature distances and putative interaction points: acceptor (\(\hbox {A}_{4}\), pink), donor (\(\hbox {D}_{7}\), blue), hydrophobic (\(\hbox {H}_{8}\), green), ring (\(\hbox {R}_{10}\), \(\hbox {R}_{12}\) orange) superposed on the most active compound 180. (Color figure online)

Interactions of 3-(5-fluoro-6-iodo-1-methyl-1H-indol-3-yl)-4-(7-methoxy-1-benzofuran-3-yl)-1H-pyrrole-2,5-dione (PDB ID: TSK) with binding site aminoacids: hydrogen bond (green), and hydrophobic (purple) [90]. (Color figure online)
A distinct structural feature of highly potent maleimides, designated to build the pharmacophore model, is a fused aromatic ring at position 3 (135 of potent maleimide compounds share a fused aromatic ring at this position, class 1 and 3, see Fig. 1) confirming the presence of two fused rings in the pharmacophore Hypo-1. The distances between pharmacophoric A, D, H, R sites of the pharmacophore hypothesis Hypo-1 are shown in Fig. 5. The pharmacophore hypothesis displaying the maximum adjusted survival score Hypo-1 was selected to generate the atom-based 3D QSAR model.
Pharmacophore validation
The resulting RMSD of 0.616 Å is suggesting that the pharmacophore fitted conformer of compound 180 is closely similar in space to the experimental receptor-bound X-ray conformation, thus validating the pharmacophore hypothesis Hypo-1 (Fig. 7).
The selected pharmacophore Hypo-1 was validated using the methodology implemented in Phase including Fischer’s randomization test at 95% significance level, scoring inactives, and 3D QSAR model.
Atom-based 3D QSAR
The best atom-based 3D QSAR model M1 (5 PLS factors) is statistically robust, internally, and externally (Table 2). The correlation coefficient for the training set (\({R}^{2})\) is higher than 0.9, suggesting that the X and Y matrices are strongly correlated. The predictive abilities of the model are satisfactory as illustrated by the value of the correlation coefficient for the test set (\({Q}^{2}>0.6\)) and Pearson’s R (\({>}0.8\)). The large values of F test (258.2) indicate a statistically significant QSAR model, which is underlined by the very low value of the significance level of variance ratio P (see Table 2). The results obtained for the test set demonstrated that our atom-based 3D QSAR model is stable and predictive. In Fig. 8, it is depicted the plot of observed versus predicted biological activities for the training and test sets obtained for atom-based 3D QSAR model M1.
The value of Spearman nonparametric rank correlation for experimental and predicted \(\hbox {IC}_{50}\) values (203 maleimides) suggests a strong correlation of the pairs. In order to analyze the obtained atom-based 3D-QSAR model and to investigate the correlation of the most important variables with biological activity, three property fields (HBD, hydrophobicity, and electron withdrawing) were generated for several most active (34, 149, 162, 180) and least potent reference ligands (191, 192, 194, 199) (Fig. 9a–c). The most favorable features are depicted in blue, whereas the detrimental features are shown in red. The blue cubes at the hydrogen bond donor sites indicate that this feature has a positive contribution for biological activity, i.e., NH belonging to maleimide ring [12]. Indeed, the NH group of maleimide ring is essential for GSK-3 inhibition since the substitution of NH by \(\hbox {CH}_{3}\) leads to a dramatical drop of affinity [12]. Another blue area situated on hydroxyethyl(methyl) substituent at position 6 on benzofuran-3-yl ring was observed for compounds 149 and 162, whose affinities are very high (\(\hbox {pIC}_{50}= 9.456\), 9.292). A detrimental hydrogen bond donor area centered on the nitrogen atom of 3-indolyl moiety was observed for weak inhibitors (Fig. 9a compounds 191, 199). Most likely, in this area the donor groups cannot make nonbonding interactions with binding site residues or steric constraints are possible.

Overlay of 3-(5-fluoro-6-iodo-1-methyl-1H-indol-3-yl)-4-(7-methoxy-1-benzofuran-3-yl)-1H-pyrrole-2,5-dione bound conformation extracted from co-crystal 3SD0 (carbon depicted in black) and its pharmacophore-derived conformation (carbon depicted in gray)
In Fig. 9b, we can observe a large, favorable hydrophobic zone (\(\hbox {H}_{8})\) on most active compounds localized mostly on indolyl ring where the presence of 1-methyl substituent increases the inhibitory activity (see compounds 149, 162, 180). An extended blue area in the same region is present also on diazepine ring of compound 34, which have a 1-ethyl substituent [43]. On the other hand, the absence of hydrophobic groups at this position decreases the activity (compounds 192, 194). Previous theoretical investigations did not identified explicitly the positive influence of the \(\hbox {CH}_{3}\) group attached to indole nitrogen (interacting with Pro-136 [70]) on affinity, although Kim et al. determined a sterically favored region with the help of comparative molecular field analysis (CoMFA) [20, 29]. Some hydrophobic areas are also situated on benzofuran ring, i.e., methyl substituent in the ligand TSK co-crystallized with GSK-3 (PDB ID: 3SD0), which interacts with phenyl ring of Phe67 [70]. Phe67 is a flexible residue located into the ATP-binding site, which participates in substrate recognition processes contributing to substrate binding and selectivity [40, 91]. The analysis of the conformational space spanned by Phe67 in the crystal structure of unphosphorylated, ligand-free \(\hbox {GSK-}3\beta \) (PDB ID: 1I09, 1H8F), and maleimide-bound \(\hbox {GSK-}3\beta \) complexes (PDB ID: 1R0E, 1Q4L, 2OW3, 3SD0), indicates that Phe67 is a flexible residue which points toward ligand (PDB ID: 1R0E, 2OW3, 3SD0) or is directed outward (PDB ID: 1Q4L, 1I09). The various positions of the Phe67 occur as a result of different conformations of the Gly-rich loop, which was experimentally observed in Ser/Thr protein kinases [92]. In unphosphorylated \(\hbox {GSK-}3\beta \) (PDB ID: 1I09), the conformation of the activation loop is similar to that detected in activated kinases [93], even in the absence of segment phosphorylation activation, the catalytically active conformation is achieved; see 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid bound into substrate binding cleft of \(\hbox {GSK-}3\beta \) (PDB ID: 1H8F) [94]. In the case of the most active compound 180, which is a close analogue of the bound ligand TSK in the \(\hbox {GSK-}3\beta \) co-crystal PDB ID: 3SD0, we noticed the same 1-methyl substituent on benzofuran ring, which undergoes hydrophobic interaction with Phe67 (distance 3.6-3.8Å) promoted by the intrinsic flexibility of Phe67 [70]. It can be observed also the presence of a red area on benzofuran ring for compounds 149, 162, and 180. Indeed, at least one of the methoxy, hydroxymethyl, 2-hydroxyethyl substituents are present at position 6 and/or 7 in the majority of the most active and selective maleimides (see Fig. 3 class 3, \({R}_{5}\) and supplementary information) [20].

Plot of experimental versus predicted \(\hbox {pIC}_{50}\) values for atom-based 3D QSAR model M1

a Hydrogen bond donor fields observed on most active (34, 149, 162, 180) and least active (191, 192, 194, 199) maleimides (order of decreasing activity). b Representation of hydrophobic fields for most active (34, 149, 162, 180) and least potent (191,192, 194, 199) maleimides (order of decreasing activity). c Electron-withdrawing regions observed on most active (34, 149, 162, 180) and least potent (191, 192, 194, 199) maleimides (order of decreasing activity)
The presence of hydrophilic groups (–OH, –CH\(_{2}\)–OH, –CH\(_{2}\)–O–CH\(_{3}\)) at positions 6, 7 of the indolyl ring increases binding affinity, i.e., the most active compound 180 bears a methoxymethyl moiety at position 7 [20]. The maleimides holding a hydoxymethyl group at position 6 of benzofuran ring exhibit increased affinity for GSK-3 as well as good selectivity against CDK-2 [20]. This fact is consistent with the electron-withdrawing chart of the compounds which show that the presence of such groups on both substituents of maleimide ring is beneficial for affinity. The positive influence of electrostatic field on benzofuran-indolyl-maleimides was also pointed out by Kim et al. [29]. This effect is noticeable on the indolyl ring of compound 149 which displays a larger unfavorable hydrophobic area at similar positions with respect to weak inhibitors. The presence of the second halogen produces a sharp decrease in biological activity, probably due to steric effects [20]. In conclusion, we identified several common pharmacophore features on the most active benzofuran-indolyl maleimides and bisarylmaleimides, which trigger maximal biological effect. Thus, the phamacophore hypothesis Hypo-1 validated by 3D atom-based QSAR and confirmed by experimental findings can be used further to prioritize novel molecules with increased susceptibility to inhibit GSK-3.
Virtual screening
The accuracy of the selected pharmacophore model (Hypo-1) was further validated by demonstrating its ability to prioritize actives (selective and nonselective) GSK-3 inhibitors against inactive compounds (see Table 3) by using actives/inactives, selectives/inactives and nonselectives/inactives validation data sets. We sought to label similar compounds and at the same time distinct Bemis–Murko chemotypes at the top of the ranking lists. Only 33 actives (61%) (20 selectives and 13 nonselectives) and 126 (6%) inactives displayed an exhaustive correspondence of their spatially mapped pharmacophoric sites with the corresponding sites of the Hypo-1. Whereas the success rate of structure based VS experiments was of 34.8% [95], our method identifies 38.46% selective and 22.03% nonselective inhibitors. The low number of inactives, which match the five-point pharmacophore Hypo-1, suggests that the Hypo-1 pattern is useful in discarding a high number of biologically inactive compounds (94%), but also nonselectives (78.97%). When the pharmacophore Hypo-1 was reduced to 2 up to 5 features (any features) hereby denoted Hypo-1.1, all 111 (100%) actives and 2097 inactives (99.57%) were mapped.

Highest scored inactives matched by Hypo-1
The overall enrichment of GSK-3 selective inhibitors measured by AUC yields satisfactory discriminative performance (0.747) as well as early enrichment metrics AROCE and eROCE, whose values higher than 0.4 are satisfactory [83, 84]. The AUC value which is based on more information content accounting for the whole hit list is higher for the target selective compounds against inactives (0.747 and 0.713), whereas the AUC values for nonselective inhibitors are very poor (0.321 and 0.566). AROCE involves adjustments to introduce early performance to AUC (five-grade weighing scheme of TP at 0.5, 1, 2, 5, 10% FPR), but this fact narrow its propensity to compare with the ROC–AUC curves above the chosen thresholds (10% FPR). eROCE which has been demonstrated as a reliable measure for the early performance of VS applications [82] is summed up on the entire ranking list. The values of AROCE and eROCE parameters are very close, showing the highest values in the case of selective compounds, but very poor discriminative power for nonselectives. The difference in terms of early enrichment between target selective versus nonselective compounds is obvious. The highest retrieval of selective compounds at 2% FP is of 50.000%, whereas the enrichment of nonselective compounds displays lower values (\(15.385 \div 17.154\%\)). The largest differences in terms of evaluation parameters between selectives and nonselectives are variable, and the highest is registered by AUC (0.426), followed by eROCE (0.363), and AROCE (0.288). We have shown that the pharmacophores Hypo-1 and Hypo-1.1 provide significant enrichment for selective GSK-3 inhibitors, but fail to retrieve nonselective compounds. The enrichments obtained by us using inactives attested in confirmatory screenings, which display a high degree of similarity to actives, might represent an extremely difficult case, but are more realistic than in the case of other benchmarking data sets where the decoys were not confirmed experimentally (DUD-E) [46] or the inactives were tested in primary screenings (MUV) [37]. However, our results can provide information about the lowest discriminative limit of the pharmacophores Hypo-1 and Hypo-1.1.

Overly of inactives on Hypo-1 (compound 180 shown in black)
A number of 38 inactives which display 19 distinct BMF scaffolds match pharmacophore Hypo-1 (see in Fig. 10 the highest ranked compounds). These molecules display a highly flexible fragment consisting of 6–10 consecutive flexible bonds; hence, they can artificially fit the Hypo-1 motif (see Fig. 11). High flexibility and large structural diversity make the application of pharmacophore difficult; therefore, recurrent structural features, which render high affinity/selectivity, have to be identified [96]. In order to reduce further the false positive rates, a careful visual analysis by the experimented chemist is needed.
The features encoded by pharmacophore Hypo-1 proved its ability to identify very potent compounds exhibiting selectivity toward GSK-3 (see Table 4).

Distinct selective chemotypes detected by Hypo-1.1 in top 10%, excluding maleimides
Generally speaking, a pharmacophore model should cover structurally diverse ligands, which are attached at the same protein binding site. Low 2D similarity of chemotypes ensures a wide range of molecular diversity associated simultaneously with a good suitability of steric and pharmacophoric features and facilitates the identification of molecular areas involved in making critical interactions. In our particular case, we mined high-affinity, target-selective compounds to derive a pharmacophore, which may retrieve active and selective compounds. The top 10% ranking lists were analyzed to evaluate the number of distinct scaffolds that were prioritized by Hypo-1 and Hypo-1.1 (Fig. 12). The more the specific pharmacophore Hypo-1 (retaining all 5 features), the lower the number of individual scaffolds detected in top 10% of ranking list and vice versa. However, it is important to retain these essential features when searching for novel hits aiming at enlarging the chemical space of GSK-3 inhibitors. In the future, we will extend the current pharmacophore investigation on highly selective inhibitors including other biological targets.
Drug repositioning
In the following, we use the pharmacophore Hypo-1 to assist “scaffold hopping” and potentiality expand the chemical space of GSK-3 inhibitors by using approved drugs from Drug Bank. Excluding marketed drugs such as lithium and valproic acid, only two clinical trials for GSK-3 inhibitor NP031112 in Phase I/II, one in Alzheimer disease and the other in progressive supranuclear palsy, were accomplished [19, 97]. The objectives pursued, including computational drug repositioning, were: (i) to identify drugs which comply with the pharmacophoric features of Hypo-1; (ii) to investigate extensively the biological activity profile and the targets of these drugs; (iii) to underline the involvement of GSK-3 in various signaling pathways where the targets of repositioned drugs are also involved; and (iv) to identify in the literature the synergies between hit drugs and GSK-3 inhibitors.
Thus, among 1510 approved drugs, we identified 30 hits, which display distinct scaffolds and match the pharmacophore sites of Hypo-1. The top hits retrieved by Hypo-1 include drugs used to treat various diseases such as: (i) leukemias (nelarabine, fludarabine, cladribine and clofarabine); (ii) nausea/vomiting (granisetron); (iii) irritable bowel syndrome (IBS) in women (alosetron); and (iv) heart diseases (betablocker bopindolol) (Fig. 13) [80]. Alosetron, which was withdrawn from the market in 2000 due to serious life-threatening adverse effects (reintroduced in 2002 with restricted use), and bopindolol which is an ester-based prodrug were excluded from the current analysis [80].
Nucleoside analogues are prodrugs, which are transformed into their active triphosphate form by means of a series of enzymatic reactions, and are subsequently incorporated into DNA [98]. Nelarabine is used to treat T-cell acute lymphoblastic leukemia (T-ALL) and T-cell acute lymphoblastic lymphoma (T-LBL) [99, 100]. Fludarabine is the most potent purine derivative used for the first- and second-line treatment in B-cell chronic lymphocytic leukemia (B-CLL) and lymphoma [101, 102]. Cladribine is a purine nucleoside prodrug used in the treatment of hairy cell leukemia (HCL) and chronic lymphocytic leukemia (CLL) [103]. Clofarabine is a second-generation purine nucleoside antimetabolite [80] which is being recommended to treat relapsed or refractory acute T-cell and B-cell lymphocytic leukemia [80].
According to fitness scores, the nucleoside analogues nelarabine, fludarabine, cladribine, clofarabine (see Figs. 13, 14) can be suggested as novel GSK-3 inhibitors.

Chemical structure of the drugs identified by pharmacophore Hypo-1

Three-dimensional structure of nelarabine superposed on pharmacophore Hypo-1 and compound 180 (carbon depicted in black)
GSK-3 biological functions related to various types of leukemia and cancer
Since the majority of drugs prioritized by Hypo-1 are used to treat blood malignancies, first we will demonstrate the potential of GSK-3 for therapeutic intervention in leukemia exemplified in experimental literature. GSK-3 is described as a potential therapeutic target in myeloid/lymphoid or mixed-lineage leukemia (MLL), chronic lymphocytic leukemia (CLL), multiple myeloma (MM), and acute myeloid leukemia (AML) as reviewed by McCubrey [104], and indicated in reference [105]. GSK-3 holds extensive regulatory ability related to cellular proliferation [11]. Evidences for therapeutic response resulting from targeting GSK-3 in various leukemia types include: (i) activation of apoptosis in CLL cells induced by GSK-3 inhibition [106]; (ii) GSK-3 inhibition in MLL murine model preclinical studies produced an effective response [107]; and (iii) innovative therapy resulted from combining GSK-3 inhibitors with imatinib in chronic myelogenous leukemia (CML) [108]. Curcumin a known GSK-3 inhibitor [109] suppresses constitutively activated targets of phosphatidylinositol-4,5-bisphosphate 3-kinas (PI3K) such as protein kinase B/Akt, forkhead box O3 (FOXO), and GSK-3 in T-ALL cells and initiates caspase-dependent apoptosis [110]. Promotion of apoptosis observed in T-ALL cells by dual calcineurin (Cn) and GSK-3 inhibitor indicates an innovative therapeutic opportunity [111]. Nevertheless, GSK-3 interacts with many substrates to induce the proapoptotic effect [112], i.e., the prosurvival B-cell lymphoma 2 (Bcl-2)-related myeloid cell leukemia sequence 1 (MCL-1) is directly inhibited by GSK-3, through an important mechanism for the regulation of apoptosis by growth factors, PI3K, and Akt [113]. However, the role of GSK-3 in cancer is confusing since GSK-3 is involved in both cell survival and apoptosis [114]. Although recent developments suggest an active role of \(\hbox {GSK-}3\beta \) in various human cancers, its role in tumourigenesis and cancer progression remains controversial [115].
Mutual biological functions of GSK-3 and antileukemic drug hits
By the virtue of the fact that GSK-3 is a key cellular protein kinase capable of interacting with a plethora of proteins to regulate a wide range of cellular functions, [116] we attempted to correlate additional biological activities of the hit drugs identified in PubChem with GSK-3 biological roles [38]. Shared biological activities observed for the antileukemic drugs identified by Hypo-1 and for GSK-3 inhibitors include: (i) tumor protein p53 agonist (fludarabine and clofarabine) [38, 117]; (ii) Wnt modulation (fludarabine and clofarabine) [38, 104]; (iii) inhibition of mouse double minute 2 homolog (Mdm2), a negative regulator of the p53 (cladribine, fludarabine) [38, 118]; (iv) inhibition of vascular endothelial growth factor receptor (VEGFR) (clofarabine) [38, 86]; (v) modulators of the neurotransmitters serotonin and dopamine which are central targets for mood disorders, depression, psychosis, and addictions [38, 116]; (vi) involvement in critical pathways which are dysregulated in cancer, i.e., transforming growth factor beta (\(\hbox {TGF-}\beta )\) [38, 119]; (vii) regulation of insulin secretion [23, 38]; (viii) inhibition of Plasmodium falciparum proliferation [37, 120]. Wherefore, our investigation suggests that there may be value in the review of the target space of these drugs. Likewise, new combinations of antileukemic drugs with GSK-3 inhibitors might bring therapeutic benefit over the existing therapies [121].
Complex disorders such as leukemias are characterized by multiple signaling abnormalities where the deregulated pathways might be redundant [122]. Therefore, it would be appropriate to develop preclinical studies to investigate the use of GSK-3 inhibitors combined with existing cytotoxic therapies, since in complex diseases it is difficult to find the right combinations of targets for just one drug [123]. In some cases, GSK-3 isoforms exhibit different tissue-specific physiological functions, suggesting therapeutic improvement arising from specific targeting of GSK-3alpha or GSK-3beta in various diseases [124]. Isoform-specific inhibitors could spawn more specific treatments, i.e., mutation of Arg96 in \(\hbox {GSK-}3\beta \) to Ala (\(\hbox {GSK-}3\alpha )\) has repercussion on the ability of \(\hbox {GSK-}3\beta \) to phosphorylate primed versus unprimed substrates [124]. \(\hbox {GSK-}3\beta \) has been investigated intensely, but some biochemical studies have demonstrated novel functions for \(\hbox {GSK-}3\alpha \) in drug resistance and cancer stem cells [125]. The current approach integrates diverse information sources such as chemical/biological properties, synergistic effects, and additional observations from the literature to assist the delivery of more steady, predictable drug repositioning model.
Conclusions
In this study, we have identified a novel GSK-3 pharmacophore Hypo-1 which can be successfully involved for “selectivity search.” The identification of a selective pharmacophore has exciting implications for GSK-3-induced diseases, specifically since protein kinases have structurally similar ATP-binding sites. The obtained 3D atom-based QSAR model M1 is robust, stable and display good correlative (\({R}^{2}>0.9\)) and satisfactory predictive (\({Q}^{2}>0.6\)) abilities. Based on these results, restricted to the current study, the straightforward pharmacophore and validation methodology can be applied to our ongoing efforts to search for novel GSK-3 inhibitors as potential therapeutic agents. The benchmarking validation data sets specifically created for GSK-3 involve experimentally attested actives and inactives in order to challenge the pharmacophore hypothesis developed. Thus, Hypo-1 was successfully involved to prioritize active and selective compounds in retrospective VS experiments. Even if the evaluation parameters display merely satisfactory values, they could be more close to real situation. Similar validation data sets involving confirmatory HTS data assembled according to the methodology developed in this paper can be extended to other targets, in order to strengthen the knowledge regarding the true, accurate potential of virtual screening methods. A computational drug repurposing experiment identified a class of purine nucleoside antileukemic drugs as potential inhibitors of GSK-3, suggesting the reassessment of the target range of these drugs. Even controversial in the case of CLL, several biological activities of the hit drugs were correlated with those of GSK-3 inhibitors suggesting potential common narrow spectra of therapeutic relevance.
Abbreviations
- GSK-3:
-
Glycogen synthase kinase-3
- CMGC:
-
Kinase group named after the initials of some members, which include key kinases: the MAPK growth and stress response kinases, the cell cycle CDK (cyclin-dependent kinases), and kinases involved in splicing and metabolic control
- CDK-2:
-
Cyclin-dependent kinase-2
- DISCO:
-
DIStance COmparison (DISCO) technique
- DUD:
-
Directory of useful decoys
- MUV:
-
Maximum unbiased validation
- HTS:
-
High-throughput screening
- NCI:
-
National Cancer Institute
- CDK-4:
-
Cyclin-dependent kinase-4
- PKC:
-
Protein kinase C
- RMSD:
-
Root mean squared deviation
- PDB:
-
Protein Data Bank
- FDA:
-
Food and Drug Administration
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- AROCE:
-
Addition of ROC enrichment
- eROCE:
-
Exponential ROC enrichment
- OPLS:
-
Optimized potentials for liquid simulations
- PLS:
-
Partial least squares
- CoMFA:
-
Comparative molecular field analysis
- NF-kappaB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- XIAP:
-
X-linked inhibitor of apoptosis protein
- MLL:
-
Myeloid/lymphoid or mixed-lineage leukemia
- CLL:
-
Chronic lymphocytic leukemia
- MM:
-
Multiple myeloma
- AML:
-
Acute myeloid leukemia
- CML:
-
Chronic myelogenous leukemia
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- T-LBL:
-
T-cell acute lymphoblastic lymphoma
- PI3K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- Akt:
-
Serine/threonine kinase Akt (also known as protein kinase B or PKB)
- mTOR:
-
Mechanistic target of rapamycin, also known as mammalian target of rapamycin (mTOR)
- FOXO:
-
Forkhead box O3
- Cn:
-
Calcineurin
- Bcl-2:
-
B-cell lymphoma 2
- MCL-1:
-
Induced myeloid leukemia cell differentiation protein Mcl-1
- B-CLL:
-
B-cell chronic lymphocytic leukemia
- p53:
-
Tumor protein p53
- HCL:
-
Hairy cell leukemia
- Wnt:
-
Wnt signaling pathway
- Mdm2:
-
Mouse double minute 2 homolog
- VEGFR:
-
Vascular endothelial growth factor receptor
- 5-HT3:
-
5-Hydroxytryptamine receptor
- IBS:
-
Irritable bowel syndrome
References
Kannan N, Neuwald AF (2004) Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2alpha. Protein Sci 13:2059–2077. doi:10.1110/ps.04637904
Hong M, Chen DC, Klein PS, Lee VM (1997) Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 272:25326–25332. doi:10.1074/jbc.272.40.25326
Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG (1998) Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett 421:125–130. doi:10.1016/S0014-5793(97)01548-2
Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P (1998) Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol 8:573–581. doi:10.1016/S0960-9822(98)70226-X
Gao C, Hölscher C, Liu Y, Li L (2011) GSK3: a key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev Neurosci 2123:1–11. doi:10.1515/rns.2011.061
Zhai P, Sadoshima J (2012) Glycogen synthase kinase-3\(\beta \) controls autophagy during myocardial ischemia and reperfusion. Autophagy 8:138–139. doi:10.4161/auto.8.1.18314
Hofmann C, Dunger N, Schölmerich J, Falk W, Obermeier F (2010) Glycogen synthase kinase 3-\(\beta \): a master regulator of tolllike receptor-mediated chronic intestinal inflammation. Inflamm Bowel Dis 16:1850–1858. doi:10.1002/ibd.21294
Schütz SV, Schrader AJ, Zengerling F, Genze F, Cronauer MV, Schrader M (2011) Inhibition of glycogen synthase kinase-3\(\beta \) counteracts ligand-independent activity of the androgen receptor in castration resistant prostate cancer. PLoS ONE 6:e25341. doi:10.1371/journal.pone.0025341
Miyashita K, Nakada M, Shakoori A, Ishigaki Y, Shimasaki T, Motoo Y, Kawakami K, Minamoto T (2009) An emerging strategy for cancer treatment targeting aberrant glycogen synthase kinase 3\(\beta \). Anticancer Agents Med Chem 9:1114–1122. doi:10.2174/187152009789734982
Thotala DK, Hallahan DE, Yazlovitskaya EM (2008) Inhibition of glycogen synthase kinase 3\(\beta \) attenuates neurocognitive dysfunction resulting from cranial irradiation. Cancer Res 68:5859–5868. doi:10.1158/0008-5472.CAN-07-6327
Mishra R (2010) Glycogen synthase kinase 3 beta: can it be a target for oral cancer. Mol Cancer 9:144–159. doi:10.1186/1476-4598-9-144
Smith DG, Buffet M, Fenwick AE, Haigh D, Ife RJ, Saunders M, Slingsby BP, Stacey R, Ward RW (2001) 3-Anilino-4-arylmaleimides: potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorg Med Chem Lett 11:635–639. doi:10.1016/S0960-894X(00)00721-6
Vougogiannopoulou K, Ferandin Y, Bettayeb K, Myrianthopoulos V, Lozach O, Fan Y, Johnson CH, Magiatis P, Skaltsounis AL, Mikros E, Meijer L (2008) Soluble 3’,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase-3 alter circadian period. J Med Chem 2351:6421–6431. doi:10.1021/jm800648y
Stukenbrock H, Mussmann R, Geese M, Ferandin Y, Lozach O, Lemcke T, Kegel S, Lomow A, Burk U, Dohrmann C, Meijer L, Austen M, Kunick C (2008) 9-Cyano-1-azapaullone (cazpaullone), a glycogen synthase kinase-3 (GSK-3) inhibitor activating pancreatic beta cell protection and replication. J Med Chem 1051:2196–2207. doi:10.1021/jm701582f
Mettey Y, Gompel M, Thomas V, Garnier M, Leost M, Ceballos-Picot I, Noble M, Endicott J, Vierfond JM, Meijer L (2003) Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects. J Med Chem 46:222–236. doi:10.1021/jm020319p
Meijer L, Thunnissen AM, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkow EM, Kim SH, Pettit GR (2000) Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem Biol 7:51–63. doi:10.1016/S1074-5521(00)00063-6
Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M (2012) Evidence for irreversible inhibition of glycogen synthase kinase-3 by tideglusib. J Biol Chem 287:893–904. doi:10.1074/jbc.M111.306472
del Ser T (2010) Phase IIa clinical trial on Alzheimer’s disease with NP12, a GSK3 inhibitor. Alzheimer’s Dement 6:S147. doi:10.1016/j.jalz.2010.05.455
del Ser T, Steinwachs KC, Gertz HJ, Andrés MV, Gómez-Carrillo B, Medina M, Vericat JA, Redondo P, Fleet D, León T (2013) Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimers Dis 33:205–215. doi:10.3233/JAD-2012-120805
Gaisina IN, Gallier F, Ougolkov AV, Kim KH, Kurome T, Guo S, Holzle D, Luchini DN, Blond SY, Billadeau DD, Kozikowski AP (2009) From a natural product lead to the identification of potent and selective benzofuran-3-yl-(indol-3-yl)maleimides as glycogen synthase kinase 3beta inhibitors that suppress proliferation and survival of pancreatic cancer cells. J Med Chem 52:1853–1863. doi:10.1021/jm801317h
Cross DAE, Cubert AA, Chalmers KA, Facci L, Skaper SD, Reith AD (2001) Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J Neurochem 77:94–102. doi:10.1046/j.1471-4159.2001.00251.x
Culbert AA, Brown MJ, Frame S, Hagen T, Cross DA, Bax B, Reith AD (2001) GSK-3 inhibition by adenoviral FRAT1 overexpression is neuroprotective and induces Tau dephosphorylation and beta catenin stabilisation without elevation of glycogen synthase activity. FEBS Lett 507:288–294. doi:10.1016/S0014-5793(01)02990-8
Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 7:793–803. doi:10.1016/S1074-5521(00)00025-9
Henriksen EJ, Dokken BB (2006) Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr Drug Targets 7:1435–1441. doi:10.2174/1389450110607011435
Shoichet BK (2004) Virtual screening of chemical libraries. Nature 432:862–865. doi:10.1038/nature03197
Dessalew N, Bharatam PV (2007) Identification of potential glycogen kinase-3 inhibitors by structure based virtual screening. Biophys Chem 128:165–175. doi:10.1016/j.bpc.2007.04.001
Prasanna S, Daga PR, Xie A, Doerksen RJ (2009) Glycogen synthase kinase-3 inhibition by 3-anilino-4-phenylmaleimides: insights from 3D-QSAR and docking. J Comput Aided Mol Des 23:113–127. doi:10.1007/s10822-008-9244-1
Dessalew N, Bharatam PV (2007) 3D-QSAR and molecular docking study on bisarylmaleimide series as glycogen synthase kinase 3, cyclin dependent kinase 2 and cyclin dependent kinase 4 inhibitors: An insight into the criteria for selectivity. Eur J Med Chem 42:1014–1027. doi:10.1016/j.ejmech.2007.01.010
Kim KH, Gaisina I, Gallier F, Holzle D, Blond SY, Mesecar A, Kozikowski AP (2009) Use of molecular modeling, docking, and 3D-QSAR studies for the determination of the binding mode of benzofuran-3-yl-(indol-3-yl)maleimides as GSK-3\(\beta \) inhibitors. J Mol Model 15:1463–1479. doi:10.1007/s00894-009-0498-x
Quesada-Romero L, Mena-Ulecia K, Tiznado W, Caballero J (2014) Insights into the interactions between maleimide derivates and GSK3b combining molecular docking and QSAR. PLoS ONE 9:e102212. doi:10.1371/journal.pone.0102212
Agrawal R, Jain P, Narayan S, Radhe D, Bahare S, Ganguly S (2013) Ligand-based pharmacophore detection, screening of potential pharmacophore and docking studies, to get effective glycogen synthase kinase inhibitors. Med Chem Res 22:5504–5535. doi:10.1007/s00044-013-0547-y
Patel DS, Bharatam PV (2006) New leads for selective GSK-3 inhibition: pharmacophore mapping and virtual screening studies. J Comput Aided Mol Des 20:55–66. doi:10.1007/s10822-006-9036-4
Pradeep H, Rajanikant GK (2012) A rational approach to selective pharmacophore designing: an innovative strategy for specific recognition of Gsk3\(\beta \). Mol Divers 16:553–562. doi:10.1007/s11030-012-9387-9
Katritzky AR, Pacureanu LM, Dobchev DA, Fara DC, Duchowicz PR, Karelson M (2006) QSAR modeling of the inhibition of glycogen synthase kinase-3. Bioorg Med Chem 14:4987–5002. doi:10.1016/j.bmc.2006.03.009
Koes DR, Camacho CJ (2012) ZINCPharmer: pharmacophore search of the ZINC database. Nucl Acids Res 40:W409–W414. doi:10.1093/nar/gks378
Huang N, Shoichet BK, Irwin JJ (2006) Benchmarking sets for molecular docking. J Med Chem 49:6789–6801. doi:10.1021/jm0608356
Rohrer SG, Baumann K (2009) Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data. J Chem Inf Model 49:169–184. doi:10.1021/ci8002649
Wang Y, Suzek T, Zhang J, Wang J, He S, Cheng T, Shoemaker BA, Gindulyte A, Bryant SH (2014) PubChem BioAssay: 2014 update. Nucleic Acids Res 42:D1075–D1082. doi:10.1093/nar/gkt978
Frey KM, Bollini M, Mislak AC, Cisneros JA, Gallardo-Macias R, Jorgensen WL, Anderson KS (2012) Crystal Structures of HIV-1 reverse transcriptase with picomolar inhibitors reveal key interactions for drug design. J Am Chem Soc 134:19501–19503. doi:10.1021/ja3092642
Urich R, Grimaldi R, Luksch T, Frearson JA, Brenk R, Wyatt PG (2014) The design and synthesis of potent and selective inhibitors of Trypanosoma brucei glycogen synthase kinase 3 for the treatment of human african trypanosomiasis. J Med Chem 57:7536–7549. doi:10.1021/jm500239b
Zhang HC, Ye H, Conway BR, Derian CK, Addo MF, Kuo GH, Hecker LR, Croll DR, Li J, Westover L, Xu JZ, Look R, Demarest KT, Andrade-Gordon P, Damiano BP, Maryanoff BE (2004) 3-(7-Azaindolyl)-4-arylmaleimides as potent, selective inhibitors of glycogen synthase kinase-3. Bioorg Med Chem Lett 14:3245–3250. doi:10.1016/j.bmcl.2004.03.090
Ye Q, Xu G, Dan L, Zhe C, Jia L, Hu Y (2009) Synthesis and biological evaluation of novel 4-azaindolyl-indolyl-maleimides as glycogen synthase kinase-3\(\beta \) (GSK-3\(\beta )\) inhibitors. Bioorg Med Chem 17:4302–4312. doi:10.1016/j.bmc.2009.05.031
Engler TA, Malhotra S, Burkholder TP, Henry JR, Mendel D, Porter WJ, Furness K, Diefenbacher C, Marquart A, Reel JK, Li Y, Clayton J, Cunningham B, McLean J, O’Toole JC, Brozinick J, Hawkins E, Misener E, Briere D, Brier RA, Wagner JR, Campbell RM, Anderson BD, Vaughn R, Bennett DB, Meier TI, Cook JA (2005) The development of potent and selective bisarylmaleimide GSK3 inhibitors. Bioorg Med Chem Lett 15:899–903. doi:10.1016/j.bmcl.2004.12.063
Kuo GH, Prouty C, DeAngelis A, Shen L, O’Neill DJ, Shah C, Connolly PJ, Murray WV, Conway BR, Cheung P, Westover L, Xu JZ, Look RA, Demarest KT, Emanuel S, Middleton SA, Jolliffe L, Beavers MP, Chen X (2003) Synthesis and discovery of macrocyclic polyoxygenated bis-7-azaindolylmaleimides as a novel series of potent and highly selective glycogen synthase kinase-3\(\beta \) inhibitors. J Med Chem 46:4021–4031. doi:10.1021/jm030115o
MarvinSketch (2013) version 6.0.6 ChemAxon. http://www.chemaxon.com. Accessed March 2014
Mysinger MM, Carchia M, Irwin JJ, Shoichet BK (2012) Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem 55:6582–6594. doi:10.1021/jm300687e
Vogel SM, Bauer MR, Boeckler FM (2011) DEKOIS: demanding evaluation kits for objective in silico screening a versatile tool for benchmarking docking programs and scoring functions. J Chem Inf Model 51:2650–2665. doi:10.1021/ci2001549
Osolodkin DI, Palyulin VA, Zefirov NS (2011) Structure-based virtual screening of glycogen synthase kinase 3b inhibitors: analysis of scoring functions applied to large true actives and decoy sets. Chem Biol Drug Des 78:378–390. doi:10.1111/j.1747-0285.2011.01159.x
Fu G, Sivaprakasam P, Dale OR, Manly SP, Cutler SJ, Doerksen RJ (2014) Pharmacophore modeling, ensemble docking, virtual screening, and biological evaluation on Glycogen Synthase Kinase-3\(\beta \). Mol Inf 33:610–626. doi:10.1002/minf.201400044
Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Krüger FA, Light Y, Mak L, McGlinchey S, Nowotka M, Papadatos G, Santos R, Overington JP (2014) The ChEMBL bioactivity database: an update. Nucleic Acids Res 42:1083–1090. doi:10.1093/nar/gkt1031
Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK (2007) BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res 35:D198–D201. doi:10.1093/nar/gkl999
Instant JChem (2013) version 5.12.4 ChemAxon. http://www.chemaxon.com. Accessed June 2014
FILTER (2009) version 2.0.2 OpenEye Scientific Software, Inc. Santa Fe, NM, USA. www.eyesopen.com
Wang R, Ying F, Lai L (1997) A new atom-additive method for calculating partition coefficients. J Chem Inf Comput Sci 37:615–621. doi:10.1021/ci960169p
Ertl P, Rohde B, Selzer P (2000) Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 43:3714–3717. doi:10.1021/jm000942e
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25. doi:10.1016/S0169-409X(00)00129-0
Egan WJ, Merz KM, Baldwin JJ (2000) Prediction of drug absorption using multivariate statistics. J Med Chem 43:3867–3877. doi:10.1021/jm000292e
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kipple KD (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2623. doi:10.1021/jm020017n
Martin YC (2005) A bioavailability score. J Med Chem 48:3164–3170. doi:10.1021/jm0492002
Fawcett T (2006) An introduction to ROC analysis. Pattern Recognit Lett 27:861–874. doi:10.1016/j.patrec.2005.10.010
Hanley JA, McNeil BJ (1982) The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143:29–36. doi:10.1148/radiology.143.1.7063747
Bemis GW, Murcko MA (1996) The properties of known drugs. 1. Molecular frameworks. J Med Chem 39:2887–2893. doi:10.1021/jm9602928
Nicholls A (2008) What do we know and when do we know it? J Comput Aided Mol Des 22:239–255. doi:10.1007/s10822-008-9170-2
Small-Molecule Drug Discovery Suite 2014-1: Phase (2014) version 3.8, Schrödinger, LLC, New York, NY, 2014 Phase, version 3.1 Schrodinger, LLC, New York. http://www.schrodinger.com
LigPrep, Schrödinger, LLC, New York, NY, (2014)
Watts KS, Dalal P, Murphy RB, Sherman W, Friesner RA, Shelley JC (2010) ConfGen: a conformational search method for efficient generation of bioactive conformers. J Chem Inf Model 50:534–546. doi:10.1021/ci100015j
Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM (2005) Integrated modeling program, applied chemical theory (IMPACT). J Comp Chem 26:1752–1780. doi:10.1002/jcc.20292
Evans DA, Doman TN, Thorner DA, Bodkin MJ (2007) 3D QSAR methods: phase and catalyst compared. J Chem Inf Model 47:1248–1251. doi:10.1021/ci7000082
Tripuraneni NG, Azam MA (2016) A combination of pharmacophore modeling, atom-based 3D-QSAR, molecular docking and molecular dynamics simulation studies on PDE4 enzyme inhibitors. J Biomol Struct Dyn 12:1–12. doi:10.1080/07391102.2015.1119732
RCSB Protein Data Bank. http://www.rcsb.org/pdb/home/home.do. Accessed Jan 2015
Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wählby A, Jones TA (2004) The Uppsala electron-density server. Acta Cryst D60:2240–2249. doi:10.1107/S0907444904013253
Shah UA, Deokar HS, Kadam SS, Kulkarn VM (2010) Pharmacophore generation and atom-based 3D-QSAR of novel 2-(4-methylsulfonylphenyl)pyrimidines as COX-2 inhibitors. Mol Divers 14:559–568. doi:10.1007/s11030-009-9183-3
Development Core Team R (2012) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Boran AD, Iyengar R (2010) Systems approaches to polypharmacology and drug discovery. Curr Opin Drug Discov Dev 13:297–309
Brown D, Superti-Furga G (2003) Rediscovering the sweet spot in drug discovery. Drug Discov Today 8:1067–1077. doi:10.1016/S1359-6446(03)02902-7
Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen-Saidon C, Liron Y, Kam Z, Cohen L, Danon T, Perzov N, Alon U (2008) Dynamic proteomics of individual cancer cells in response to a drug. Science 322:1511–1516. doi:10.1126/science.1160165
Miller JR, Dunham S, Mochalkin I, Banotai C, Bowman M, Buist S, Dunkle B, Hanna D, Harwood HJ, Huband MD, Karnovsky A, Kuhn M, Limberakis C, Liu JY, Mehrens S, Mueller WT, Narasimhan L, Ogden A, Ohren J, Prasad JV, Shelly JA, Skerlos L, Sulavik M, Thomas VH, VanderRoest S, Wang L, Wang Z, Whitton A, Zhu T, Stover CK (2009) A class of selective antibacterials derived from a protein kinase inhibitor pharmacophore. Proc Natl Acad Sci USA 106:1737–1742. doi:10.1073/pnas.0811275106
Medina-Franco JL, Giulianotti MA, Welmaker GS, Houghten RA (2013) Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today 18:495–501. doi:10.1016/j.drudis.2013.01.008
Csermely P, Korcsmáros T, Kiss HJM, London G, Nussinov R (2013) Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol Ther 138:333–408. doi:10.1016/j.pharmthera.2013.01.016
Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 36:D901–D906. doi:10.1093/nar/gkm958
PubChem Substance Database. http://www.ncbi.nlm.nih.gov/pcsubstance. Accessed Jan 2016
Jain AN, Nicholls A (2008) Recommendations for evaluation of computational methods. J Comput Aided Mol Des 22:133–139. doi:10.1007/s10822-008-9196-5
Avram S, Pacureanu LM, Seclaman E, Bora A, Kurunczi L (2011) PLS-DA-Docking Optimized Combined Energetic Terms (PLSDA-DOCET) protocol: a brief evaluation. J Chem Inf Model 51:3169–3179. doi:10.1021/ci2002268
Avram SI, Crisan L, Bora A, Pacureanu LM, Avram S, Kurunczi L (2013) Retrospective group fusion similarity search based on eROCE evaluation metric. Bioorg Med Chem 21:1268–1278. doi:10.1016/j.bmc.2012.12.041
Kramer T, Schmidt B, Lo Monte F (2012) Small-molecule inhibitors of GSK-3: structural insights and their application to Alzheimer’s disease models. Int J Alzheimers Dis. doi:10.1155/2012/381029
Bertrand JA, Thieffine S, Vulpetti A, Cristiani C, Valsasina B, Knapp S, Kalisz HM, Flocco M (2003) Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J Mol Biol 333:393–407. doi:10.1016/j.jmb.2003.08.031
Patel DS, Dessalew N, Iqbal P, Bharatam PV (2007) Structure-based approaches in the design of GSK-3 selective inhibitors. Curr Protein Pept Sci 8:352–364. doi:10.2174/138920307781369409
Feng L, Geisselbrecht Y, Blanck S, Wilbuer A, Atilla-Gokcumen GE, Filippakopoulos P, Kräling K, Celik MA, Harms K, Maksimoska J, Marmorstein R, Frenking G, Knapp S, Essen LO, Meggers E (2011) Structurally sophisticated octahedral metal complexes as highly selective protein kinase inhibitors. J Am Chem Soc 133:5976–5986. doi:10.1021/ja1112996
Pande V, Ramos MJ (2005) Structural basis for the GSK-\(\beta \) binding affinity and selectivity against CDK-2 of 1-(4-aminofurazan-3yl)-5-dialkylaminomethyl-1\(H\)-1,2,3] triazole-4-carboxylic acid derivatives. Bioorg Med Chem Lett 15:5129–5135. doi:10.1016/j.bmcl.2005.08.077
Discovery Studio Visualizer-Accelrys (2009) version 2.5 San Diego, CA. http://accelrys.com
Ilouz R, Kowalsman N, Eisenstein M, Eldar-Finkelman H (2006) Identification of novel glycogen synthase kinase-3 substrate-interacting residues suggests a common mechanism for substrate recognition. J Biol Chem 281:30621–30630. doi:10.1074/jbc.M604633200
Hubbard SR (2000) Protein tyrosine kinase structure and function. Annu Rev Biochem 69:373–398. doi:10.1146/annurev.biochem.69.1.373
ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, Jain J (2001) Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat Struct Biol 8:593–596. doi:10.1038/89624
Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH (2001) Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 15105:721–732. doi:10.1016/S0092-8674(01)00374-9
Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK (2002) Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem 2345:2213–2221. doi:10.1021/jm010548w
Verdonk ML, Berdini V, Hartshorn MJ, Mooij WTM, Murray CW, Taylor RD, Watson P (2004) Virtual screening using protein–ligand docking: avoiding artificial enrichment. J Chem Inf Comput Sci 44:793–806. doi:10.1021/ci034289q
Cheng H, Woodgett J, Maamari M, Force T (2011) Targeting GSK-3 family members in the heart: a very sharp double-edged sword. J Mol Cell Cardiol 51:607–613. doi:10.1016/j.yjmcc.2010.11.020
Rodriquez CO, Mitchell BS, Ayres M, Eriksson S, Gandhi V (2002) Arabinosylguanine is phosphorylated by both cytoplasmic deoxycytidine kinase and mitochondrial deoxyguanosine kinase. Cancer Res 62:3100–3105
Alvarado Y, Welch MA, Swords R, Bruzzi J, Schlette E, Giles FJ (2007) Nelarabine activity in acute biphenotypic leukemia. Leuk Res 31:1600–1603. doi:10.1016/j.leukres.2006.12.013
Gandhi V, Mineishi S, Huang P, Chapman AJ, Yang Y, Chen F, Nowak B, Chubb S, Hertel LW, Plunkett W (1995) Cytotoxicity, metabolism, and mechanisms of action of 2’,2’-difluorodeoxyguanosine in Chinese hamster ovary cells. Cancer Res 55:1517–1524
Brockman RW, Schabel FM Jr, Montgomery JA (1997) Biological activity of 9-\(\beta \)-D-arabinofuranosyl-2-fluoroadenine, a metabolically stable analog of 9-\(\beta \)-D-arabinofuranosyladenine. Biochem Pharmacol 26:2193–2196. doi:10.1016/0006-2952(77)90275-1
Ricci F, Tedeschi A, Morra E, Montillo M (2009) Fludarabine in the treatment of chronic lymphocytic leukemia: a review. Ther Clin Risk Manag 5:187–207
Robak T (2001) Cladribine in the treatment of chronic lymphocytic leukemia. Leuk Lymphoma 40:551–564. doi:10.3109/10428190109097654
McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Cocco L, Cervello M, Martelli AM (2014) Multifaceted roles of GSK-3 and Wnt/\(\beta \)-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 28:15–33. doi:10.1038/leu.2013.184
Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, Kakoza RM, Chow KT, Ross L, Alexe G, Tolliday N, Inguilizian H, Galinsky I, Stone RM, DeAngelo DJ, Roti G, Aster JC, Hahn WC, Kung AL, Stegmaier K (2012) The intersection of genetic and chemical genomic screens identifies GSK-3\(\alpha \) as a target in human acute myeloid leukemia. J Clin Invest 122:935–947. doi:10.1172/JCI46465
Mirlashari MR, Randen I, Kjeldsen-Kragh J (2012) Glycogen synthase kinase-3 (GSK-3) inhibition induces apoptosis in leukemic cells through mitochondria-dependent pathway. Leuk Res 36:499–508. doi:10.1016/j.leukres.2011.11.013
Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TCP, Cleary ML (2008) Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 455:1205–1209. doi:10.1038/nature07284
Reddiconto G, Toto C, Palamà I, De Leo S, de Luca E, De Matteis S, Dini L, Passerini CG, Di Renzo N, Maffia M, Coluccia AM (2012) Targeting of GSK-3\(\beta \) promotes imatinib-mediated apoptosis in quiescent CD34+ chronic myeloid leukemia progenitors, preserving normal stem cells. Blood 119:2335–2345. doi:10.1182/blood-2011-06-361261
Bustanji Y, Taha MO, Almasri IM, Al-Ghussein MA, Mohammad MK, Alkhatib HS (2009) Inhibition of glycogen synthase kinase by curcumin: Investigation by simulated molecular docking and subsequent in vitro/in vivo evaluation. J Enzyme Inhib Med Chem 24:771–778. doi:10.1080/14756360802364377
Hussain AR, Al-Rasheed M, Manogaran PS, Al-Hussein KA, Platanias LC, Al Kuraya K, Uddin S (2006) Curcumin induces apoptosis via inhibition of PI3’-kinase/AKT pathway in acute T cell leukemias. Apoptosis 11:245–254. doi:10.1007/s10495-006-3392-3
Tosello V, Bordin F, Yu J, Agnusdei V, Indraccolo S, Basso G, Amadori A, Piovan E (2016) Calcineurin and GSK-3 inhibition sensitizes T-cell acute lymphoblastic leukemia cells to apoptosis through X-linked inhibitor of apoptosis protein (XIAP) degradation. Leukemia 30:812–822. doi:10.1038/leu.2015.335
Frame S, Cohen P (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359:1–16. doi:10.1042/bj3590001
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 1721:749–760. doi:10.1016/j.molcel.2006.02.009
Beurel E, Jope RS (2006) The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol 79:173–189. doi:10.1016/j.pneurobio.2006.07.006
Luo J (2009) Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett 18273:194–200. doi:10.1016/j.canlet.2008.05.045
Kaidanovich-Beilin O, Beaulieu JM, Jope RS, Woodgett JR (2012) Neurological Functions of the Masterswitch Protein Kinase GSK-3. Front Mol Neurosci 5:48. doi:10.3389/fnmol.2012.00048
Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS (2003) Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem 278:48872–48879. doi:10.1074/jbc.M305870200
Kulikov R, Boehme KA, Blattner C (2005) Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol 25:7170–80. doi:10.1128/MCB.25.16.7170-7180.2005
Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE (2009) A negative feedback control of TGF-\(\beta \) signaling by GSK3-mediated Smad3 linker phosphorylation at Ser204. J Biol Chem 284:19808–19816. doi:10.1074/jbc.M109.016667
Masch A, Kunick C (2015) Selective inhibitors of Plasmodium falciparum glycogen synthase-3 (PfGSK-3): new antimalarial agents? Biochim Biophys Acta 54:1644–1649. doi:10.1016/j.bbapap.2015.03.013
Porta C, Paglino C, Mosca A (2014) Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 4:64. doi:10.3389/fonc.2014.00064
Matsuda S, Nakanishi A, Wada Y, Kitagishi Y (2013) Roles of PI3K/AKT/PTEN pathway as a target for pharmaceutical therapy. Open Clin Chem J 7:23–29. doi:10.2174/1874104501307010023
Chen EY, DeRan MT, Ignatius MS, Grandinetti KB, Clagg R, McCarthy KM, Lobbardi RM, Brockmann J, Keller C, Wu X, Langenau DM (2014) Glycogen synthase kinase 3 inhibitors induce the canonical WNT/\(\beta \)-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc Natl Acad Sci USA 111:5349–5354. doi:10.1073/pnas.1317731111
McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, Gizak A, Demidenko ZN, Cocco L, Martelli AM, Cervello M (2014) GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 305:2881–2911. doi:10.18632/oncotarget.2037
Thamilselvan V, Menon M, Thamilselvan S (2011) Anticancer efficacy of deguelin in human prostate cancer cells targeting glycogen synthase kinase-3 \(\beta /\beta \)-catenin pathway. Int J Cancer 129:2916–2927. doi:10.1002/ijc.25949
Acknowledgements
We thank Chemaxon Ltd., OpenEye Ltd., and Accelrys Inc. for providing academic license. This project was financially supported by the Project No. 1.2/2016 of the Institute of Chemistry of Romanian Academy, Timisoara.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors indicate no potential conflicts of interest.
Additional information
Dedicated to the 150th anniversary of the Romanian Academy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Crisan, L., Avram, S. & Pacureanu, L. Pharmacophore-based screening and drug repurposing exemplified on glycogen synthase kinase-3 inhibitors. Mol Divers 21, 385–405 (2017). https://doi.org/10.1007/s11030-016-9724-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-016-9724-5