
Abstract
The Tyro3, Axl and Mertk (TAM) subfamily of receptor protein tyrosine kinases functions in cell growth, differentiation, survival, and most recently found, in the regulation of immune responses and phagocytosis. All three receptors and their ligands, Gas6 (growth arrest-specific gene 6) and protein S, are expressed in the central nervous system (CNS). TAM receptors play pivotal roles in adult hippocampal neurogenesis. Loss of these receptors causes a comprised neurogenesis in the dentate gyrus of adult hippocampus. TAM receptors have a negative regulatory effect on microglia and peripheral antigen-presenting cells, and play a critical role in preventing overproduction of pro-inflammatory cytokines detrimental to the proliferation, differentiation, and survival of adult neuronal stem cells (NSCs). Besides, these receptors also play an intrinsic trophic function in supporting NSC survival, proliferation, and differentiation into immature neurons. All these events collectively ensure a sustained neurogenesis in adult hippocampus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurogenesis is a process by which new neurons differentiate from neural stem cells (Gage 2000; Emsley et al. 2005). This event was initially considered to occur only during embryonic or early postnatal development, but numerous evidence has demonstrated that it also constantly takes place in the adult brain, predominantly in two locations, i.e., the subventricular zone (SVZ) lining the lateral ventricles (Reynolds and Weiss 1992; Richards et al. 1992) and the subgranular zone (SGZ) of dentate gyrus in the hippocampal complex (Gage et al. 1995; Palmer et al. 1997). Injury or degenerative changes in the CNS may induce neurogenesis in adult brains. Newborn neurons and neuronal differentiation have been observed in other areas throughout the adult brain, such as the amygdal (Bernier et al. 2002), brainstem (St-John 1998), neocortex (Magavi et al. 2000), substantia nigra (Zhao et al. 2003), tegmentum (Hermann et al. 2006), and spinal cord (S-i et al. 2001), particularly in responses to injury or degenerative changes. Differentiation of neuronal stem cells to mature neurons is composed of several steps, from stem cell proliferation, migration, survival, commitment to neuronal lineage, and finally to the integration of the newly differentiated mature neurons into the existing circuits, even as far as to the spinal cord (Chen et al. 2004).
Adult neurogenesis in hippocampal subgranular zone is a well-studied model system. Figure 1 shows a typical hippocampal neurogenesis pattern in adult mouse brain (Fig. 1). In general, NSCs proliferate and give rise to transient amplifying (TA) progenitors that may subsequently differentiate into immature neurons, some of which migrate to the granule cell layer, where they further maturate into granular neurons. These newly differentiated neurons can receive input from the entorhinal cortex and send projections to CA3 and hilar regions (Markakis and Gage 1999; Laplagne et al. 2007; Toni et al. 2008; Yan et al. 2007). Similarly, NSCs in the SVZ can also proliferate and generate TA progenitors, which differentiate into immature neurons that subsequently migrate in chains along the rostral migratory stream (RMS) to the olfactory bulb (OB), where they differentiate into interneurons. It is well accepted that adult hippocampal neurogenesis is involved in spatial learning and memory (Deng et al. 2010; Aimone et al. 2011), while neurogenesis in the SVZ generates new neurons destined for the olfactory bulb to function in fine olfaction discrimination (Conover and Shook 2011).

Adult neurogenesis in the dentate gyrus of hippocampal complex. (a) A representative picture of dentate gyrus (DG) shows the BrdU labeled proliferative NSCs (green), doublecortin immunostained immature neurons (red), and granular cell layer (GCL) stained with DAPI (blue). (b) An enlarged picture shows details in the dashed square of (a). A newborn NSC was labeled with BrdU along the subgranular zone of the dentate gyrus (green, by open triangle), and the newly differentiated immature neurons were doubly stained with doublecortin and BrdU (white arrow). These immature neurons migrate into the granule cell layer, and differentiate into mature neurons. This pattern represents neurogenic process in adult hippocampus. Scale bar: 50 μm for (a) and 10 μm for (b)
Neurogenesis is a precisely regulated process. Many intrinsic and extrinsic regulatory mechanisms have been identified, including a number of morphogens that are critical for embryonic development of the nervous system, such as Notch (Morrison et al. 2000), Sonic hedgehog homolog (SHH) (Lai et al. 2003), Wnts (Lie et al. 2005; Wexler et al. 2009), and Bone morphogenetic proteins (BMPs) (Lillien and Raphael 2000). These regulatory systems are conserved and continue to function during the adult neurogenesis. In addition, many other neurotransmitters, growth factors, neurotrophins, cytokines, and hormones may play critical regulatory roles at different phases of adult neurogenesis. Many intrinsic factors, such as micro RNA (miRNAs), transcription factors, cell-cycle regulators, and epigenetic factors also exhibit cell-autonomous regulation of adult NSCs proliferation, differentiation, and survival (Suh et al. 2009; G-l and Song 2011).
Neurogenesis is dramatically affected by inflammatory factors (Ekdahl et al. 2003; Monje et al. 2003; Rolls et al. 2007). Microglia and astrocytes, both play roles in regulation of immune responses in the inflamed brain, are involved in the onset of inflammation and major immune defenses in the infected and damaged CNS. Microglia, like its counterpart in the peripheral immune system, macrophage, contribute to the immune responses by acting as antigen-presenting and innate immune responsive cells that secrete cytokines and other signaling molecules (Carson et al. 1998; Shrikant and Benveniste 1996). However, chronic inflammation is recognized as a major negative contributor to adult neurogenesis (Ekdahl et al. 2003; Monje et al. 2003), although there are several evidences that show the activated microglia promoting neurogenesis through secreting anti-inflammatory cytokines (Butovsky et al. 2005, 2006; Gómez-Nicola et al. 2011; Walton et al. 2006). Chronic inflammation or acute inflammation induced by infection or lipopolysaccharide (LPS) administration promotes microglial pro-inflammatory responses, which are clearly detrimental to neurogenesis (Ekdahl et al. 2003; Monje et al. 2003; Cacci et al. 2008; Iosif et al. 2006; Kuzumaki et al. 2010; Liu et al. 2005; Picard-Riera et al. 2002). We have recently demonstrated that the Tyro3, Axl and Mertk (TAM) subfamily of receptor tyrosine kinases functions as important homeostatic regulators and sets appropriate guidance for microglial response to infection and tissue damages. Mice lacking all three receptors (TAM TKO) produced increased level of pro-inflammatory cytokines, especially IL-6, which inhibits neuronal stem cell (NSC) proliferation and differentiation (Ji et al. 2013). This review will focus on the recent progress of TAM functions in glial and NSC cells and their contribution in the regulation of adult neurogenesis.
TAM receptors, their cognate ligands and functions
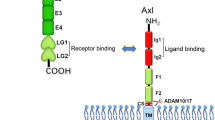
The Tyro3, Axl and Mertk proteins have been identified as a distinct subfamily of receptor-type protein tyrosine kinase (PTK) (Janssen et al. 1991; Jia and Hanafusa 1994; Lai et al. 1994; Lai and Lemke 1991; Mark et al. 1994; Rescigno et al. 1991; Taylor et al. 1995; O’Bryan et al. 1991). As shown in Fig. 2, this subfamily of proteins contains a single transmembrane domain and is expressed on the cell plasma membrane with the N-terminus located on the outside of the cell surface, serving as an extracellular ligand-binding domain. The C-terminus of protein is oriented intracellularly and harbors a large intracellular kinase domain followed by a short sequence containing a few tyrosine residues. When activated, the tyrosine residues on the C-terminal short tail are phosphorylated and act as docking sites for downstream signaling molecules (Lai et al. 1994; O’Bryan et al. 1991; Heiring et al. 2004; Sasaki et al. 2006). Two structurally close-related proteins, namely growth-arrest-specific 6 (GAS6) and protein S, have been identified as TAM receptor ligands (Stitt et al. 1995). The binding of those ligands to TAM receptors triggers receptor dimerization and activation. As a result, it causes the recruitment, phosphorylation, and activation of multiple downstream signaling proteins, which ultimately changes gene expression or biological responses.

schematic diagram of TAM receptors. Extracellular domain consists of two immunoglobulin domains (Ig) and two fibronectin type III domains (FN III) are located outside of the cell surface. Intracelluar domain is composed of a large kinase domain (KD) followed by a carboxyl terminal tail housing a few tyrosine residues. The numbers indicate the tyrosine location on the mouse Tyro3 protein
TAM receptors and their ligands are broadly expressed in immune, nervous, reproductive, and vascular systems. They were initially identified as growth trophic receptors for cell survival in numerous cell types involving many cell lineages, including mesangial and glomerular cells (Yanagita et al. 1999; Yin et al. 2002), fibroblasts (Bellosta et al. 1997; Goruppi et al. 1996), vascular smooth muscle cells (Valverde et al. 2004; Nakano et al. 1996; Son et al. 2006), endothelial cells (D’Arcangelo et al. 2002; O’Donnell et al. 1999), testicular cells (Chan et al. 2000), lens epithelial cells (Valverde et al. 2004), and peripheral macrophages (Anwar et al. 2009). The growth trophic and anti-apoptotic effects of the TAM signaling pathway have also been demonstrated in a variety of cell types in the nerve system, including neurons (Allen et al. 1999; Pierce et al. 2008; Yagami et al. 2002), Schwann cells (R-h et al. 1996), and oligodendrocytes (Binder et al. 2008; Shankar et al. 2003, 2006).
Many lines of evidence show that phosphatidylinositol-3-kinase (PI3K) is a critical signaling pathway initiated by TAM receptor activation (Konishi et al. 2004; Hasanbasic et al. 2010; Goruppi et al. 1997; Weinger et al. 2008; Chen et al. 2014). The phosphorylated TAM receptors are able to activate PI3K by either directly binding the p85 subunit of PI3K (Weinger et al. 2008; Braunger et al. 1997) or via the growth factor receptor-bound 2 (Grb2) adaptor protein to indirectly recruit and activate the PI3K (Weinger et al. 2008). The activated PI3K causes the phosphorylation of Akt or nuclear translocation of NF-κB, and eventually leads to anti-apoptosis response and cell survival. In addition, the binding of the Grb2 adaptor protein to the phosphorylated TAM receptors can also recruit the son of sevenless (SOS), which activates the extracellular signal-related kinase (Erk)-signaling pathway to promote cell proliferation (Goruppi et al. 1996, 1997, 1999; Fridell et al. 1996; Brown et al. 2012) (Fig. 3a). Such a TAM receptor-mediated signaling pathway has been found in a number of cell types including cardiac fibroblasts (Stenhoff et al. 2004), Schwann cells (R-h et al. 1996), and vascular smooth muscle cells (Benzakour et al. 1995).

Simplified TAM signaling pathways in supporting cell proliferation and survival (a) and inhibiting inflammation (b). (a) The phosphorylated TAM receptors activate phosphatidylinositol-3-kinase (PI3K) by either directly binding the p85 subunit of PI3K; or via the growth factor receptor-bound 2 (Grb2) adaptor protein to indirectly recruit and activate the PI3K, which in turn causes the phosphorylation of Akt and nuclear translocation of NF-κB, eventually leading to anti-apoptosis response and cell survival. In addition, the activation of the Grb2 adaptor protein can also recruit the son of sevenless (SOS) that subsequently activates the extracellular signal-related kinase (ERK)-signaling pathway to promote cell proliferation. (b) Activation of TAM receptors upregualtes the transcription of both suppressor of cytokine signal-ling (SOCS) 1 and 3 via interferon-α receptor (IFNAR) and transducer and activator of transcription 1 (Stat1) signal transduction pathway. As a result, the increased SOCS-1 and −3 suppress the cytokine receptor signal transduction cascades
Creation of the Tyro3, Axl or Mertk single gene knockout mice and subsequent generation of different combinations of the compound double or triple knockout mice uncovered many unexpected biological functions of TAM receptors (Scott et al. 2001; Lu et al. 1999; Lu and Lemke 2001). The major discoveries from the knockout mice are the participation of the TAM receptors in phagocytic regulation, mainly through Mertk and Axl (Scott et al. 2001; Zagorska et al. 2014; Tsou et al. 2014), and their precise regulation of both adaptive and innate immune responses (Lu and Lemke 2001; Rothlin et al. 2007; Carrera Silva et al. 2013). Mice without these three receptors exhibit systemic autoimmune disorders, due to hyperactivation of the antigen-presenting cells (APC) such as macrophage and dendritic cell (Lu and Lemke 2001; Ye et al. 2011). In these APC cells, the TAM receptors adopt a different signaling mechanisms involving the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway (Rothlin et al. 2007). Pathogenic activation of pattern recognition receptors, such as Toll-like receptors (TLRs), triggers a wide spectrum of cytokine production and, interestingly, an upregulation of TAM expression as well (Zagorska et al. 2014; Rothlin et al. 2007). These newly expressed TAM receptors stimulate transcriptional expression of suppressor of cytokine signaling (SOCS)-1 and −3, which function as negative regulators for the TLRs and cytokine receptor signaling cascades (Lemke and Rothlin 2008) (Fig. 3b). Such TAM-mediated negative regulation of cytokine/chemokine production ensures the immune system will mount appropriate innate immune responses to infection or tissue damages.
TAM receptors, especially Mertk, play an essential role in regulating phagocytic functions for many professional and non-professional phagocytes; the well-studied of which are Sertoli cells in testis, retinal pigment epithelial (RPE) cells in eye, and the macrophages and dendritic cells in immune system. Mice lacking all three TAM receptors are sterile and blind due to impaired spermatogenesis (Lu et al. 1999; Lu and Lemke 2001) and postnatal photoreceptor degeneration (Feng et al. 2002; Prasad et al. 2006). The death of developing germ cells in testis is a direct consequence of the dysfunctional Sertoli cells that are unable to perform a normal phagocytic clearance of the abandon cytoplasmic debris from the maturing germ cells (Lu et al. 1999). The blindness observed in these TAM triple knockout mice is caused by a defective Mertk-mediated phagocytic clearance of the spent photoreceptor out-segments by the adjacent RPE cells that normally express Mertk receptor (Lu et al. 1999; Lu and Lemke 2001; Prasad et al. 2006). Similar to their function in Sertoli and RPE cells, this subfamily of receptors plays a very important role in phagocytic clearance of the apoptotic cells by macrophages and dendritic cells (Scott et al. 2001; Seitz et al. 2007). Mounting evidence shows that the TAM cognate ligands, Gas6, and protein S can simultaneously bind phosphatidylserine (PS) on the surface of apoptotic cell and the TAM receptors on phagocyte, which triggers phagocytic uptake of the engulfed dead cell (Zagorska et al. 2014; Tsou et al. 2014; Nakano et al. 1997; Anderson et al. 2003). Although the molecular mechanism underlying the TAM-mediated signaling pathway in regulation of phagocytosis is not clearly elucidated, the TAM involvement in regulation of cytoskeletal rearrangement during phagocytic process is convincing. Several TAM-mediated signaling models have been proposed (Wu et al. 2005; Grommes et al. 2008; Nandrot et al. 2004; Ji et al. 2014), but most likely, these signal transduction pathways will trigger cytoskeletal reorganization and phagocytosis.
TAM receptors protect neurogenesis by maintaining BBB integrity and inhibiting peripheral inflammation
Brain is normally separated from the peripheral immune system by the brain–blood barrier (BBB), which is formed by capillary endothelial cells, a thick basement membrane, and the endfeet of astrocytes. TAM receptors, especially Tyro3 and Axl, are expressed and function on microvessel endothelial and vascular smooth muscle cells (Zhu et al. 2010; Melaragno et al. 2004; Holland et al. 2005; Korshunov et al. 2006). Loss of these receptors, the brain microvessel integrity, cause endothelial cell adhesion to be interrupted (Zhu et al. 2010; Li et al. 2013). When BBB is disrupted, it becomes more permeable and allows lymphocytes and large molecules, such as proinflammatory cytokines and neurotoxic molecules, to freely cross the BBB to impair neurogenesis in the adult brain.
On the other hand, TAM receptors play a critical regulatory role in the peripheral immune system. Loss of TAM receptors on dendritic cells (DC)s and macrophage (Seitz et al. 2007; Lemke and Lu 2003; Cohen et al. 2002), or other non-professional phagocytes (Prasad et al. 2006; Xiong et al. 2008), causes defective phagocytosis and hence, accumulation of apoptotic debris which is not normally presented. TAM receptors also negatively regulate cytokine production and without these receptors, the activated DCs and macrophages produce increased amount of proinflammatory cytokines. Constant encountering self-antigens released from apoptotic cells and unrestricted production of proinflammatory cytokines lead to chronic inflammation and systemic autoimmune disorders seen in TAM triple knockout mice (Lu and Lemke 2001; Rothlin et al. 2007; Ye et al. 2011).
Systemic or local chronic inflammation in the CNS is detrimental to normal neural functions (Lehnardt et al. 2003), as well as to the adult neurogenesis and neuronal differentiation. The bacterial LPS can provoke microglial inflammation and elicit the release of proinflammatory cytokines that affect NSC activity (Ekdahl et al. 2003; Monje et al. 2003; Carpentier and Palmer 2009), but using immunosuppressive drugs to hinder inflammation will restore hippocampal neurogenesis (Monje et al. 2003). Systemic autoimmune disorders usually produce increased level of proinflammatory cytokines and autoreactive lymphocytes, which are able to drive peripheral autoimmunity into the CNS. TNF-α has been shown to increase the permeability of brain microvascular endothelial cells (Dickstein et al. 2000; Jacob et al. 2010; Ozaki et al. 1999; Tsao et al. 2001; Wong and Dorovini-Zis 1992; Yang et al. 1999). A higher level of circulating TNF-α was found in TKO blood and caused abnormal activation of endothelial cells (Lu and Lemke 2001; Li et al. 2013). TNF-α increases BBB permeability and induces intracellular adhesion molecule (ICAM) expression on endothelial cells (Nishioku et al. 2010; McHale et al. 1999). Such cytokine-driven and exaggerated ICAM-dependent leukocyte-endothelial interactions were found in the brains of MRL–lpr mice (James et al. 2006), a mouse model with a systemic autoimmune disease that spontaneously develops a manifestation of human multiple sclerosis. In addition, CD3 positive T cells were constantly detected in the parenchymal tissue in the TKO brains. T-cell mediated inflammation has been considered as a major factor of demyelination and damage to oligodendrocytes in multiple sclerosis disorders. Disruption of the BBB in TKO mice increases permeability of proinflammatory cytokines and autoreactive T cells that cause inflammatory damage to the CNS, where the most vulnerable region is the hippocampal complex (Li et al. 2013). This is probably one important cause for the impaired neurogenesis in the TAM TKO mice. This observation is in agreement with other findings that the removal of T cells, especially the CD4 population, causes an increase in NSC proliferation, followed by an escalated improvement in functional recovery after cortical infarction (Wolf et al. 2009). It is conceivable that depletion of T cells in the TAM TKO mice might provide insights regarding the causes of the impaired neurogenesis. These observations demonstrate that systemic autoimmunity and neuroinflammation in the TAM TKO mice may contribute significantly to hippocampal damage and the inhibition of adult neurogenesis.
TAM receptors maintain neurogenesis by the negative regulation of microglia and astrocyte activation
Brain inflammation has been recognized as major negative contributor to adult neurogenesis (Monje et al. 2003; Carpentier and Palmer 2009). TAM receptors have been postulated to inhibit prolonged and unrestricted innate immune responses of macrophages and DCs by regulating expression of SOCSs or Twist proteins, which terminates cytokine receptors-mediated signaling or inhibits NF-κB transcriptional activity respectively (Rothlin et al. 2007; Lemke and Rothlin 2008; Sharif et al. 2006). As resident macrophages, microglia act as immunocompetent cells in the brain and spinal cord and are responsible for CNS protection against a variety of pathogenic factors including damaged neurons, aging–related protein aggregates, and infectious agents. However, chronic activation of microglia causes neuronal damage through the release of neurotoxic molecules, such as proinflammatory cytokines, reactive oxygen species, and complement proteins. These factors are detrimental not only to normal neural function (Lehnardt et al. 2003), but also to neurogenesis and differentiation of neuronal stem cells (NSCs) into immature neurons (Ekdahl et al. 2003, 2009; Monje et al. 2003; Rolls et al. 2007; Iosif et al. 2006; Kuzumaki et al. 2010). Microglia, expressing a wide spectrum of cytokine receptors, TLRs, and all components of the IKK-NF-κB signaling pathway (Rivest 2009; Olson and Miller 2004; Hanisch 2002) can be activated by a variety of factors including proinflammatory cytokines, TNF-α, LPS, etc. After activation, they can produce numerous proinflammatory mediators. LPS-elicited microglial inflammation has been shown to induce the release of proinflammatory cytokines affecting NSC proliferation in vitro and inhibiting hippocampal neurogenesis and neuronal differentiation in vivo (Ekdahl et al. 2003; Monje et al. 2003; Cacci et al. 2008; Iosif et al. 2006; Kuzumaki et al. 2010; Liu et al. 2005; Picard-Riera et al. 2002).
We have recently shown that microglia express all three members of TAM receptors (Ji et al. 2013). Similar to their negative regulatory role on DCs and macrophages, the TAM receptors restrain microglia from hyper response to activation. Microglia lacking TAM receptors produced increased amounts of proinflammatory cytokines upon stimulation by LPS, poly I:C, and CpG, which act through TLR4, TLR3, and TLR9 accordingly. This suggests that TAM receptors, as negative mediators, participate in the regulation of microglial innate immune responses. The conditioned medium from LPS-stimulated TAM TKO microglia cultures consistently exhibited more severe neurotoxic for NSCs, as shown by increased apoptosis, decreased proliferation, and neuronal differentiation. The major component in the TKO microglial conditioned medium was identified as IL-6, since either the IL-6 neutralizing antibody or the conditioned medium from the Il6 −/−TKO microglial culture were able to reverse the neurotoxic effects of the TKO microglial conditioned medium. Furthermore, the neurotoxic effect of the high concentration of IL-6 from the TKO microglia was further verified in vivo by knocking out the Il6 gene in the TAM TKO background, in which decreased adult hippocampal neurogenesis was seen in the TKO mice to reverse the level compatible to that of the WT control mice. This result indicates that IL-6 is a major player upregulated in the TKO microglia and renders neurotoxic to adult hippocampal neurogenesis in the TKO mice. Another proinflammatory cytokine, TNF-α, was also upregulated in the activated TKO microglia. However, neither the neutralizing antibody nor the TNF receptor knockout in the TKO background restored normal NSC proliferation and neuronal differentiation, except for the TNF-α when used as a mediator linking hyperreactive TKO microglia and halted neurogenesis.
In addition to microglia, astrocyte is another type of immune modulating cells in the CNS that is capable of producing inflammatory neurotoxic mediators (Saijo et al. 2009). Astrocytes express TLRs, IL-1β receptors (Olson and Miller 2004; Saijo et al. 2009; Carpentier et al. 2005; Gorina et al. 2011; Glass et al. 2010; Farina et al. 2007; Meng et al. 2014), and all three members of the TAM receptors (Ji et al. 2013). In response to LPS activation, the cultured TKO astrocytes released higher levels of IL-6 than the WT cells did, indicating that TAM receptors also play a negative regulatory role in astrocyte. Interestingly, since the astrocytes also express IL-1β receptor (Saijo et al. 2009), IL-1β stimulation on the TKO astrocytes elicited a stronger expression of IL-6 and IL-1β than compared to the WT cells. It is likely that the enhanced production of IL-1β by TKO microglia may further stimulate astrocytes to generate more proinflammatory cytokines detrimental to neurogenesis in adult hippocampus.
The negative impact of TAM receptors on the activated microglia and astrocytes is likely caused by the overwhelmingly activated MAP kinases in response to stimulation by pathogenic reagents. P38 has been shown to be a major MAP kinase in microglia for regulation of proinflammatory cytokine production (Bachstetter et al. 2011). TKO microglia also exhibited stronger p38 activation upon TLRs activation than did the WT cells. This enhanced signaling by p38, possibly in collaboration with other MAP kinases such as pErk1/2, is likely responsible for the increased production of proinflammatory cytokines.
TAM receptors maintain neurogenesis by supporting NSCs
As discussed above, hyperreactive microglia in the TKO mice produced increased level of proinflammatory cytokines that were detrimental to neuronal stem cell proliferation and differentiation (Ji et al. 2013). However, when compared with the number of β-tubulin III+ neurons differentiated from the TKO and WT NSCs pretreated with LPS-treated microglia-conditioned medium, the TKO NSCs showed even more decreased neuronal differentiation. The in vivo study in which the TKO brain showed further decline in regards to NSC proliferation and differentiation into the BrdU+/DCX+ neuronal progenies than did the WT brains that had undergone the LPS-induced inflammation (Ji et al. 2013) also reflected this. The data implies that TAM receptors may play an intrinsic functional role in NSC proliferation and neuronal differentiation.
Since they were originally cloned from many fast growing or transformed cells, TAM receptors were initially considered as growth trophic receptors based on their upregulated presence in these cells (Lai and Lemke 1991; O’Bryan et al. 1991). They sustain cell growth and survival and support PC12 cell neuronal differentiation upon neuronal growth factor stimulation (Zheng et al. 2009). Genome-wide analysis of the differential expressed genes in the neuronal progenitor versus the differentiated neuronal cells reveals that all three members of the TAM family are expressed in the embryonic cortical neuronal progenitor cells (Wang et al. 2011). Mice lacking both Axl and Mertk caused early differentiation and migration of the SVZ NSCs (Wang et al. 2011) and knockout of their common ligand, Gas6, reduced the NSC numbers in the SVZ (Gely‐Pernot et al. 2012). This evidence indicates that TAM receptors may play important intrinsic roles in the maintenance of the cortical neuronal progenitor cell identity, in the regulation of NSCs survival, proliferation, and differentiation.
We have recently observed that the primary culture NSC expressed all three members of the TAM receptors that provided trophic support for NSCs survival, proliferation, and differentiation into immature neurons in vitro. NSCs lacking TAM receptors manifested in slow growth, reduced proliferation, and a smaller extent of neuronal differentiation, but also in increased cell death. For the underlined mechanism, we have found the TKO NSCs expressed less NGF, but showed a compensational increased expression of TrkA, TrkB and TrkC, implying that the TAM receptors may function in coordination with neurotrophins in NSCs. According to some other studies, the ERK pathway might be the main factor TAM receptors use to regulate NSCs proliferation and differentiation. Without TAM receptors, phosphorylation of ERK will be upregulated (Ji et al. 2013) and will suppress the Krüppel-like factor (Klf4), inducing NSCs to stop self-renewal and start differentiation (Kim et al. 2012). Moreover, some studies pointed out that the ERK pathway might be important to regulating NSCs’ differentiation towards neurons (Samuels et al. 2008). Taken together, TAM receptors may regulate NSCs’ proliferation, differentiation, and survival in coordination with neurotrophins or through regulating the ERK pathway.
Summary and perspectives
In summary, TAM receptors are expressed in the CNS, including NSCs, microglia, and astrocytes. They affect adult neurogenesis by negative regulation of the glial cell hyperactivation in the CNS, maintenance of BBB integrity, and homeostatic prevention of overreactive inflammatory responses by peripheral immune system. These processes depend on cooperative interaction between TAM receptors and cytokine receptor signaling systems. Notably, they also play an intrinsic supporting role in the survival, proliferation, and differentiation of adult NSCs.
It is noteworthy that the expression patterns of three TAM members are sharply different in the brain. Tyro3 is abundantly expressed in neurons, Axl is mainly restricted to the rostral migratory stream (RMS), and Mertk transcript is widespread and likely to be glial concentrated (Prieto et al. 2000, 2007). Each member of the TAM receptors plays a unique role in regulating adult neurogenesis. However, according to our unpublished results, deletion of a single receptor is not enough to cause changes in neurogenesis. Further studies are still needed to examine the specific role for each TAM receptor. Nevertheless, it is likely that loss of TAM receptors on immunocompetent cells and on NSCs has a unique impact on different stages of neurogenesis. Enhanced inflammation affects preferentially on the NSC proliferation, whereas loss of TAM on NSCs dominantly affects the differentiation stage between the proliferating NCS to immature neurons, as summarized in Fig. 4. TAM plays such an important regulatory role in the CNS suggesting that the TAM receptor signaling system may offer a novel target for the treatment of chronic neuroinflammtion and aging-related neurodegenerative disease.

Effects of TAM triple knockout on the adult hippocampal neurogenesis. Both type 1 and type 2 NSC are able to proliferate for self-renewal; and type 2 can exit from cell cycle and differentiate into immature neuron, which further differentiate into mature neurons. (a) Loss of TAM receptors from NSCs affects proliferation and differentiation with an increased effect from proliferating stem cell stage to the neuronal differentiation into immature neurons. (b) Loss of TAM receptors on macroglia (MG) and astrocytes (AS) increases proinflammatory mediators, such as IL-6; and this effect is more dramatic on the proliferating NSCs then on the neuronal differentiation stage. (c) Loss of TAM receptors on the peripheral dendritic cells (DC) and macrophages (MΦ) enhances release of Il6 and other proinflammatory mediators that in turn inhibit neurogenesis
Chronic inflammation has been long known as a risky factor for the development of neurodegenerative diseases. Non-steroidal anti-inflammatory drugs (NSAID) have been used to attenuate inflammation in the patients with neurodegenerative diseases. Early epidemiological studies highlighted a reduced incidence of Alzheheimer’s disease (Stewart et al. 1997) and Parkinson’s disease (Samii et al. 2009; Gao et al. 2011) in NSAID users, but more recent results from clinical trials provided disappointing results (Jaturapatporn et al. 2012; Rees et al. 2011) because of cardiovascular risks (Finckh and Aronson 2005) and the timing of NSAID administration. By the time a neurodegenerative disease is diagnosed, neuronal loss has already happened. Some studies showed NSAID appeared to lessen cognitive decline in the early stages of the diseases, but it also accelerated decline in the late stages of the diseases (Breitner et al. 2011; Leoutsakos et al. 2012).
Whether or not the expression profile of TAM receptors is altered in the neurodegenerative disease patients is currently not clear. Activation of the TAM receptors by their cognate ligands, Gas6 and Protein S, is postulated to suppress inflammation and improve neurogenesis in treatment of neurodegenerative diseases. Gas6 has been shown to activate several downstream signaling pathways including MAPK/ERK, PI3K/AKT, and JAK/STAT (Linger et al. 2008). Activation of TAM receptors is a double-bladed sword: all these pathways can inhibit severe inflammation observed in neurodegenerative patients’ brains, but these pathways also exist in the activation of classic oncogenic networks (Linger et al. 2008). Therefore, the long term goal is to selectively activate TAM receptors in an attempt to reduce chronic inflammation and to promote adult neurogenesis in the treatment of aging related diseases, without activating oncogenic signal transduction networks.
Abbreviations
- NSC:
-
Neural stem cell
- DG:
-
Dentate gyrus
- TAM:
-
Tyro3, Axl, and Mertk
- TKO:
-
Triple knockout
- BrdU:
-
5-bromo-2′-deoxyuridine
References
Aimone JB, Deng W, Gage FH (2011) Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron 70:589–596
Allen MP, Zeng C, Schneider K, Xiong X, Meintzer MK et al (1999) Growth arrest-specific gene 6 (Gas6)/adhesion related kinase (Ark) signaling promotes gonadotropin-releasing hormone neuronal survival via extracellular signal-regulated kinase (ERK) and Akt. Mol Endocrinol 13:191–201
Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H et al (2003) Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol 4:87–91
Anwar A, Keating AK, Joung D, Sather S, Kim GK et al (2009) Mer tyrosine kinase (MerTK) promotes macrophage survival following exposure to oxidative stress. J Leukoc Biol 86:73–79
Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM et al (2011) Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta). J Neuroinflammation 8:79
Bellosta P, Zhang Q, Goff SP, Basilico C (1997) Signaling through the ARK tyrosine kinase receptor protects from apoptosis in the absence of growth stimulation. Oncogene 15
Benzakour O, Formstone C, Rahman S, Kanthou C, Dennehy U et al (1995) Evidence for a protein S receptor (s) on human vascular smooth muscle cells. analysis of the binding characteristics and mitogenic properties of protein S on human vascular smooth muscle cells. Biochem J 308:481–485
Bernier PJ, Bédard A, Vinet J, Lévesque M, Parent A (2002) Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc Natl Acad Sci 99:11464–11469
Binder MD, Cate HS, Prieto AL, Kemper D, Butzkueven H et al (2008) Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J Neurosci 28:5195–5206
Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, et al. (1997) Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 14
Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG et al (2011) Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement 7:402–411
Brown JE, Krodel M, Pazos M, Lai C, Prieto AL (2012) Cross-phosphorylation, signaling and proliferative functions of the Tyro3 and Axl receptors in Rat2 cells. PLoS One 7:e36800
Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M (2005) Activation of microglia by aggregated β-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-γ and IL-4 render them protective. Mol Cell Neurosci 29:381–393
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE et al (2006) Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci 31:149–160
Cacci E, Ajmone-Cat MA, Anelli T, Biagioni S, Minghetti L (2008) In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia 56:412–425
Carpentier PA, Palmer TD (2009) Immune influence on adult neural stem cell regulation and function. Neuron 64:79–92
Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ et al (2005) Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 49:360–374
Carrera Silva EA, Chan PY, Joannas L, Errasti AE, Gagliani N et al (2013) T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 39:160–170
Carson MJ, Reilly CR, Sutcliffe JG, Lo D (1998) Mature microglia resemble immature antigen‐presenting cells. Glia 22:72–85
Chan MC, Mather JP, McCray G, Lee WM (2000) Identification and regulation of receptor tyrosine kinases Rse and Mer and their ligand Gas6 in testicular somatic cells. J Androl 21:291–302
Chen J, Magavi SS, Macklis JD (2004) Neurogenesis of corticospinal motor neurons extending spinal projections in adult mice. Proc Natl Acad Sci U S A 101:16357–16362
Chen X, Ji R, Jiang X, Yang R, Liu F, et al. (2014) Iterative type I polyketide synthases involved in enediyne natural product biosynthesis. Iubmb Life
Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC et al (2002) Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med 196:135–140
Conover JC, Shook BA (2011) Aging of the subventricular zone neural stem cell niche. Aging Dis 2:49
D’Arcangelo D, Gaetano C, Capogrossi MC (2002) Acidification prevents endothelial cell apoptosis by Axl activation. Circ Res 91:e4–e12
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11:339–350
Dickstein JB, Moldofsky H, Hay JB (2000) Brain–blood permeability: TNF-alpha promotes escape of protein tracer from CSF to blood. Am J Physiol Regul Integr Comp Physiol 279:R148–R151
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100:13632–13637
Ekdahl CT, Kokaia Z, Lindvall O (2009) Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158:1021–1029
Emsley JG, Mitchell BD, Kempermann G, Macklis JD (2005) Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog Neurobiol 75:321–341
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145
Feng W, Yasumura D, Matthes MT, LaVail MM, Vollrath D (2002) Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J Biol Chem 277:17016–17022
Finckh A, Aronson MD (2005) Cardiovascular risks of cyclooxygenase-2 inhibitors: where we stand now. Ann Intern Med 142:212–214
Fridell Y, Jin Y, Quilliam LA, Burchert A, McCloskey P et al (1996) Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol Cell Biol 16:135–145
Gage FH (2000) Mammalian neural stem cells. Science 287:1433–1438
Gage FH, Coates PW, Palmer TD, Kuhn HG, Fisher LJ et al (1995) Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proc Natl Acad Sci 92:11879
Gao X, Chen H, Schwarzschild MA, Ascherio A (2011) Use of ibuprofen and risk of Parkinson disease. Neurology 76:863–869
Gely‐Pernot A, Coronas V, Harnois T, Prestoz L, Mandairon N et al (2012) An endogenous vitamin K‐dependent mechanism regulates cell proliferation in the brain subventricular stem cell niche. Stem Cells 30:719–731
Ming G-l, Song H (2011) Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70:687–702
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934
Gómez-Nicola D, Valle-Argos B, Pallas-Bazarra N, Nieto-Sampedro M (2011) Interleukin-15 regulates proliferation and self-renewal of adult neural stem cells. Mol Biol Cell 22:1960–1970
Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM (2011) Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59:242–255
Goruppi S, Ruaro E, Schneider C (1996) Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene 12:471–480
Goruppi S, Ruaro E, Varnum B, Schneider C (1997) Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3 T3 fibroblasts. Mol Cell Biol 17:4442–4453
Goruppi S, Ruaro E, Varnum B, Schneider C (1999) Gas6-mediated survival in NIH3T3 cells activates stress signalling cascade and is independent of Ras. Oncogene 18
Grommes C, Lee CD, Wilkinson BL, Jiang Q, Koenigsknecht-Talboo JL et al (2008) Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J Neuroimmune Pharm 3:130–140
Hanisch UK (2002) Microglia as a source and target of cytokines. Glia 40:140–155
Hasanbasic I, Cuerquis J, Varnum B, Blostein MD (2010) Intracellular signaling pathways involved in. J Immunol 185:5859–5868
Heiring C, Dahlback B, Muller YA (2004) Ligand recognition and homophilic interactions in Tyro3: structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J Biol Chem 279:6952–6958
Hermann A, Maisel M, Wegner F, Liebau S, Kim DW et al (2006) Multipotent neural stem cells from the adult tegmentum with dopaminergic potential develop essential properties of functional neurons. Stem Cells 24:949–964
Holland SJ, Powell MJ, Franci C, Chan EW, Friera AM et al (2005) Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res 65:9294–9303
Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S et al (2006) Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci 26:9703–9712
Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ et al (2010) C5a alters blood–brain barrier integrity in experimental lupus. FASEB J 24:1682–1688
James W, Hutchinson P, Bullard D, Hickey M (2006) Cerebral leucocyte infiltration in lupus‐prone MRL/MpJ‐faslpr mice − roles of intercellular adhesion molecule‐1 and P‐selectin. Clin Exp Immunol 144:299–308
Janssen JW, Schulz AS, Steenvoorden AC, Schmidberger M, Strehl S et al (1991) A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene 6:2113–2120
Jaturapatporn D, Isaac M, McCleery J, Tabet N (2012) Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst Rev 2
Ji R, Tian S, Lu HJ, Lu Q, Zheng Y et al (2013) TAM receptors affect adult brain neurogenesis by negative regulation of microglial cell activation. J Immunol 191:6165–6177
Ji R, Yan F, Yang R, Meng L, Ma X (2014) The preparation and evaluation of DNA microarray probes for detecting Bacillus anthracis. J Mıcrobıol Mıcrobıal Res 2:8–16
Jia R, Hanafusa H (1994) The proto-oncogene of v-eyk (v-ryk) is a novel receptor-type protein tyrosine kinase with extracellular Ig/GN-III domains. J Biol Chem 269:1839–1844
Kim MO, Kim S-H, Cho Y-Y, Nadas J, Jeong C-H et al (2012) ERK1 and ERK2 regulate embryonic stem cell self-renewal through phosphorylation of Klf4. Nat Struct Mol Biol 19:283–290
Konishi A, Aizawa T, Mohan A, Korshunov VA, Berk BC (2004) Hydrogen peroxide activates the Gas6-Axl pathway in vascular smooth muscle cells. J Biol Chem 279:28766–28770
Korshunov VA, Mohan AM, Georger MA, Berk BC (2006) Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ Res 98:1446–1452
Kuzumaki N, Ikegami D, Imai S, Narita M, Tamura R et al (2010) Enhanced IL-1beta production in response to the activation of hippocampal glial cells impairs neurogenesis in aged mice. Synapse 64:721–728
Lai C, Lemke G (1991) An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron 6:691–704
Lai C, Gore M, Lemke G (1994) Structure, expression, and activity of Tyro 3, a neural adhesion-related receptor tyrosine kinase. Oncogene 9:2567–2578
Lai K, Kaspar BK, Gage FH, Schaffer DV (2003) Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci 6:21–27
Laplagne DA, Kamienkowski JE, Espósito MS, Piatti VC, Zhao C et al (2007) Similar GABAergic inputs in dentate granule cells born during embryonic and adult neurogenesis. Eur J Neurosci 25:2973–2981
Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R et al (2003) Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A 100:8514–8519
Lemke G, Lu Q (2003) Macrophage regulation by Tyro 3 family receptors. Curr Opin Immunol 15:31–36
Lemke G, Rothlin CV (2008) Immunobiology of the TAM receptors. Nat Rev Immunol 8:327–336
Leoutsakos JMS, Muthen BO, Breitner J, Lyketsos CG (2012) Effects of non‐steroidal anti‐inflammatory drug treatments on cognitive decline vary by phase of pre‐clinical Alzheimer disease: findings from the randomized controlled Alzheimer’s disease anti‐inflammatory prevention trial. Int J Geriatr Psychiatry 27:364–374
Li Q, Lu Q, Lu H, Tian S, Lu Q (2013) Systemic autoimmunity in TAM triple knockout mice causes inflammatory brain damage and cell death. PLoS One 8:e64812
Li R-h, Chen J, Hammonds G, Phillips H, Armanini M et al (1996) Identification of Gas6 as a growth factor for human Schwann cells. J Neurosci 16:2012–2019
Lie D-C, Colamarino SA, Song H-J, Désiré L, Mira H et al (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437:1370–1375
Lillien L, Raphael H (2000) BMP and FGF regulate the development of EGF-responsive neural progenitor cells. Development 127:4993–5005
Linger RM, Keating AK, Earp HS, Graham DK (2008) TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res 100:35–83
Liu YP, Lin HI, Tzeng SF (2005) Tumor necrosis factor-alpha and interleukin-18 modulate neuronal cell fate in embryonic neural progenitor culture. Brain Res 1054:152–158
Lu Q, Lemke G (2001) Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 293:306–311
Lu Q, Gore M, Zhang Q, Camenisch T, Boast S et al (1999) Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 398:723–728
Magavi SS, Leavitt BR, Macklis JD (2000) Induction of neurogenesis in the neocortex of adult mice. Nature 405:951–955
Mark MR, Scadden DT, Wang Z, Gu Q, Goddard A et al (1994) rse, a novel receptor-type tyrosine kinase with homology to Axl/Ufo, is expressed at high levels in the brain. J Biol Chem 269:10720–10728
Markakis EA, Gage FH (1999) Adult‐generated neurons in the dentate gyrus send axonal projections to field CA3 and are surrounded by synaptic vesicles. J Comp Neurol 406:449–460
McHale JF, Harari OA, Marshall D, Haskard DO (1999) TNF-α and IL-1 sequentially induce endothelial ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J Immunol 163:3993–4000
Melaragno MG, Cavet ME, Yan C, Tai L-K, Jin Z-G et al (2004) Gas6 inhibits apoptosis in vascular smooth muscle: role of Axl kinase and Akt. J Mol Cell Cardiol 37:881–887
Meng L, Jiang X, Ji R (2014) Role of IL6 and TNFα in hippocampal neurogenesis of TAM triple knockout mice. Int J Adv Innovat Thoughts Ideas 3:109
Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302:1760–1765
Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C et al (2000) Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell 101:499–510
Nakano T, Kawamoto K, Higashino K-I, Arita H (1996) Prevention of growth arrest-induced cell death of vascular smooth muscle cells by a product of growth arrest-specific gene,< i > gas6</i> FEBS Lett 387:78–80
Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K et al (1997) Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J Biol Chem 272:29411–29414
Nandrot EF, Kim Y, Brodie SE, Huang X, Sheppard D et al (2004) Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking alphavbeta5 integrin. J Exp Med 200:1539–1545
Nishioku T, Matsumoto J, Dohgu S, Sumi N, Miyao K et al (2010) Tumor necrosis factor-α mediates the blood–brain barrier dysfunction induced by activated microglia in mouse brain microvascular endothelial cells. J Pharmacol Sci 112:251
O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B et al (1991) axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol 11:5016–5031
O’Donnell K, Harkes IC, Dougherty L, Wicks IP (1999) Expression of receptor tyrosine kinase Axl and its ligand Gas6 in rheumatoid arthritis: evidence for a novel endothelial cell survival pathway. Am J Pathol 154:1171–1180
Olson JK, Miller SD (2004) Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173:3916–3924
Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T et al (1999) Cutting edge: combined treatment of TNF-alpha and IFN-gamma causes redistribution of junctional adhesion molecule in human endothelial cells. J Immunol 163:553–557
Palmer TD, Takahashi J, Gage FH (1997) The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci 8:389–404
Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B et al (2002) Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci U S A 99:13211–13216
Pierce A, Bliesner B, Xu M, Nielsen-Preiss S, Lemke G et al (2008) Axl and Tyro3 modulate female reproduction by influencing gonadotropin-releasing hormone neuron survival and migration. Mol Endocrinol 22:2481–2495
Prasad D, Rothlin CV, Burrola P, Burstyn-Cohen T, Lu Q et al (2006) TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci 33:96–108
Prieto AL, Weber JL, Lai C (2000) Expression of the receptor protein‐tyrosine kinases Tyro‐3, Axl, and Mer in the developing rat central nervous system. J Comp Neurol 425:295–314
Prieto AL, O’Dell S, Varnum B, Lai C (2007) Localization and signaling of the receptor protein tyrosine kinase Tyro3 in cortical and hippocampal neurons. Neuroscience 150:319–334
Rees K, Stowe R, Patel S, Ives N, Breen K, et al. (2011) Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: evidence from observational studies. Cochrane Database Syst Rev 11
Rescigno J, Mansukhani A, Basilico C (1991) A putative receptor tyrosine kinase with unique structural topology. Oncogene 6:1909–1913
Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255:1707
Richards L, Kilpatrick T, Bartlett P (1992) De novo generation of neuronal cells from the adult mouse brain. Proc Natl Acad Sci 89:8591
Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9:429–439
Rolls A, Shechter R, London A, Ziv Y, Ronen A et al (2007) Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol 9:1081–1088
Rothlin CV, Ghosh S, Zuniga EI, Oldstone M, Lemke G (2007) TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131:1124–1136
Saijo K, Winner B, Carson CT, Collier JG, Boyer L et al (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137:47–59
Samii A, Etminan M, Wiens MO, Jafari S (2009) NSAID use and the risk of Parkinson’s DISEASE. Drugs Aging 26:769–779
Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K et al (2008) Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci 28:6983–6995
Sasaki T, Knyazev PG, Clout NJ, Cheburkin Y, Gohring W et al (2006) Structural basis for Gas6-Axl signalling. EMBO J 25:80–87
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R et al (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411:207–211
Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK (2007) Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol 178:5635–5642
Shankar SL, O’Guin K, Cammer M, McMorris FA, Stitt TN et al (2003) The growth arrest-specific gene product Gas6 promotes the survival of human oligodendrocytes via a phosphatidylinositol 3-kinase-dependent pathway. J Neurosci 23:4208–4218
Shankar SL, O’Guin K, Kim M, Varnum B, Lemke G et al (2006) Gas6/Axl signaling activates the phosphatidylinositol 3-kinase/Akt1 survival pathway to protect oligodendrocytes from tumor necrosis factorα-induced apoptosis. J Neurosci 26:5638–5648
Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G et al (2006) Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med 203:1891–1901
Shrikant P, Benveniste EN (1996) The central nervous system as an immunocompetent organ: role of glial cells in antigen presentation. J Immunol 157:1819–1822
Yamamoto S-I, Nagao M, Sugimori M, Kosako H, Nakatomi H et al (2001) Transcription factor expression and Notch-dependent regulation of neural progenitors in the adult rat spinal cord. J Neurosci 21:9814–9823
Son B-K, Kozaki K, Iijima K, Eto M, Kojima T et al (2006) Statins protect human aortic smooth muscle cells from inorganic phosphate-induced calcification by restoring Gas6-Axl survival pathway. Circ Res 98:1024–1031
Stenhoff J, Dahlbäck B, Hafizi S (2004) Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth of cardiac fibroblasts. Biochem Biophys Res Commun 319:871–878
Stewart WF, Kawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer’s disease and duration of NSAID use. Neurology 48:626–632
Stitt TN, Conn G, Goret M, Lai C, Bruno J et al (1995) The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80:661–670
St-John WM (1998) Neurogenesis of patterns of automatic ventilatory activity. Prog Neurobiol 56:97–117
Suh H, Deng W, Gage FH (2009) Signaling in adult neurogenesis. Annu Rev Cell Dev 25:253–275
Taylor IC, Roy S, Yaswen P, Stampfer MR, Varmus HE (1995) Mouse mammary tumors express elevated levels of RNA encoding the murine homology of SKY, a putative receptor tyrosine kinase. J Biol Chem 270:6872–6880
Toni N, Laplagne DA, Zhao C, Lombardi G, Ribak CE et al (2008) Neurons born in the adult dentate gyrus form functional synapses with target cells. Nat Neurosci 11:901–907
Tsao N, Hsu HP, Wu CM, Liu CC, Lei HY (2001) Tumour necrosis factor-alpha causes an increase in blood–brain barrier permeability during sepsis. J Med Microbiol 50:812–821
Tsou WI, Nguyen KQ, Calarese DA, Garforth SJ, Antes AL et al (2014) Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of Ligand-induced activation. J Biol Chem 289:25750–25763
Valverde P, Obin M, Taylor A (2004) Role of Gas6/Axl signaling in lens epithelial cell proliferation and survival. Exp Eye Res 78:27–37
Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM et al (2006) Microglia instruct subventricular zone neurogenesis. Glia 54:815–825
Wang J, Zhang H, Young AG, Qiu R, Argalian S et al (2011) Transcriptome analysis of neural progenitor cells by a genetic dual reporter strategy. Stem Cells 29:1589–1600
Weinger JG, Gohari P, Yan Y, Backer JM, Varnum B et al (2008) In brain, Axl recruits Grb2 and the p85 regulatory subunit of PI3 kinase; in vitro mutagenesis defines the requisite binding sites for downstream Akt activation. J Neurochem 106:134–146
Wexler EM, Paucer A, Kornblum HI, Palmer TD, Geschwind DH (2009) Endogenous Wnt signaling maintains neural progenitor cell potency. Stem Cells 27:1130–1141
Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C et al (2009) CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol 182:3979–3984
Wong D, Dorovini-Zis K (1992) Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J Neuroimmunol 39:11–21
Wu Y, Singh S, Georgescu M-M, Birge RB (2005) A role for Mer tyrosine kinase in αvβ5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci 118:539–553
Xiong W, Chen Y, Wang H, Wu H, Lu Q et al (2008) Gas6 and the Tyro 3 receptor tyrosine kinase subfamily regulate the phagocytic function of Sertoli cells. Reproduction 135:77–87
Yagami T, Ueda K, Asakura K, Sakaeda T, Nakazato H et al (2002) Gas6 rescues cortical neurons from amyloid β protein-induced apoptosis. Neuropharmacology 43:1289–1296
Yan X-B, Wang S-S, Hou H-L, Ji R, Zhou J-N (2007) Lithium improves the behavioral disorder in rats subjected to transient global cerebral ischemia. Behav Brain Res 177:282–289
Yanagita M, Ishii K, Ozaki H, Arai H, Nakano T et al (1999) Mechanism of inhibitory effect of warfarin on mesangial cell proliferation. J Am Soc Nephrol 10:2503–2509
Yang GY, Gong C, Qin Z, Liu XH, Lorris Betz A (1999) Tumor necrosis factor alpha expression produces increased blood–brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res Mol Brain Res 69:135–143
Ye F, Han L, Lu Q, Dong W, Chen Z et al (2011) Retinal self-antigen induces a predominantly Th1 effector response in Axl and Mertk double-knockout mice. J Immunol 187:4178–4186
Yin JL, Pilmore HL, Yan YQ, McCaughan GW, Bishop GA et al (2002) Expression of growth arrest-specific gene 6 and its receptors in a rat model of chronic renal transplant rejection. Transplantation 73:657–660
Zagorska A, Traves PG, Lew ED, Dransfield I, Lemke G (2014) Diversification of TAM receptor tyrosine kinase function. Nat Immunol 15:920–928
Zhao M, Momma S, Delfani K, Carlén M, Cassidy RM et al (2003) Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci 100:7925–7930
Zheng Y, Zhang L, Lu Q, Wang X, Yu F et al (2009) NGF-induced Tyro3 and Axl function as survival factors for differentiating PC12 cells. Biochem Biophys Res Commun 378:371–375
Zhu D, Wang Y, Singh I, Bell RD, Deane R et al (2010) Protein S controls hypoxic/ischemic blood–brain barrier disruption through the TAM receptor Tyro3 and sphingosine 1-phosphate receptor. Blood 115:4963–4972
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ji, R., Meng, L., Li, Q. et al. TAM receptor deficiency affects adult hippocampal neurogenesis. Metab Brain Dis 30, 633–644 (2015). https://doi.org/10.1007/s11011-014-9636-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-014-9636-y