
Abstract
Baicalin is one bioactive flavone with anti-inflammatory and neuroprotective activities. The neuroprotective effects of baicalin on pathological changes and behavioral deficits were explored in a mouse model of amyloid β (Aβ) 1–42 protein-induced Alzheimer’s disease (AD). Mice received a bilateral injection of Aβ1–42 protein into the hippocampus, then they were treated with baicalin (30, 50 and 100 mg/kg body weight, orally) or Tween 80. The therapeutic effects of baicalin were monitored by Morris water maze trial and probe test. Then mice were sacrificed for immunohistochemistry and western blot analysis. After a relatively short-term treatment of 14 days, 100 mg/kg of baicalin significantly ameliorated memory impairment in the Morris water maze test and probe test, and also attenuated glial cell activations and increase of TNF-α and IL-6 expressions induced by Aβ1–42 protein. These results suggest that baicalin ameliorated Aβ1–42 protein-related pathology and cognitive dysfunction via its anti-neuroinflammatory activity, and may be a potential candidate for the treatment of AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a multifactorial neurological disorder, clinically characterized by a progressive loss of cognitive function, neuropsychiatric and behavioral disorders (Rong et al. 2013; Wimo et al. 2003). Neuropathological examination of the brains of AD patients reveals massive accumulation of extracellular amyloid plaques composed of aggregated amyloid beta (Aβ) peptide and intraneuronal neurofibrillary tangles consisted of paired filaments of abnormally phosphorylated tau protein (Mattson 2004; Reddy et al. 2010).
Aβ peptides are generated after sequential proteolytic cleavage of APP by two different proteases, β- and γ-secretase (Querfurth and LaFerla 2010). APP is cleaved by beta-site APP cleaving enzyme-1 (BACE1) and β-secretase to produce the secreted sAPPβ ectodomain and the membrane-bound C-terminal fragment C99. Then, C99 is cleaved by γ-secretase, which releases Aβ40 and Aβ42 (Selkoe 1998). Aβ1–42 aggregates faster (Jarrett et al. 1993; Meral and Urbanc 2013), is genetically more strongly linked to AD (Sawamura et al. 2000) and forms oligomers that are more toxic (Dahlgren et al. 2002). Injection of Aβ1–42 into hippocampal region of mouse have been widely used as an animal model of Alzheimer’s disease (Lee et al. 2012; McLarnon and Ryu 2008).
Based on the “amyloid cascade hypothesis” of AD, which purports that cerebral Aβ peptide accumulation sets a neurotoxic cascade into motion (Hardy and Allsop 1991; Selkoe 2001), a great deal of focus has been directed toward anti-amyloid therapies that reduce production or enhance clearance of cerebral Aβ (Kukar et al. 2008; Schenk et al. 1999; Tan et al. 2002; Weggen et al. 2001). However, the hypothesis fails to explain how Aβ in any of its forms causes neuronal death. This is why an extension to an amyloid cascade-inflammatory hypothesis is appropriate. Chronic neuroinflammation associated with persistent glial activation is a major disease process evoked by Aβ and intimately associated with the progress of AD pathologies (Akiyama et al. 2000a; Wyss-Coray and Mucke 2002). The neuroinflammatory response could be involved in neural dysfunction, cell death and other neurodegenerative changes, ultimately creating a vicious cycle. Multiple epidemiological and animal model studies present that NSAIDs, the most widely used anti-inflammatory agents, have a substantial sparing effect on AD (Aisen et al. 2003; Choi et al. 2013; Heneka et al. 2005).
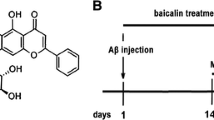
Traditional Chinese medicine (TCM) has played a critical role in the promotion of health, prevention of disease, and treatment of illness for thousands of years in China and other Asian countries (Bochorakova et al. 2003; Wang et al. 2011). Baicalin (Fig. 1) is one of the main bioactive flavone glucuronides derived as a medical herb from the dried roots of Scutellaria baicalensis Georgi, and it is widely used for the treatment of fever, inflammation, and other conditions (Zhao et al. 2013). Recent studies revealed that baicalin could protect against neuronal cell death and enhance neurological function following cerebral ischemia (Cheng et al. 2013; Jung et al. 2008; Tu et al. 2009). Even though the exact mechanism was not reported, Zhang et al. (2006) demonstrated that baicalin passed through the blood–brain barrier and distributed within brain tissue, specifically in the hippocampus, striatum, cortex, and thalamus. Baicalin was also detectable in cerebral spinal fluid in the rabbit model of brain edema (Huang et al. 1999). Thus, we hypothesized that baicalin would exhibit neuroprotection against neuronal injuries.

Molecular structure of baicalin
The present study evaluated the neuroprotective effects of baicalin on AD-like pathological changes and behavioral deficits induced by an intrahippocampal injection of Aβ1–42 protein.
Methods
Animals
50 male ICR mice (25–30 g, 6 weeks old) at the start of the experiment were used. The animals were housed in controlled room temperature (22 ± 2 °C) and humidity (50―60 %) under a 12 h:12 h light–dark cycle (lights on 06:00 h). Accessible water and food was provided ad libitum. All procedures used in the present study followed the “National Institutes of Health Guide for Care and Use of Laboratory Animals” (Publication No. 85–23, revised 1985) and were approved by the local Animal Ethics Committee. Also, Efforts were made to minimize animal suffering and to reduce the number of animal used.
Aβ1–42 protein injection
Human Aβ1–42 protein (Tocris, Ellisville, MO, USA) was prepared as stock solution at a concentration of 1 mg/ml in sterile 0.1 M phosphate-buffered saline (PBS), and aliquots were stored at −20 °C. Aβ solution was aggregated by incubation at 37 °C for 4 days before use. Mice were injected with aggregated Aβ1–42 protein into the hippocampus as described beforehand (Lee et al. 2012). In brief, mice were anesthetized with isoflurane (2.5 %). The sterilization of the injection site was carried out using gauze embedded in 70 % ethanol. A volume of 1 μl of Aβ1–42 solution was injected into the hippocampus bilaterally at the following stereotaxic coordinates using Hamilton's microsyringe: anteroposterior, −2.00 mm from bregma; mediolateral, ±1.50 mm from midline; and dorsoventral, −1.00 mm from dura. The needle was removed after 5 min using three intermediate steps with a 1-min inter-step delay to minimize backflow. Mice were placed on a thermal pad (32–33 °C) until they awakened (Cai et al. 2011).
Drug treatment
Baicalin was purchased from Ankang Dongke Maidisen Nature Pharmaceutical Co., Ltd. (Shaanxi, P.R. China). It is suspended in Tween 80, which is a nanostructured carrier that has been demonstrated to improve oral bioavailability of baicalin and prolong its retention time in blood (Zhao et al. 2013).
50 mice were randomly divided into 5 groups; In baicalin 30 (n = 10), baicalin 50 (n = 10), and baicalin 100 (n = 10) groups, baicalin was dissolved in 10 % Tween 80 and was administrated immediately after Aβ1–42 protein injection and once daily for 14 days at a concentration of 30, 50 and 100 mg/kg body weight by gavage, respectively. The doses were based on some previous reports (Liu et al. 2013; Xue et al. 2010), which were proved to be safe in the mouse model of neurologic diseases. In control group, animals (n = 10) were injected with Aβ1–42 protein, and received 10 % Tween 80 solution orally. 10 sham animals were injected with the same amount of sterile saline (1 μl), and received 10 % Tween 80 solution orally.
Morris Water Maze Test
The Morris water maze procedure was used for evaluating the memory of mouse. The experimental apparatus consisted of a circular tank (diameter = 120 cm, height = 50 cm) that was divided into four quadrants, filled with water and maintained at 22 ± 2 °C. At first, a visible platform test was performed, which confirmed that there were no significant differences in sensory, motor or motivational activities among these five groups. Then, hidden platform and reverse hidden platform tests were performed in succession. For the hidden platform test, a round platform (diameter = 9 cm) was placed at the midpoint of the fourth quadrant, 2 cm below the water surface. A training trial was conducted once a day for 5 days. During each trial, the mice were placed in the water at a fixed position, opposite the platform and at the edge of the pool. The mice were allowed to swim freely until they escaped onto the platform. In each trial, the swimming pathway and latency locating the hidden platform was recorded with a camera. The reverse hidden platform test was identical to the hidden platform test except the locations of the platform and the swim start were reversed; the reverse hidden platform trial lasted for 3 days.
Probe test
To assess memory consolidation, a probe test was performed 24 h after the Morris water maze test. For the probe test, the platform was removed and the mice were allowed to swim freely. The swimming pattern of every mouse was recorded with a camera. Consolidated spatial memory was estimated by the time spent in the target quadrant area.
Immunohistochemistry
Mice were deeply anesthetized with ether and sacrificed. Their brains were taken and post-fixed in PBS containing 4 % paraformaldehyde overnight at 4 °C. Post-fixed brains were cut into two hemispheres; hemispheres were embedded in paraffin, serially sectioned (3 μm) and mounted on silan-covered slides. Immunohistochemistry was carried on these sections using Iba-1 antibody (1:200; Abcam, Cambridge, UK) for activated microglia and GFAP antibody (1:200; santa cruz, Dallas, USA) for activated astrocytes. Following quenching of endogenous peroxidase with 1.5 % hydrogen peroxide in methanol (v/v) for 20 min, high temperature antigen retrieval was performed by immersion of the slides in a water bath at 95–98 °C in 10 mM trisodium citrate buffer pH 6.0, for 45 min. After overnight incubation at 4 °C with primary antibodies, the slides were washed with PBS and incubated with the appropriate biotinylated secondary antibody (Abcam, Cambridge, UK) for 50 min at room temperature. The sections were washed in PBS, and the detection was performed with horseradish peroxidase–streptavidin solution (Abcam, Cambridge, UK). After 30 min of incubation at room temperature, sections were washed with PBS and the detection was completed by using DAB (3,30-diaminobenzidine, Abcam, Cambridge, UK) in chromogen solution, followed by counter-staining with Harris’s hematoxylin. Control and experimental tissues were placed on the same slide and processed under the same conditions (Passos et al. 2010).
Image analysis
After immunostaining, hemispheres sections were examined by light microscopy (Nikon, Shanghai, China). Iba-1 and GFAP immunostaining were evaluated at cross-sections of hemispheres, especially focused on hippocampus. Iba-1+ and GFAP+ cells in hippocampus were counted by cellular nuclear.
Western blot analysis
Western blot analysis were carried out to detect the expression of TNF-α and IL-6 in the brains. Isolated hippocampal tissues from both hemispheres were lysed briefly with protein lysis buffer (50 mmol/l Tris–HCl, pH 7.4, 150 mmol/l NaCl, 1 % NP-40, 0.1 % sodium dodecyl sulfate, 1 mmol/l dithiothreitol, 0.01 g/l aprotinin, 0.01 g/l leupeptin, 1 mmol/l phenylmethylsulfonyl fluoride) and lysed in 4 °C for 1 h. Samples of homogenates were injected to SDS-PAGE (8 % gel). Proteins were transferred to PVDF membranes in transfer buffer [25 mM Tris–HCl buffer (pH 7.4) containing 192 mM glycine and 20 % v/v methanol] at 400 mA for 2 h at 4 °C. Western blots were incubated for 3 h with a blocking solution (5 % skim milk) at room temperature followed by incubation in anti-TNF-α and anti-IL-6 antibody for 24 h at 4 °C. Then, it was incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Each membrane was developed using a chemiluminescence system Fluorchem HD2 (Alpha, Innotech Corporation, Miami, FL, USA). The blots were stripped and incubated with an anti-β-actin antibody. TNF-α and IL-6 expression levels were normalized to the actin levels on the same membranes.
Statistical analysis
The distribution of all continuous variables that followed a normal Gaussian distribution are presented as mean ± SEM and compared using one-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. Repeated measure analysis of variance would be conducted for evaluating the difference among groups over time in the Morris water maze test. Statistical significance was defined as P values < 0.05. All statistical analyses were performed with the SPSS statistical software program package (SPSS version 15.0 for windows, SPSS Inc., Chicago, Illinois, USA).
Results
Baicalin ameliorated Aβ1–42 protein-induced cognitive dysfunction
In the hidden platform test, the escape latency time (Fig. 2a) was dependent on both the time effect (P < 0.05) and the group effect (P < 0.01); the sham and baicalin 100 groups escaped significantly faster than the control group (both P < 0.05). A similar result was observed for the swimming distance (P < 0.05 for the time effect; P < 0.01 for the group difference; both P < 0.01 for the comparisons of the sham group and baicalin 100 group to the control group. Fig. 2b). Swimming speed was not significantly different among these groups (Fig. 2c).

Effects of baicalin on cognitive function evaluated with the Morris water maze test. Escape latency time, swimming distance and swimming speed from the hidden platform test (a, b and c, respectively) and the reverse hidden platform test (d, e and f, respectively) were shown. *P < 0.05 compared to control group
In the reverse hidden platform test, the time effect and difference among these groups were both significant in the escape latency time (P < 0.01 and P < 0.01 respectively, Fig. 2d) and swimming distance (P < 0.05 and P < 0.01 respectively, Fig. 2e). Compared to control group, the escape latency time and swimming distance in the sham group and baicalin 100 group were significantly shorter (all P < 0.01). No significant differences in the swimming speed were observed (Fig. 2f).
After the Morris water maze test, we conducted a probe test to analyze maintenance of memory. During the probe test, the time spent in the target quadrant by the mice treated with baicalin 100 was significantly increased compared with control mice (Fig. 3, P < 0.01).

Effect of baicalin on improvement of memory impairment evaluated with the probe test. 24 h after the Morris water maze, a probe test was performed and the time spent in the target quadrant by the mice treated with 100 mg/kg of baicalin was significantly increased. *P < 0.05 compared to control group
However, the changes in the water maze test and in the probe test in baicalin 30 and baicalin 50 groups were not significant. Thus, we adopted 100 mg/kg of baicalin for further studies.
Baicalin attenuated Aβ1–42 protein induced glia activation
In this present study, HE staining was used for identifying the different macroscopic pathological features in the hippocampus. Representative coronal sections from hippocampus were showed in Fig. 4. The injection of Aβ1–42 protein increased the number of Iba-1 and GFAP positive cells. After 14 days treatment of baicalin at the concentration of 100 mg/kg, the number of Iba-1+ microglia was reduced, compared to control group (P < 0.01, Fig. 4d). Similar results were observed in astrocytes, which were detected by an anti-GFAP antibody. It is obvious that administration of 100 mg/kg of baicalin significantly attenuated astrocyte activation (P < 0.01, Fig. 4h).

Effects of baicalin on pathological changes in the hippocampus of mice after Aβ1–42 injection. a-c photomicrographs of Iba-1 positive cells in the hippocampus of sham, control and baicalin 100 group; d Numbers of Iba-1 positive microglia in sham and baicalin 100 groups were lower than that in control group; e-g GFAP staining in the hippocampus of sham, control and baicalin 100 group; h The bar charts showed that the number of GFAP positive astrocytes in the sham and baicalin 100 groups was lower than that in control group. *P < 0.05 compared to control group
Baicalin administration reduced Aβ1–42 protein induced proinflammatory cytokines expression
To further identify the effect of baicalin on hippocampal neuroinflammation involved in AD, the expression of pro-inflammatory cytokines were examined. It has been demonstrated that the expression pro-inflammatory cytokines would increase significantly in the amyloid β1–42-induced mice. Our results showed that TNF-α (P < 0.01, Fig. 5a) and IL-6 (P < 0.05, Fig. 5b) were significantly reduced in baicalin 100 group, compared to control group.

Effects of baicalin on the expressions of pro-inflammatory cytokines. The bar charts showed that TNF-α a and IL-6 b levels in the sham and baicalin 100 groups were significantly decreased, compared to control group. *P < 0.05 compared to control group
Discussion
The inflammatory reaction induced by Aβ involves the release of damaging factors such as TNF-α and IL-6, which promote activation of intracellular pathways that contribute to the progression of AD (Walsh and Selkoe 2004). The development of therapies for this neurodegenerative disorder represents a major challenge to academic, biotechnology, and pharmaceutical researchers. Epidemiological data demonstrated that flavonoids might reduce the risk rate for the incidence of AD. Meanwhile, the anti-inflammatory ability of the flavonoid family have been considered to provide rational for this hypothesis (Letenneur et al. 2007). Baicalin, a flavone derived from the dried roots of Scutellaria baicalensis Georgi, has multiple effects such as antioxidant, anticancer and anti-inflammatory effects (Ikemoto et al. 2000; Li et al. 2000; Shieh et al. 2000). Moreover, recent studies revealed that baicalin possessed neuroprotective effects in the ischemic cerebral injury (Xue et al. 2010; Zhou et al. 2013), which raised the possibility of using baicalin as a potential agent for neurodegenerative disorders such as AD. Our results firstly demonstrated that 100 mg/kg of baicalin treatment might ameliorated memory impairment, and also attenuated glial cell activations and increase of TNF-α and IL-6 expressions induced by Aβ1–42 protein.
Previous studies have demonstrated that Aβ peptide administrated induced behavioural changes specific to learning and memory (Colaianna et al. 2010; Passos et al. 2010). In the hidden platform and reverse platform tests, the escape latency time and swim distance both significantly decreased as the training proceeded, indicating an ongoing spatial learning and memory in the mice; however, the extent of the decreases in the baicalin 100 group was significantly higher in comparison to control group. There was no significant difference in the swimming speed among these groups, which indicates that 100 mg/kg of baicalin might attenuate the Aβ1–42 induced impairment in spatial learning and memory. In the probe test, the time spent in the target quadrant was also significantly increased in the mice treated with 100 mg/kg of baicalin. Hippocampus is one of the pivotal regions of learning and memory, especially temporary memory, and its injury is recognized as a key procedure for AD (Reilly et al. 2003). Thus, the effects of 100 mg/kg of baicalin on the pathological changes of hippocampus would be examined.
Previous studies demonstrated that increased inflammatory components contributed to the progression and acceleration of AD by activating plaque-associated microglia and astrocytes. It is now well established that most types of injury or pathological process within the CNS lead to activation of those glia from their resting state (Gao and Hong 2008; Kreutzberg 1996). Microglial and astrocyte activation have been previously demonstrated in the brains of AD patients (Akiyama et al. 2000b). Therefore, the inhibition of microglia and astrocyte is a therapeutic target for the treatment of neurodegenerative disorders. In this study, the numbers of Iba-1 positive microglia and GFAP positive astrocytes were significantly decreased by 100 mg/kg of baicalin administration, indicating that baicalin attenuated Aβ1–42 protein-induced activation of microglia and astrocytes in hippocampus.
In general, under physiological conditions, pro-inflammatory cytokines such as TNF-α and IL-6 are absent or expressed at very low level in the brain. However, they can be induced by amyloid peptide in astrocytes, microglia, neurons and endothelial cells, leading to neurodegeneration. The inflammation is mediated by pro-inflammatory cytokines and would create a chronic and self-sustaining inflammatory interaction between activated microglia and astrocytes, stressed neurons and Aβ plaques (Rubio-Perez and Morillas-Ruiz 2012). In the present study, we found that 100 mg/kg of baicalin statistically reduced the intracellular expressions of TNF-α and IL-6 in the hippocampus of the mouse brain.
In conclusion, our results demonstrated that 100 mg/kg of baicalin restored Aβ1–42 protein-induced cognitive function, attenuated the activation of microglia and astrocytes, and reduced the expression of proinflammatory cytokines, suggesting that baicalin might be a potential therapeutic option of AD and related neurodegeneration. However, as the mouse model we used can not fully represent human AD pathology and some additional elements such as diet, social as well as environmental factors is difficult to mimic in animals, the bioavailability extend of baicalin is still need to be evaluated in further research.
References
Aisen PS et al (2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289:2819–2826. doi:10.1001/jama.289.21.2819
Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K (2000a) Cell mediators of inflammation in the Alzheimer disease brain Alzheimer disease and associated disorders 14 Suppl 1:S47-53
Akiyama H et al (2000b) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Bochorakova H, Paulova H, Slanina J, Musil P, Taborska E (2003) Main flavonoids in the root of Scutellaria baicalensis cultivated in Europe and their comparative antiradical properties. Phytother Res 17:640–644. doi:10.1002/ptr.1216
Cai M et al (2011) Neuroprotective effects of a traditional herbal prescription on transient cerebral global ischemia in gerbils. J Ethnopharmacol 138:723–730. doi:10.1016/j.jep.2011.10.016
Cheng F et al. (2013) Baicalin’s Therapeutic Time Window of Neuroprotection during Transient Focal Cerebral Ischemia and Its Antioxidative Effects In Vitro and In Vivo Evidence-based complementary and alternative medicine : eCAM 2013:120261 doi:10.1155/2013/120261
Choi SH et al (2013) Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer’s disease. J Neurochem 124:59–68
Colaianna M et al (2010) Soluble beta amyloid (1–42): a critical player in producing behavioural and biochemical changes evoking depressive-related state? Br J Pharmacol 159:1704–1715. doi:10.1111/j.1476-5381.2010.00669.x
Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ (2002) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J biol Chem 277:32046–32053. doi:10.1074/jbc.M201750200
Gao HM, Hong JS (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 29:357–365. doi:10.1016/j.it.2008.05.002
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Heneka MT et al (2005) Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain :J neurol 128:1442–1453
Huang Z, Yang Y, Li X, Chen X (1999) Plasma and cerebrospinal fluid pharmacokinetics of baicalin in the rabbit model of brain edema. Chin J Contemp Paediatr 11:146–148
Ikemoto S, Sugimura K, Yoshida N, Yasumoto R, Wada S, Yamamoto K, Kishimoto T (2000) Antitumor effects of Scutellariae radix and its components baicalein, baicalin, and wogonin on bladder cancer cell lines. Urology 55:951–955
Jarrett JT, Berger EP, Lansbury PT Jr (1993) The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochem 32:4693–4697
Jung SH, Kang KD, Ji D, Fawcett RJ, Safa R, Kamalden TA, Osborne NN (2008) The flavonoid baicalin counteracts ischemic and oxidative insults to retinal cells and lipid peroxidation to brain membranes. Neurochem Int 53:325–337. doi:10.1016/j.neuint.2008.09.004
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318
Kukar TL et al (2008) Substrate-targeting gamma-secretase modulators. Nature 453:925–929. doi:10.1038/nature07055
Lee HE et al (2012) Neuroprotective effect of sinapic acid in a mouse model of amyloid beta (1–42) protein-induced Alzheimer’s disease. Pharmacol Biochem Behav 103:260–266
Letenneur L, Proust-Lima C, Le Gouge A, Dartigues JF, Barberger-Gateau P (2007) Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol 165:1364–1371
Li BQ et al (2000) The flavonoid baicalin exhibits anti-inflammatory activity by binding to chemokines. Immunopharmacol 49:295–306
Liu Y, Chi L, Gao F, Meng X, Zhang Y, Jiang W (2013) Protective effect of baicalin against convulsion induced by pentylenetetrazol in mice Tianjin. J Tradit Chin Med 30:492–494
Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430:631–639. doi:10.1038/nature02621
McLarnon JG, Ryu JK (2008) Relevance of abeta1-42 intrahippocampal injection as an animal model of inflamed Alzheimer’s disease brain. Curr Alzheimers Res 5:475–480
Meral D, Urbanc B (2013) Discrete molecular dynamics study of oligomer formation by N-terminally truncated amyloid beta-protein. J Mol Biol 425:2260–2275. doi:10.1016/j.jmb.2013.03.010
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease The New England. J Med 362:329–344. doi:10.1056/NEJMra0909142
Passos GF et al (2010) Involvement of phosphoinositide 3-kinase gamma in the neuro-inflammatory response and cognitive impairments induced by beta-amyloid 1–40 peptide in mice. Brain Behav Immun 24:493–501
Reddy PH, Manczak M, Mao P, Calkins MJ, Reddy AP, Shirendeb U (2010) Amyloid-beta and mitochondria in aging and Alzheimer’s disease: implications for synaptic damage and cognitive decline Journal of Alzheimer’s disease : JAD 20 Suppl 2:S499-512 doi:10.3233/JAD-2010-100504
Reilly JF et al (2003) Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc National Acad Sci United States of Am 100:4837–4842. doi:10.1073/pnas.0330745100
Rong Z, Pan R, Xu Y, Zhang C, Cao Y, Liu D (2013) Hesperidin pretreatment protects hypoxia-ischemic brain injury in neonatal rat Neuroscience doi:S0306-4522 (13) 00802-610.1016/j.neuroscience.2013.09.030
Rubio-Perez JM, Morillas-Ruiz JM (2012) A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal 2012:756357
Sawamura N et al (2000) Mutant presenilin 2 transgenic mice. A large increase in the levels of Abeta 42 is presumably associated with the low density membrane domain that contains decreased levels of glycerophospholipids and sphingomyelin. J Biol Chem 275:27901–27908
Schenk D et al (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177. doi:10.1038/22124
Selkoe DJ (1998) The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Biol 8:447–453
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Shieh DE, Liu LT, Lin CC (2000) Antioxidant and free radical scavenging effects of baicalein, baicalin and wogonin. Anticancer Res 20:2861–2865
Tan J et al (2002) Role of CD40 ligand in amyloidosis in transgenic Alzheimer’s mice. Nat Neurosci 5:1288–1293
Tu XK, Yang WZ, Shi SS, Wang CH, Chen CM (2009) Neuroprotective effect of baicalin in a rat model of permanent focal cerebral ischemia. Neurochem Res 34:1626–1634. doi:10.1007/s11064-009-9953-4
Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44:181–193. doi:10.1016/j.neuron.2004.09.010
Wang CZ, Mehendale SR, Calway T, Yuan CS (2011) Botanical flavonoids on coronary heart disease. The American journal of Chinese medicine 39:661–671. doi:10.1142/S0192415X1100910X
Weggen S et al (2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414:212–216
Wimo A, Winblad B, Aguero-Torres H, von Strauss E (2003) The magnitude of dementia occurrence in the world. Alzheimer Dis Assoc disord 17:63–67
Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35:419–432
Xue X et al (2010) Baicalin attenuates focal cerebral ischemic reperfusion injury through inhibition of nuclear factor kappaB p65 activation. Biochem Biophys Res Commun 403:398–404
Zhang L, Xing D, Wang W, Wang R, Du L (2006) Kinetic difference of baicalin in rat blood and cerebral nuclei after intravenous administration of Scutellariae Radix extract. J Ethnopharmacol 103:120–125. doi:10.1016/j.jep.2005.07.013
Zhao L, Wei Y, Huang Y, He B, Zhou Y, Fu J (2013) Nanoemulsion improves the oral bioavailability of baicalin in rats: in vitro and in vivo evaluation. Int J Nanomedicine 8:3769–3779. doi:10.2147/IJN.S51578
Zhou QB, Duan CZ, Jia Q, Liu P, Li LY (2013) Baicalin attenuates focal cerebral ischemic reperfusion injury by inhibition of protease-activated receptor-1 and apoptosis Chinese. J Integr Med. doi:10.1007/s11655-013-1441-7
Acknowledgements
This work was supported by the National Nature Scientific Fund of China (81,271,392, 81,301,050) and the Fund of Logistics University of Chinese People’s Armed Police Forces (WHTD201306-1). The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Chong Chen and Xiaohong Li contributed equally to this study.
Rights and permissions
About this article

Cite this article
Chen, C., Li, X., Gao, P. et al. Baicalin attenuates Alzheimer-like pathological changes and memory deficits induced by amyloid β1–42 protein. Metab Brain Dis 30, 537–544 (2015). https://doi.org/10.1007/s11011-014-9601-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-014-9601-9