
Abstract
Ground squirrel torpor during winter hibernation is characterized by numerous physiological and biochemical changes, including alterations to fuel metabolism. During torpor, many tissues switch from carbohydrate to lipid catabolism, often by regulating key enzymes within glycolytic and lipolytic pathways. This study investigates the potential regulation of pyruvate kinase (PK), a key member of the glycolytic pathway, within the skeletal muscle of hibernating ground squirrels. PK was purified from the skeletal muscle of control and torpid Richardson’s ground squirrels, and PK kinetics, structural stability, and posttranslational modifications were subsequently assessed. Torpid PK displayed a nearly threefold increase in K m PEP as compared to control PK when assayed at 5 °C. ProQ Diamond phosphoprotein staining as well as phospho-specific western blots indicated that torpid PK was significantly more phosphorylated than the euthermic control. PK from the torpid condition was also shown to possess nearly twofold acetyl content as compared to control PK. In conclusion, skeletal muscle PK from the Richardson’s ground squirrel may be regulated posttranslationally between the euthermic and torpid states, and this may inhibit PK functioning during torpor in accordance with the decrease in glycolytic rate during dormancy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pyruvate kinase (PK; 2.7.1.40) is the cytosolic enzyme that catalyzes the irreversible and terminal reaction of the glycolytic pathway within mammalian cells. It mediates the transfer of the phosphate group on phosphoenolpyruvate to ADP producing ATP and pyruvate, which is typically shuttled into the mitochondria for further oxidation. Being at a point of energy production within the cell, PK is a common regulatory site in many mammalian systems, rat liver [1, 2] and mouse liver [3] PK being regulated by reversible phosphorylation. However, this form of regulation has not been shown previously in normal mammalian muscle cells.
Reversible phosphorylation and other posttranslational modifications ensure that the flux of cellular metabolites can be quickly and tightly controlled in response to changing environmental conditions. An extreme example of this is the significant metabolic reorganization necessary for survival during mammalian winter hibernation. Hibernation consists of extended bouts of torpor where body temperature (T b) drops to near ambient (2–10 °C) and metabolic rate drops to as much as 2–4% of the normal rates [4]. During these dormant periods, physiological processes are slowed greatly or stopped completely, including heart rate dropping from 200–300 to 3–5 beats per minute [5], breathing is reduced from 100–200 to 4–6 breaths/min [6], and renal function typically ceases [5]. These processes increase to near-euthermic levels during the many short (12–24 h) arousal periods that interrupt the periods of torpor.
Of particular importance to this study are the many molecular changes that occur during torpor. In order to conserve the fixed amount of metabolic fuel and maintain a deep hypometabolic state, ATP-consuming processes, such as protein synthesis [7] and ion transport via ion-motive ATPases [8], are typically suppressed during hibernation. Furthermore, fuel utilization during dormancy changes to feature lipid catabolism rather than carbohydrate catabolism as the energy source during torpor [9, 10]. This suppression of glucose metabolism during hibernation is thought to be mediated by the posttranslational modification of key enzymes within the glycolytic pathway [11]; however, this regulation appears to be tissue specific. For instance, while many mammalian glycolytic enzymes are known to be regulated by reversible phosphorylation, few mammalian skeletal muscle glycolytic enzymes have been found to be controlled by reversible phosphorylation. This study proposes that skeletal muscle PK from the hibernating ground squirrel, Urocitellus richardsonii, is regulated posttranslationally between the euthermic and torpid states, with increased PK phosphorylation and/or acetylation acting to decrease PK functioning during torpor.
Materials and methods
Animals
Richardson’s ground squirrels were captured in the summer near Calgary, Alberta and held in an animal care facility at the University of Calgary. The squirrels were housed at 22 °C in separate cages and exposed to a fall photoperiod (10-h light: 14-h dark) for an 8-week feeding period to optimize their body lipid reserves. Following this time, some squirrels were removed from their cages and placed in a cold room (maintained at 4 °C) without food but with access to water. This room was held in a state of constant darkness for the duration of the experiments. Squirrels were allowed to enter hibernation, and after 1 month of torpor cycles, they were sampled in their last cycle after a minimum of 2 days of constant torpor (rectal temperature 6.4–7.4 °C); the minimum length of continuous torpor was confirmed visually by monitoring the disturbance of sawdust bedding. Euthermic squirrels, which were fed and maintained at a room temperature of 22 °C, remained alert and active prior to sampling and had rectal temperatures between 36 and 37 °C. At the time of sampling, all squirrels were decapitated and tissues were removed and frozen in liquid nitrogen. Samples were then sent to Carleton University where they were subsequently stored at − 80 °C.
PK purification
Frozen skeletal muscle samples were homogenized using a Polytron Homogenizer 1:5 in buffer A (25 mM MES, pH 6, 1.25 mM EDTA, 1.25 mM EGTA, 12.5 mM β-glycerophosphate (β-GP), 5 mM β-mercaptoethanol, and 5% glycerol). Prior to homogenization, a few crystals of phenylmethylsulfonyl fluoride (PMSF) were added to inhibit serine/threonine proteases. Homogenates were centrifuged at 13,500×g at 5 °C, after which the supernatant was decanted and held on ice.
Following homogenization and centrifugation, carboxymethyl (CM−) Sephadex beads were used to fill a column to the approximate dimensions of 3 × 1.8 cm (h × d). These beads were subsequently equilibrated in 15 mL of buffer A and 2 mL of crude muscle extract was added onto the column. After allowing the extract to seep into the column, the column was washed with 30 mL of buffer A and fractions were collected using a Gilson FC203B Fraction Collector. An endpoint spectrophotometric reading of the wash fractions at 280 nm indicated that this was sufficient to clear the column of any unbound protein. PK was then eluted using a 0–1 M KCl gradient (KCl solution was made in buffer A). Fractions were assayed for PK activity and the four most active fractions were pooled (~ 2 mL) and applied to a similarly sized column of blue-agarose beads pre-equilibrated in buffer B (25 mM imidazole, pH 7, 1.25 mM EDTA, 1.25 mM EGTA, 12.5 mM β-GP, 5 mM β-mercaptoethanol, and 5% glycerol).
After allowing the pooled fractions from the CM− Sephadex column to seep into the blue-agarose column, the beads were washed with 30 mL of buffer B. Again, this was a sufficient amount of washing to rid the column of unbound protein, as determined by assay at 280 nm. PK was again eluted with a 0–1 M KCl gradient (KCl solution was made in buffer B) and subsequently assayed under V max conditions.
PK assay
PK activity was monitored spectrophotometrically at 340 nm using a Thermo Labsystems Multiskan spectrophotometer. The optimal conditions for muscle PK were 1 mM phosphoenolpyruvate (PEP), 2 mM ADP, 0.15 mM NADH, 1 Unit of lactate dehydrogenase (LDH) activity (Sigma, St. Louis, MO, USA), 5 mM MgCl2, 50 mM KCl, and 50 mM imidazole, pH 7. The fraction with the most activity was diluted 10× in buffer B to obtain an appropriate amount of enzyme activity to characterize PK kinetics. Reactions were initiated by adding 5 µL of dilute purified PK in a total volume of 200 µL. Enzyme activity is in U/mg of purified protein.
K m values were determined at optimal cosubstrate concentrations. K a and I 50 values associated with the effects of various metabolites and salts (Mg-ATP, alanine, aspartate, fructose-1,6-bisphosphate (F16P2), MgCl2, and KCl) on PK activity were determined at suboptimal cosubstrate concentrations (0.05 mM PEP, 0.3 mM ADP, 0.15 mM NADH, and 1 Unit of LDH activity).
Temperature effects on PK
For assays at 5 °C, the spectrophotometer, purified enzyme sample, and microplates prepared for the desired assay (without the purified PK) were placed into a temperature-controlled incubator previously cooled to 5 °C. A random microplate well was monitored by a telethermometer until the solution reached 5 °C, during which 10 µL of purified (undiluted) PK was quickly added to the appropriate microplate wells to begin the assay. Following the assay, the temperature of the same microplate well was reassessed by a telethermometer to determine if there was any temperature change during the assay.
Similarly, for assays at 36 °C microplates were prepared (without purified PK) and placed on an EchoTherm chilling/heating plate (Torrey Pines Scientific, Carlsbad, CA, USA) set to 37 °C. In the meantime, the Thermo Labsystems spectrophotometer was set to heat the microplate measurement chamber to 37 °C. Once the temperature of the measurement chamber and a random microplate well reached the desired temperature, 5 µL of 10× diluted purified PK was added to each well (PK was held at room temperature (~ 25 °C) prior to the assay). The temperature of a random microplate well was quickly checked after the addition of enzyme and following the assay to ensure assay temperature stability.
Physical stability of PK
Investigation into the physical stability of PK from euthermic and torpid conditions was undertaken by exposing purified PK to various concentrations of known denaturants, urea, and guanidine hydrochloride. The concentrations used depended on the assay temperature and the denaturant, with urea concentrations ranging from 0 to 2 M for assays at 5 °C and RT, and 0 to 1 M for assays at 35 °C. Guanidine hydrochloride, on the other hand, ranged in concentration from 0 to 0.75 M at 5 °C and RT, and 0 to 0.2 M for assays at 35 °C. Purified extracts were incubated with the denaturants for 24 h prior to assaying for maximal PK activity.
SDS polyacrylamide gel electrophoresis and immunoblotting
SDS resolving gels [10% v/v acrylamide, 400 mM Tris, pH 8.8, 0.1% w/v SDS, 0.2% w/v ammonium persulfate (APS), 0.04% v/v tetramethylethylenediamine (TEMED)] were prepared with a 5% stacking gel (5% acrylamide, 190 mM Tris, pH 6.8, 0.1% w/v SDS, 0.15% w/v APS, 0.1% v/v TEMED). Purified euthermic and torpid PK were loaded onto these gels and separated electrophoretically in SDS-PAGE running buffer (25 mM Tris-base, 190 mM glycine, and 0.1% w/v SDS) at 180 V for 45 min. 3 µL of Spectra™ Multicolor Broad Range Protein Ladder was added to one lane of every gel to act as molecular weight markers. Following electrophoresis, proteins were electroblotted onto polyvinylidene difluoride (PVDF) membranes (Millipore) by wet transfer using transfer buffer (25 mM Tris, pH 8.5, 192 mM glycine, and 20% v/v methanol). The electroblotting was run at room temperature for 1.5 h at 160 mA.
Following protein transfer, the PVDF membranes were incubated overnight at 4 °C with either: (1) pan-acetyl primary antibody (Santa Cruz Biotechnology), (2) phosphoserine primary antibody (Calbiochem), (3) phosphotyrosine primary antibody (Cell Signalling Technology), or (4) anti-ubiquitin primary antibody (BIOMOL affiniti). All primary antibodies were diluted 1:1000 in Tris-buffered saline with Tween-20 (TBST; 20 mM Tris-base, 140 mM NaCl, 0.05% Tween-20) with the addition of a small amount of sodium azide. After the overnight incubation, membranes were washed with TBST three times for 5 min each, followed by incubation with the appropriate secondary antibody conjugated with horseradish peroxidase (Bioshop Canada) at a dilution of 1:4000 in TBST. Membranes were incubated at 4 °C for 1.5 h. Membranes were then washed three times for 5 min each time, and signal was then detected using enzymatic chemiluminescence (ECL) and the ChemiGenius Bioimaging System (Syngene, MD, USA). The membranes were then stained with Coomassie blue (0.25% w/v Coomassie Brilliant Blue R in 50% methanol, 7.5% acetic acid) and destained with destaining solution (60% v/v methanol, 20% v/v acetic acid in ddH2O) until bands were clearly seen. Band densities were analyzed using the associated Bioimaging software and normalized with the corresponding Coomassie blue-stained band.
ProQ diamond phosphoprotein staining
To investigate the total phosphorylation of PK from euthermic and torpid states, purified PK samples were added to the wells of a 10% SDS-PAGE gel. The gel was run at 180 V for 45 min in running buffer. The gel was then washed three times in fixing solution (50% v:v methanol, 10% v:v acetic acid) and left overnight in this solution at ~ 4 °C. The following day, the gel was washed three times with ddH2O for 10 min each and then stained with ProQ Diamond phosphoprotein stain (Invitrogen, Eugene, OR, USA) for 30 min. Following staining, the gel was washed three times with ddH2O for 5 min each. Fluorescent bands on the gel were visualized using the ChemiGenius Bioimaging System (Syngene, Frederick, MD, USA) and intensities were quantified using the associated GeneTools software.
Once quantified, the gel was then stained for 30 min with Coomassie blue and destained for 10 min with destaining solution. PK band intensities from ProQ Diamond chemiluminescence were normalized against the corresponding Coomassie blue-stained band to normalize for any variations in sample loading.
Alkaline phosphatase incubations
Crude skeletal muscle PK was incubated 1:1 in phosphatase incubation buffer (50 mM imidazole, pH 7, 10% glycerol, 10 mM β-mercaptoethanol, 20 mM MgCl2, 10 mM EDTA, and 15 Units of alkaline phosphatase activity) for 24 h at 4 °C. Following the incubation, PK was purified as outlined above and a portion of the purified extracts from euthermic and torpid conditions were stained with ProQ Diamond phosphoprotein stain (details are the same as outlined above). The remaining solution was assayed to determine the K m ADP at room temperature.
Data and protein determination
Enzyme activity was analyzed with a Microplate Analysis Program [12] and kinetic parameters were determined using the Kinetics v.3.5.1 program [13]. Graphs and histograms were created using the SigmaPlot 11 program. Soluble protein concentration was determined by the Coomassie blue dye-binding method using the Bio-Rad-prepared reagent with bovine serum albumin as the standard.
Results
PK purification
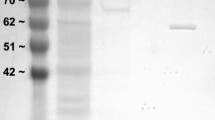
PK was purified from Richardson’s ground squirrel skeletal muscle through the use of an ion exchange column (CM− Sephadex) and an affinity column (Blue-agarose). PK was purified 390-fold, with an overall yield of the final PK preparation of 21% and a specific activity of 390 U/mg (Table 1; Fig. 1). The molecular weight of PK was approximately equal to that of rabbit muscle PK (shown in Fig. 1) at a value of approximately 57 kDa.

Purified euthermic PK from the skeletal muscle of U. richardsonii. Lane 1 shows the protein ladder; Lane 2 shows commercial rabbit muscle pyruvate kinase (Sigma); Lane 3 shows the purified PK sample following CM− and blue-agarose columns; Lane 4 shows a crude skeletal muscle sample prior to any purification
Purified skeletal muscle PK kinetics
Purified squirrel skeletal muscle PK displayed markedly different kinetics between euthermic and torpid states. One distinct change in kinetics was in the K m PEP, where torpid PK displayed a significantly higher K m PEP at both room temperature and 35 °C as compared to the euthermic condition (Table 2). Euthermic PK displayed its highest K m PEP at the temperature closest to T b during torpor, while torpid PK K m PEP remained relatively constant at all assay temperatures with a slight increase in K m at the temperature experienced during euthermia. Conversely, K m ADP for torpid PK was 61 and 54% lower at RT and 35 °C, respectively, than the corresponding values for euthermic PK. Interestingly, torpid PK displayed its highest K m ADP at the T b typically found during torpor, while euthermic PK K m PEP was the highest at RT and lowest at the two temperature extremes (5 and 35 °C). Both euthermic and torpid PK displayed predictable behaviors at the various assay temperatures, with increasing temperature leading to increased enzyme activity. However, the degree to which enzyme activity increased varied between the two conditions. Torpid PK V max was 45% higher at 5 °C and ~ 50% lower at room temperature and 35 °C as compared to the corresponding euthermic V max at those temperatures. Similar changes were observed for K cat where the turnover number for torpid PK was significantly higher at the low temperature and lower at room temperature and 35 °C as compared to the euthermic control.
Effectors of PK
The response of muscle PK to various metabolites commonly encountered in a cell was investigated in this study. One consistent inhibitor of PK activity was ATP, which inhibited euthermic and torpid forms of PK at subsaturating substrate levels and at all temperatures. Torpid PK was significantly less susceptible to ATP inhibition at 5 and 35 °C, with ~ 5 fold and twofold higher I 50 ATP, respectively, as compared to the euthermic values. Furthermore, at the same temperatures, torpid PK’s fold inactivation caused by ATP were 70 and 50% less, respectively, as compared to the values for euthermic PK (Table 3).
KCl and MgCl2 activated PK from both conditions at all three temperatures. PK from the torpid condition was markedly less sensitive to MgCl2 activation at 5 °C as compared to PK from the euthermic condition, with K a MgCl2 of 0.7 ± 0.1 and 4 ± 1 mM, respectively (Table 3). At room temperature, torpid PK displayed a significantly lower K a at 1.2 ± 0.3 mM as compared to PK from the euthermic condition with a K a of 3.1 ± 0.1 mM. Lastly, at 35 °C, there was no difference between euthermic and torpid PK responses to MgCl2. Conversely to the situation with MgCl2 at 5 °C, torpid PK was significantly more sensitive to KCl with a K a that was 69% less than the corresponding K a from the euthermic animal (Table 3). At room temperature and 35 °C, K a KCl and the fold activation by the monovalent cation were similar for PK from both conditions.
Several different metabolites were tested to see if they would affect PK activity at suboptimal substrate concentrations. The amino acids alanine and aspartate did not affect PK activity significantly at concentrations up to 10 mM. Similarly, the Krebs cycle intermediate, citrate, did not have any effect on PK activity at concentrations up to 10 mM. F16P2 also showed little propensity to effect PK activity under the conditions of this experiment up to a concentration of 10 mM.
Physical stability of PK
PK stability was assessed by exposing the purified enzyme to various concentrations of the chemical denaturants, urea and guanidine hydrochloride. At 5 °C, torpid PK was significantly less susceptible to urea denaturation as compared to the euthermic enzyme (Table 4). This difference in structural stability was lost at room temperature with euthermic and torpid PK displaying similar I 50 urea. Lastly, at 35 °C, the I 50 urea could not be determined for euthermic PK as the purified enzyme was unstable and lost all activity after an overnight incubation at 35 °C. The torpid form of PK, however, was able to withstand the prolonged exposure to higher temperatures and displayed an I 50 urea of 0.60 ± 0.03 M.
Guanidine hydrochloride-induced denaturation of euthermic and torpid PK was nearly identical at 5 °C and varied only slightly at room temperature, with torpid PK displaying a 24% higher I 50 GnHCl. Again, euthermic PK was unable to withstand incubation at high temperature overnight, and thus the I 50 GnHCl could not be determined at 35 °C. PK from the torpid condition, however, had an I 50 GnHCl of 0.13 ± 0.01 M at the same temperature.
Phosphorylation of PK
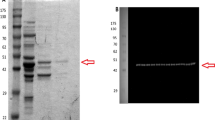
Investigation into the phosphorylation state of squirrel muscle PK began with using ProQ Diamond phosphoprotein stain to quantify the overall level of enzyme phosphorylation. Figure 2 indicates that torpid PK was ~ 40% more phosphorylated than euthermic PK, and that when the purified extracts were acted upon by commercial alkaline phosphatase there was a dramatic decrease in protein phosphorylation. Subsequent western blot analysis indicated that torpid PK had a greater degree of tyrosine and serine phosphorylation, ~ 50 and ~ 40%, respectively, as compared to the control condition. Although not statistically significant, dephosphorylation of torpid PK also appeared to alter PK kinetics, as is evident in Fig. 3 where torpid PK K m ADP increased nearly twofold, to a value similar to that seen for euthermic PK.

Phosphorylation state of euthermic and torpid PK from the skeletal muscle of U. richardsonii. Histogram bars that are indicated as ‘post-phosphatase’ are the ProQ Diamond relative band intensities following incubation of the samples with alkaline phosphatase (see “Materials and Methods” for details). Data are mean ± SEM, n = 4 determinations on independent enzyme samples. a represents significant difference from the euthermic relative band intensity without any phosphatase treatment. b represents significant difference from the corresponding value without phosphatase treatment. The statistical test used for these determinations was the Holm–Sidak method, p < 0.05

Alkaline phosphatase incubations to alter PK K m ADP. K m values are mean ± SEM, n = 4 independent determinations on different incubated samples. (a) represents significant difference from the corresponding euthermic PK value using the Holm–Sidak method, p < 0.05
Other posttranslational modifications
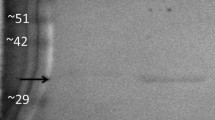
In addition to phosphorylation, other posttranslational modifications that might potentially be important to the role of this enzyme during torpor were investigated. It can be observed from Fig. 4 that PK from the two conditions may be ubiquitinated, but they are ubiquitinated to the same degree. Interestingly, torpid PK appears to be more highly acetylated, with a ~ 70% greater signal detected as compared to euthermic PK.

Posttranslational modification of skeletal muscle PK from U. richardsonii as assessed through western blots. Relative band intensities are mean ± SEM, n = 4 determinations on independently purified PK samples. (a) represents significant difference from the corresponding euthermic value using Student’s t test, p < 0.05
Discussion
Entrance into torpor during winter mammalian hibernation is characterized by a drastic reduction in metabolic rate [14] and, typically, a reorganization of fuel usage within most body tissues to favor lipid oxidation [15]. Skeletal muscle metabolism is no exception, with various studies indicating significant decreases in muscle glycolytic activity during hibernation [11, 16, 17]. Glycolytic suppression during hibernation is usually mediated through the regulation of one or more key enzymes within the pathway, and this study suggests that PK might be a key regulatory point, which aids in tempering skeletal muscle glucose metabolism during torpor.
Previous studies on Spermophilus lateralis skeletal muscle PK did not show any differences between euthermic and hibernator kinetics from crude preparations [11]. While that study touched the surface of several glycolytic enzymes, this study aimed to look deeper at the behavior of PK during hibernation. PK from U. richardsonii was purified to electrophoretic homogeneity (Fig. 1), and then its kinetics at high and low temperatures were assessed. Generally speaking, regardless of temperature, ground squirrel muscle PK behaved similarly to an M1-type PK typically found in skeletal muscle [18]. For instance, ground squirrel PK displayed a K m PEP that ranged from 0.03 to 0.1 mM with a Hill coefficient of ~ 1 (data not shown), as well as K m ADP and I 50 ATP that are fairly close to typical M1-type PK values [18].
Although euthermic and torpid PK displayed properties that were typical of M1-type enzymes, there were significant differences between PK kinetics from the two conditions. At 35 °C, or near-normal T b, numerous kinetic changes were observed between euthermic and torpid PK. These included significant increases in K m PEP and decreases in K m ADP, V max, and K cat for torpid PK as compared to the euthermic form of the enzyme. In deciphering the possible significance of these changes to the ground squirrel, it is important to look at the endogenous levels of metabolites and substrates typically found during euthermia. Endogenous level of ADP within resting mammalian muscle is approximately 1 mM [19], which is well above the K m ADP values for either enzyme form at 35 °C (Table 2). This suggests that the decrease in K m ADP for torpid PK at 35 °C may be inconsequential as ADP levels are nearing saturating conditions. Conversely, mammalian skeletal muscle PEP concentrations under normal conditions range from 0.03 to 0.06 mM [20, 21], which is well within the K m values determined in this study, potentially making this a key kinetic parameter for determining PK activity. Thus, the lower K m PEP for euthermic PK as compared to torpid PK indicates that the torpid form of the enzyme is less able to function at high temperature, while the euthermic form appears suitable for high-temperature function.
Assays at 5 °C revealed significant kinetic differences between torpid and euthermic PK; however, these differences did not decidedly indicate an activation or inhibition of PK activity at low temperatures. Torpid PK showed a significant increase in K m PEP, V max, and K cat when compared to the euthermic condition (Table 2). Although contradictory, it is unlikely that PK would function at V max capacity since the muscle PEP concentration is known to decrease from the above-stated range during torpor [21]. This suggests that the increase in K m PEP may be more significant during torpor and, thus, may indicate a decrease in PK functioning during dormancy.
Decreased PK function at the low temperature typically found during torpor coincides well with the overall muscle metabolic landscape found during dormancy. During torpor, most organs, including the skeletal muscle, switch from carbohydrate to lipid oxidation for energy [15]. While suppression of carbohydrate metabolism typically entails decreasing glycogen catabolism (through regulation of glycogen phosphorylase) [3], and/or decreasing glucose catabolism (through regulating hexokinase) [17], PK typically plays an important regulatory role in slowing the glycolytic rate further along the glycolytic pathway. PK as a glycolytic control point during hibernation has been almost exclusively associated with mammalian liver [3]. However, studies in the jumping mouse, Zapus hudsonius, showed that skeletal muscle PEP concentrations rose during torpor but concentrations of pyruvate decreased during dormancy, suggesting that muscle PK may be an important control point within mammalian muscle during hibernation [21]. This study essentially coincides with the previous study, showing that skeletal muscle PK displays decreased functioning during torpor.
PK is typically susceptible to a variety of metabolites depending on the tissue-specific isoform being observed. An important regulatory molecule for PK is ATP. At both high and low temperatures, torpid PK was significantly less susceptible to ATP inhibition than euthermic PK (Table 3). Looking at the endogenous levels of ATP during euthermia (~ 6 mM) and hibernation (~ 3.5 mM), it is clear that PK from this ground squirrel may be sufficiently susceptible to ATP inhibition to make it a potentially important regulatory molecule throughout the life of the squirrel [19]. The fact that euthermic PK is particularly more sensitive to ATP inhibition likely reflects the fact that at a normal Tb energy flux can be much greater and rapid changes in ATP levels would need to temper glycolytic output more forcefully.
All isoforms of PK require monovalent and divalent cations, and in vivo these cations are most likely potassium and magnesium, respectively [22, 23]. In this study, increasing concentrations of potassium and magnesium activated both euthermic and torpid PK; however, the sensitivity to activation by these ions varied between the two enzyme forms. With regard to magnesium, torpid PK at 5 °C was significantly less sensitive to the divalent cation and also showed substantially lower fold activation as compared to euthermic PK under the same conditions (Table 3). Judging by the known intracellular concentration of magnesium within skeletal muscle (0.5–3 mM) [24], the insensitivity of torpid PK at low temperature likely contributes to the decreased PK functioning during dormancy. With respect to potassium, torpid PK displayed a significantly lower K a KCl at 5 °C as compared to euthermic PK (Table 3). However, the approximate potassium ion concentration within mammalian muscle is ~ 200 mM [25], and even with a slight reduction in concentration during hibernation [26], KCl levels are likely to be well above the K a values observed here. Thus, the most relevant kinetic parameter in regards to potassium would be its resulting fold activation of PK, which is not significantly different between the two enzyme forms at any temperature studied (Table 3).
F16P2 is a common allosteric regulator of most PK isozymes; however, it is not known to affect the most common skeletal muscle isozyme, PKM1. The binding site for FBP is thought to be coded for by the spliced portion of the PKM mRNA (unspliced mRNA is responsible for the production of PKM2) [27]. Consistent with these facts, F16P2 did not affect euthermic or torpid PK at any temperature in this study (Table 3). Several other common cellular molecules were tested for their effects on PK activity, including alanine, aspartate, and citrate. Alanine is a known inhibitor of the liver isozyme of PK, but is typically not a key regulatory molecule for the muscle isozyme [28]. The results shown here (Table 3) support this claim, as alanine did not affect euthermic or torpid PK activity at concentrations as high as 10 mM. Alternatively, aspartate is known to activate plant-derived PK [29], as well as PK from a variety of anoxia-tolerant molluscs [30]. This activation was not found in ground squirrel muscle PK which is likely a product of these enzymes being somewhat distantly related. Citrate, on the other hand, has not previously been known to affect PK; however, it is a common product of lipid oxidation and can accumulate to appreciable amounts as hibernators favor lipid catabolism during torpor [31]. Citrate has been shown to affect several metabolic enzymes, such as phosphofructokinase [32] and glucose-6-phosphate dehydrogenase [33], in estivating land snails; however, it does not appear to affect PK within the hibernating ground squirrel (Table 3).
The kinetics changes noted above suggest that there may be structural variations between the PK from the skeletal muscle of euthermic animals and PK from animals that are in torpor. Two common methods used to elucidate structural differences between two enzyme forms are chemical and thermal denaturation. With regard to chemical denaturation, both urea and guanidine hydrochloride were used. It can be observed from Table 4 that torpid PK is significantly less susceptible to urea denaturation as compared to euthermic PK at 5 °C. Furthermore, as the incubation temperature increased, torpid PK became increasingly more susceptible to urea denaturation. The opposite was true for euthermic PK whose I 50 urea increased as the incubation temperature increased. The changes observed for I 50 GnHCl were less pronounced than those with urea, with torpid PK being 7% more susceptible to denaturation at 5 °C, and ~ 20% less susceptible to denaturation at RT (Table 4). Generally speaking, it appears that torpid PK is significantly more stable at low temperatures as compared to euthermic PK, with euthermic PK becoming increasingly more stable as the temperature increased. This may be significant during torpor as protein turnover is required to stay low as it is impossibly costly to maintain euthermic rates of protein synthesis and degradation. Thus, a more chemically stable enzyme may be able to withstand greater insult by the cellular environment without unfolding and requiring recycling. It is important to note that denaturation studies at 35 °C could not be completed as the enzyme was unstable overnight at this high temperature.
Based on the kinetic and stability variations between euthermic and torpid PK, it appears that there may be significant structural differences between PK from the two conditions. Structural variations in proteins are common in animals that transition into hypometabolic states, and usually occur in the form of posttranslational modifications to the proteins. Principle among these modifications is reversible protein phosphorylation, which was investigated in this study through ProQ Diamond phosphoprotein staining and phosphoserine and phosphotyrosine western blots on purified PK. It can be observed from Fig. 2 that the relative intensity of torpid PK bands following ProQ Diamond phosphoprotein staining was ~ 40% more than the band intensity for euthermic PK. Subjecting both enzyme forms to alkaline phosphatase treatment for 48 h resulted in a drastic decrease in ProQ staining. Furthermore, phosphoserine western blots indicated that torpid PK contained ~ 50% more serine phosphorylation than PK from the control condition (Fig. 4). Conversely, phosphotyrosine levels remained relatively constant between euthermic and torpid conditions.
Pyruvate kinase has been known to be phosphorylated in a variety of systems, such as rat, chicken, and human liver [2, 34,35,36], rat pancreatic islet [37], human and rat erythrocyte cells [38], and many human cancer cells [39, 40]. Phosphorylation of muscle PK is far less common and has only been found in the channeled whelk and the spadefoot toad [41, 42]. The only experimental evidence for mammalian muscle PK phosphorylation comes from work on human cancer cells, which showed increased phosphorylation of pyruvate kinase muscle isoform 2 (PKM2). Although this isoform is not found in mammalian skeletal muscle, it has an amino acid sequence that is only 4% dissimilar to the predominant muscle isoform (i.e., PKM1). Indeed, PKM1 and PKM2 are expressed from alternative RNA splicing of the same gene [43], and the 43 amino acids that are present in PKM2 do not appear to contain any obvious phosphorylation sequence [39]. Furthermore, computational analysis of mammalian muscle PK does show considerable evidence for serine, threonine, and tyrosine phosphorylation sites on rat, mouse, and human muscle PK (phosphorylation predictions were done with PHOSIDA and NetPhos 2.0 sites; data not shown). PHOSIDA is a web-based system that encompasses both already identified phosphosites as well as phosphosite predictions for your protein of interest [44]. NetPhos 2.0, on the other hand, is a prediction-based system that uses an artificial neural network to predict specific serine, threonine, and tyrosine phosphorylation sites [45]. Taken together, these applications indicate that it is possible to phosphorylate mammalian muscle PK and this is the first study that shows this to be true.
The known effects of reversible phosphorylation on PK functioning are inconsistent, with some reports indicating that it has no effect on PK activity [37], and others showing that phosphorylation inhibits PK [2, 40, 46, 47]. In an effort to determine whether reversible PK phosphorylation had an effect on enzyme function in this study, crude muscle extracts were incubated with exogenous alkaline phosphatase prior to purification, and the K m ADP was reassessed. It can be observed from Fig. 3 that incubation of euthermic PK with alkaline phosphatase did not affect its affinity for ADP to a significant degree; however, incubation of torpid PK with alkaline phosphatase increased the K m ADP by approximately twofold to a value similar to that seen for euthermic PK. Thus, it appears that reversible phosphorylation may regulate changes in PK kinetics.
In addition to phosphorylation, ubiquitination and acetylation were investigated in this study to discern other key posttranslational modifications for skeletal muscle PK during hibernation. First, western blots detecting ubiquitin-conjugated proteins indicated that at least a portion of the PK population within euthermic and torpid PK from the skeletal muscle was ubiquitinated; however, there was little difference in the degree of ubiquitination between euthermic and torpid conditions (Fig. 4). This result may represent some level of protein turnover that may be occurring under both conditions; however, ubiquitination does not appear to be a critical form of regulation during the transition into torpor.
An analysis of the level of acetylation of muscle PK through western blot indicated that torpid PK was significantly more acetylated than the euthermic form of the enzyme (Fig. 4). Acetylation is emerging as a crucial and varied regulatory mechanism that is being found on an array of proteins within the cell. Recently, Zhao and colleagues [48] found that many enzymes of glycolysis (including PK), gluconeogenesis, TCA cycle, urea cycle, fatty acid metabolism, and glycogen metabolism were found to be acetylated in human liver tissue. The effect of protein acetylation is varied, and studies have shown that acetylation both positively and negatively affects protein–protein interactions [49, 50] and protein stability or half-life [51, 52]. Few studies have been conducted to reveal the potential function of PK acetylation, but one study shows that acetylation of the M2 isoform of PK, found only in embryonic and tumor cells, targets this protein for lysosome-dependent degradation [53]. While possible for squirrel skeletal muscle PK, it may be unlikely that acetylation targets this enzyme for degradation as it is a crucial component of glycolysis that is necessary for arousal. During hibernation, the ground squirrel switches to using lipid oxidation for their primary fuel source, as evidenced by a respiratory quotient of ~ 0.7 [9]. However, during the many periodic arousals that occur during hibernation the respiratory quotient quickly ramps up to ~ 0.8–1 indicating a rapid switch back to carbohydrate oxidation [4]. It would be uneconomical with the ground squirrel’s fixed fuel reserves to degrade PK during torpor only to have to synthesize it again upon arousal. Acetylation could potentially regulate protein–protein interactions between PK and another cellular protein, or it could result in some of the kinetic or physical changes that were seen between euthermic and torpid PK. Clearly, there are many possible functional outcomes to PK acetylation and the role of ground squirrel muscle PK acetylation will require further investigation.
Conclusion
Purified skeletal muscle PK from Richardson’s ground squirrels showed distinctly different kinetic properties between euthermic and torpid conditions, with torpid PK appearing to be less active at a low temperature that is typically experienced by the squirrels during dormancy as compared to euthermic PK. PK from the euthermic condition was however well suited to function at normal mammalian T b. ProQ Diamond phosphoprotein staining and western blot analyses indicate that skeletal muscle PK from the torpid animal was significantly more phosphorylated than the enzyme from the euthermic condition. Moreover, incubations that stimulated alkaline phosphatase were able to alter PK kinetics, suggesting that this reversible modification is linked to some of the kinetic changes observed in this study. In addition to phosphorylation, skeletal muscle PK was found to be differentially acetylated, with torpid PK showing greater levels of acetylation. The role of PK acetylation is unknown but may represent an important covalent modification during torpor.
References
Ljungstrom O, Hjelmquist G, Engstrom L (1974) Phosphorylation of purified rat liver pyruvate kinase by cyclic-3′,5′-AMP-stimulated protein kinase. Biochem Biophys Acta 358:289–298
Riou JP, Claus TH, Pilkis SJ (1978) Stimulation of glucagon of an in vivo phosphorylation of rat hepatic pyruvate kinase. J Biol Chem 253:656–659
Storey KB (1987) Regulation of liver metabolism by enzyme phosphorylation during mammalian hibernation. J Biol Chem 262(4):1670–1673
Carey HV, Andrews MT, Martin SL (2003) Mammalian hibernation: cellular and molecular response to depressed metabolism and low temperature. Physiol Rev 83:1153–1181
Zatzman ML (1984) Renal and cardiovascular effects of hibernation and hypothermia. Cryobiology 21(6):593–614
McArthur MD, Milsom WK (1991) Changes in ventilation and respiratory sensitivity associated with hibernation in Columbian (Spermophilus columbianus) and golden-mantled (Spermophilus lateralis) ground squirrels. Physiol Zool 64:940–959
Van Breukelen F, Sonenberg N, Martin SL (2004) Seasonal and state-dependent changes of eIF4E and 4E-BP1 during mammalian hibernation: implications for the control of translation during torpor. Am J Physiol Regul Integr Comp Physiol 287:349–353
MacDonald JA, Storey KB (1999) Regulation of ground squirrel Na + K + ATPase activity by reversible phosphorylation during hibernation. Biochem Biophys Res Commun 254:424–429
South FE, House WA (1967) Energy metabolism in hibernation. In: Fisher KC, Dawe AR, Lyman CP, Schonbaum E, South FE (eds) Mammalian hibernation III. Oliver and Boyd, Ltd., Edinburgh, pp 305–324
Tashima LS, Adelstein SJ, Lyman CP (1970) Radioglucose utilization by active, hibernating, and arousing ground squirrels. Am J Physiol 218:303–309
Brooks SBJ, Storey KB (1992) Mechanisms of glycolytic control during hibernation in the ground squirrel Spermophilus lateralis. J Comp Physiol B 162:23–28
Brooks SPJ (1992) A simple computer program with statistical tests for the analysis of enzyme kinetics. Biotechniques 13:906–911
Brooks SPJ (1994) A program for analyzing enzyme rate data obtained from a microplate reader. Biotechniques 17:1155–1161
Geiser F (1988) Reduction of metabolism during hibernation and daily torpor in mammals and birds: temperature effect or physiological inhibition? J Comp Phys B 158:25–37
Hochachka PW, Guppy M (1987) Metabolic arrest and the control of biological time. Harvard University Press, Cambridge
Hachimi ZE, Tijane M, Boissonnet G, Benjouad A, Desmadril M, Yon JM (1990) Regulation of the skeletal muscle metabolism during hibernation of Jaculus orientalis. Comp Biochem Physiol B 96(3):457–459
Abnous K, Storey KB (2008) Skeletal muscle hexokinase: regulation in mammalian hibernation. Mol Cell Biochem 319:41–50
Hall ER, Cottam GL (1978) Isozymes of pyruvate kinase in vertebrates: their physical, chemical kinetic and immunological properties. Int J Biochem 9:785–794
English TE, Storey KB (2000) Enzymes of adenylate metabolism and their role in hibernation of the white-tailed prairie dog Cynomys leucurus. Arch Biochem Biophys 376:91–100
Dohm GL, Patel VK, Kasperek GJ (1986) Regulation of muscle pyruvate metabolism during exercise. Biochem Med Metabol Biol 35(3):260–266
Storey KB, Kelly DA (1995) Glycolysis and energetics in organs of hibernating mice (Zapus hudsonius). Can J Zool 73:202–207
Larson TM, Laughlin LT, Holden HM, Rayment I, Reed GH (1994) Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemisty 33(20):6301–6309
Frey PA, Hegeman AD (2007) Phosphotransfer and nucleotidyltransfer. Enzymatic reaction mechanisms. Oxford University Press, New York, pp 495–496
MacDermott M (1990) The intracellular concentration of free magnesium in extensor digitorum longus muscles of the rat. Exp Physiol 75:763–769
Bijlani RL, Manjunatha S (2010) Membrane potential at rest and during activity. In: Understanding medical physiology: a textbook for medical students. Jaypee Brothers Medical Publishers Ltd. New Delhi, India p 118
Willis JS, Goldman SS, Foster RF (1971) Tissue K concentration in relation to the role of the kidney in hibernation and the cause of periodic arousal. Comp Biochem Physiol A Physiol 39(3):437–445
Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL (1998) The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 6(2):195–210
Carbonell J, Marco R, Feliu JE, Sols A (1973) Pyruvate kinase: classes of regulatory isozymes in mammalian tissues. FEBS J 37:148–156
Smith CR, Knowles VL, Plaxton WC (2000) Purification and characterization of cytosolic pyruvate kinase from Brassica napus (rapeseed) suspension cell cultures. Eur J Biochem 267:4477–4486
Storey KB (1986) Aspartate activation of pyruvate kinase in anoxia tolerant molluscs. Comp Biochem Physiol B 83:807–812
Storey KB (1989) Integrated control of metabolic rate depression via reversible phosphorylation of enzymes in hibernating mammals. In: Malan A, Canguilhem AB (eds) Living in the cold II Colloque INSERM. John Libbey Eurotext Ltd, Montrouge, pp 309–319
Whitwam RE, Storey KB (1991) Regulation of phosphofructokinase during estivation and anoxia in the land snail, Otala Lactea. Physiol Zool 64:595–610
Ramnanan CJ, Storey KB (2006) Glucose-6-phosphate dehydrogenase regulation during hypometabolism. Biochem Biophys Res Commun 339:7–16
Titanji VPK, Zetterqvist O, Engstrom L (1976) Regulation in vitro of rat liver pyruvate kina se by phosphorylation-dephosphorylation reactions, catalyzed by cyclic-AMP dependent protein kinases and a histone phosphatase. Biochim Biophys Acta 422(1):98–108
Boivin P, Galand C, Estrada M (1980) Phosphorylation of human red cell and liver pyruvate kinase. Differences between liver and erythrocyte L-type subunits. Cell Mol Life Sci 36(8):900–901
Fister P, Eigenbrodt E, Presek P, Reinacher M, Schoner W (1983) Pyruvate kinase type M2 is phosphorylated in the intact chicken liver cell. Biochem Biophys Res Commun 115(2):409–414
MacDonald MJ, Kowluru A (1985) Evidence for calcium enhanced phosphorylation of pyruvate kinase by pancreatic islets. Mol Cell Biochem 68(2):107–114
Nakashima K, Fujii S, Kaku K, Kaneko T (1982) Calcium-calmodulin dependent phosphorylation of erythrocyte pyruvate kinase. Biochem Biophys Res Commun 104(1):285–289
Weernink PA, Rijksen G, van der Heijden MC, Staal GE (1990) Phosphorylation of pyruvate kinase type K in human gliomas by a cyclic adenosine 5′-monophosphate-independent protein kinase. Cancer Res 50(15):4604–4610
Hitosugi T, Kang S, Vander Heiden MG, Chung T, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu T, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2(97):ra73
Plaxton WC, Storey KB (1984) Phosphorylation in vivo of red-muscle pyruvate kinase from the channelled whelk, Busycotpus canaliculatum, in response to anoxic stress. Eur J Biochem 143:267–272
Cowan KJ, Storey KB (1999) Reversible phosphorylation of skeletal muscle pyruvate kinase and phosphofructokinase during estivation in the spadefoot toad, Scaphiopus couchii. Mol Cell Biochem 195:173–181
Noguchi T, Inoue H, Tanaka T (1986) The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem 261:13807–13812
Gnad F, Ren S, Cox J, Olsen JV, Macek B, Oroshi M, Mann M (2007) PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol 8:R250
Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294:1351–1362
Eigenbrodt E, Mostafa MA, Schoner W (1977) Inactivation of pyruvate kinase type M2 from chicken liver by phosphorylation, catalyzed by a cAMP-independent protein kinase. Hoppe Seylers Z Physiol Chem 358:1047–1055
Presek P, Reinacher M, Eigenbrodt E (1988) Pyruvate kinase type M2 is phosphorylated at tyrosine residues in cells transformed by Rous sarcoma virus. FEBS Lett 242(1):194–198
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan K (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004
Walter L, Bienz M (1998) Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature 395(6701):521–525
Dhalluin C, Carlson JE, Zeng L, He C, Aggarwai AK, Zhou MM (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399(6735):491–496
Caron C, Boyault C, Knochbin S (2005) Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioassays 27(4):408–415
Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW (2002) Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 111(5):709–720
Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xoing Y, Guan KL, Lei QY (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 42(6):719–730
Acknowledgements
The research was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (OPG6793) to K.B. Storey and by an NSERC CGS postgraduate scholarship to R.A.V. Bell.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bell, R.A.V., Storey, K.B. Purification and characterization of skeletal muscle pyruvate kinase from the hibernating ground squirrel, Urocitellus richardsonii: potential regulation by posttranslational modification during torpor. Mol Cell Biochem 442, 47–58 (2018). https://doi.org/10.1007/s11010-017-3192-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3192-9