
Abstract
The intertidal marine snail, Littorina littorea, has evolved to survive bouts of anoxia and extracellular freezing brought about by changing tides and subsequent exposure to harsh environmental conditions. Survival in these anoxic conditions depends on the animals entering a state of metabolic rate depression in order to maintain an appropriate energy production-consumption balance during periods of limited oxygen availability. This study investigated the kinetic, physical, and regulatory properties of pyruvate kinase (PK), which catalyzes the final reaction of aerobic glycolysis, from foot muscle of L. littorea to determine if the enzyme is differentially regulated in response to anoxia and freezing exposure. PK purified from foot muscle of anoxic animals exhibited a lower affinity for its substrate phosphoenolpyruvate than PK from control and frozen animals. PK from anoxic animals was also more sensitive to a number of allosteric regulators, including alanine and aspartate, which are key anaerobic metabolites in L. littorea. Furthermore, PK purified from anoxic and frozen animals exhibited greater stability compared to the non-stressed control animals, determined through high-temperature incubation studies. Phosphorylation of threonine and tyrosine residues was also assessed and demonstrated that levels of threonine phosphorylation of PK from anoxic animals were significantly higher than those of PK from control and frozen animals, suggesting a potential mechanism for regulating PK activity. Taken together, these results suggest that PK plays a role in suppressing metabolic rate in these animals during environmental anoxia exposure.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Survival in extreme environments often requires drastic changes to an animal’s physiology and underlying biochemistry. Species that reside in the intertidal zone demonstrate a high degree of metabolic flexibility that enables them to withstand the continuously changing environmental conditions associated with the changing tide. Alternating between submergence in sea water at high tide and exposure to an aerial environment at low tide, the animals must cope with rapid changes in oxygen availability, osmolality, and environmental temperatures. At high tide, respiration in marine invertebrates occurs normally, providing the animals with an abundance of oxygen for metabolic processes. During low tide, however, the animals are exposed to an aerial environment and can no longer efficiently carry out gas exchange across their gills. Additionally, if isolated in a tidal pool, hyper-saline conditions can develop as seawater evaporates, while animals living at certain latitudes may be exposed to subzero temperatures when tides recede. Because of this, many intertidal invertebrates exhibit metabolic and physiological adaptations to survive in these environments [1,2,3].
Freezing and anoxia tolerance are common adaptations observed in invertebrates inhabiting the intertidal zone. Littorina littorea is one such animal capable of surviving periodic bouts of anoxia and, depending on the climate, freezing. In fact, laboratory experiments have shown that it can survive at least 6 days of anoxia exposure and up to 8 days of freezing at − 8 °C [3,4,5]. One of the most important biochemical adaptations supporting anoxia/freeze tolerance is metabolic rate depression. A lack of oxygen deprives animals of the ability to produce sufficient energy to maintain their normal metabolic state, requiring that a new energy generation-consumption equilibrium be established. Non-essential processes are therefore shut down allowing only the most vital pathways to proceed. Indeed, transcription and translation have both been shown to be suppressed during anoxia exposure in the periwinkle [6, 7], while stress-responsive miRNA expression patterns suggest that the cell cycle is also arrested [8]. Metabolic rate depression is often regulated in part by enzymes at key points in cellular metabolism and is commonly accomplished via reversible protein phosphorylation (RPP) which has been previously observed in L. littorea [9] as well as other anoxia-tolerant species [10,11,12]. In L. littorea, enzymes governing carbohydrate metabolism have been the focus of study due to the importance of regulating the rate of carbohydrate fermentation and anaerobic ATP generation. Indeed, previous studies have demonstrated anoxia and freezing-induced kinetic changes in the kinetic properties of various enzymes of carbohydrate metabolism which were consistent with covalent modification and a general curtailing of carbohydrate metabolism [13,14,15]. By inhibiting the activity of these enzymes, the rate of carbohydrate metabolism can be reduced significantly, supporting metabolic reorganization during environmental stress exposure.
Pyruvate kinase (PK; E.C. 2.7.1.40) is a key enzyme of glucose metabolism catalyzing the terminal reaction of aerobic glycolysis; the transfer of a phosphate group from phosphoenolpyruvate (PEP) to ADP, producing ATP and pyruvate. Its product pyruvate can be further metabolized in the mitochondria to produce ATP via oxidative phosphorylation or fermented in the cytosol by lactate dehydrogenase (LDH) to produce lactic acid. PK is one of two enzymes operating at the PEP branch point, where PEP is oxidized by PK. Alternatively, in some animals conducting anaerobic metabolism, PEP can be converted to oxaloacetate by the gluconeogenic enzyme phosphoenolpyruvate carboxykinase (PEPCK). Thus, the inhibition of PK is thought to be a mechanism to promote gluconeogenesis [16, 17]. Given that PK sits at a metabolic branch point, directly produces ATP, and that its product has multiple potential metabolic fates, it should come as no surprise that PK is one of the main control points of glycolysis.
Our group has extensively investigated PK as a potential regulatory locus of carbohydrate metabolism in animals surviving in extreme environmental conditions, including both invertebrates and vertebrates. In all of these animals, environmental stress-induced phosphorylation of PK was associated with a less active form of the enzyme [18,19,20,21,22,23]. Interestingly, however, earlier work on PK in L. littorea suggested the exact opposite effect was at work—that PK was dephosphorylated resulting in a less active form of the enzyme in response to anoxia [13]. This led to the hypothesis that this earlier work may not completely describe PK regulation in this animal. The research presented here therefore explores the stress-responsive regulation of PK in L. littorea in significantly greater detail than before by purifying PK from animals exposed to control, anoxic, and freezing conditions, and then comparing the kinetic parameters and phosphorylation states of the enzyme. In doing so, this research seeks to establish greater clarity around stress-responsive regulation of PK in marine invertebrates and add to the body of evidence showing that PK can act as a key control point in mediating metabolic rate depression.
2 Materials and Methods
2.1 Animals
Littorina littorea were purchased from the Kowloon market in Ottawa, Canada. The animals were first rinsed quickly with tap water, followed by quick rinsing in sea water. The snails were then transferred to a 30 L tub of aerated sea water and held at 9 °C in an incubator in complete darkness in order to acclimatize for 5 days. The water was changed periodically during acclimation. Control animals were sampled directly from the acclimatized conditions. Anoxic conditions were simulated by placing snails in sealed jars (previously equilibrated with nitrogen gas (N2) which contained a small amount of deoxygenated sea water. The jars were gassed for 15 min with N2 upon adding the snails, after which time the jars were sealed with parafilm and returned to 9 °C for 24 h. To simulate freezing conditions, snails were placed in containers lined with damp paper towel and then held in incubators for 24 h at -6 °C. Freezing occurred within the first hour of the experiment. Following experimental procedures, animals were rapidly dissected and tissues were flash frozen in liquid nitrogen before being held at -80 °C until use.
2.2 Preparations of Tissue
Tissue from control, frozen and anoxic samples of foot muscle of L. littorea were rapidly weighed and homogenized 1:10 w:v in ice-cold homogenization buffer A (10 mM MES buffer, pH 6.0, 2 mM ethylenediaminetetraacetic acid (EDTA), 2 mM ethylene glycol tetraacetic acid (EGTA), 15 mM β-glycerophosphate, 15 mM β-mercaptoethanol (β-MeSH), 10% v/v glycerol). Immediately before homogenization, a few crystals of phenylmethylsulphonyl fluoride (PMSF) were added. A Polytron homogenizer (Brinkmann Instruments, Westbury, NY, USA) was used to completely homogenize the samples, which were then spun in an Eppendorf 5810R centrifuge (22331 Hamburg, GER) for 30 min at 13,500×g at 4 °C. The supernatant (crude homogenate) was carefully pipetted off and held on ice until use.
2.3 Pyruvate Kinase Assay
PK was assayed as previously described [21]. Briefly, the PK-catalyzed reaction was coupled to the consumption of NADH by LDH, which was measured by detecting absorbance at 340 nm using a microplate reader (Multiskan Spectrum, Thermo Labsystems, Finland). Optimal assay conditions were 50 mM Tris at pH 7.2, 20 mM KCl, 2 mM ADP, 5 mM MgCl2, 1 mM PEP, 1 U of LDH and 0.15 mM NADH. Assay reactions were started by adding PK to a total volume of 200 μL. Vmax and Km values were determined at optimal assay conditions. I50 and Ka values were determined at suboptimal assay conditions (0.2 mM PEP, 0.15 mM NADH, 2 mM ADP, 2 mM MgCl2 and 1 U of LDH). All assays were performed at 22 °C unless otherwise stated.
2.4 Purification of Pyruvate Kinase
A 2 mL aliquot of crude homogenate was added to a CM− (Carboxymethyl cation exchange material) column (0.75 cm × 10 cm) that had been previously equilibrated with 20 mL of buffer A. The column was then washed with 15 mL of buffer A. PK was eluted from the column with 20 mL of buffer B (same as buffer A, but pH 6.5). 1.2 mL fractions were collected using an automated fraction collector (Gilson Medical Electronics, Inc., Middleton, WI, USA). To determine which fractions contained PK, 10 μL of each fraction was taken and used to measure PK activity spectrophotometrically. The highest-activity fractions were pooled and loaded on to a hydroxyapatite (HA) column that had been previously equilibrated with buffer C (10 mM MOPS, pH 7.0, 2 mM EDTA, 2 mM EGTA, 15 mM β-glycerophosphate, 15 mM β-MeSH and 10% v/v glycerol). PK was eluted from the column with a 0–500 mM potassium phosphate gradient made in buffer C and collected in 1.2 mL fractions which were then assayed for PK activity. Peak activity fractions were pooled for use in further experiments. Protein concentrations were determined according to manufacturer instructions using Coomassie blue G-250 dye-binding reagent (Bio-Rad, Hercules, CA, USA) with bovine serum albumin used as a standard [24].
2.5 Activator/Inhibitor Treatments
The activity of PK was tested in the presence of various activators and inhibitors. In the experiment shown in Fig. 2, the conditions were as follows: In the negative treatment, suboptimal assay conditions were used which included 0.2 mM PEP, 0.15 mM NADH, 2 mM ADP, 2 mM MgCl2 and 1 U of LDH in Tris assay buffer (pH 7.2). The alanine condition was the same as the control condition but also included 0.5 mM alanine. The Fructose-1,6-bisphosphate (FBP), isoleucine and aspartate conditions were the same as the alanine condition but also included 0.065 mM FBP, 1.5 mM isoleucine and 0.5 mM aspartate, respectively. The relative Vmax of all treatments were compared to the Vmax value obtained from the negative treatment which was set to 1.0.
2.6 Assessment of Thermal Stability
Temperature incubation experiments were performed to determine and compare the stability of PK from control, anoxic and frozen animals at high temperature (60 °C). 750 μL aliquots of purified enzyme were incubated for up to 90 min at 60 °C using a dry block heater (Fischer Scientific, Dubuque, IA, USA). 25 μL aliquots of sample were removed from the stock sample at specified time intervals and immediately placed on ice. Following removal of the final aliquot after 90 min, samples were left to equilibrate at room temperature before being assayed at optimal conditions.
2.7 SDS Polyacrylamide Gel Electrophoresis and Immunoblotting
Purity of PK was confirmed using SDS-PAGE as previously described [21]. The purified PK samples were mixed 1:1 (v/v) with and SDS-PAGE loading buffer (100 mM Tris-base, pH 6.8, 4% w/v SDS, 20% v/v glycerol, 0.2% w/v bromophenol blue and 10% v/v β-MeSH) and then boiled for 5 min before being stored at 20 °C until use. A 10% SDS-PAGE gel (10% v/v acrylamide, 400 mM Tris, pH 8.8, 0.1% w/v SDS, 0.2% w/v ammonium persulfate (APS), 0.04% v/v TEMED) with a 5% stacking gel (5% acrylamide, 190 mM Tris, pH 6.8, 0.1% w/v SDS, 0.15% w/v APS, and 0.1% v/v TEMED) run at a constant voltage of 180 V was used to separate the proteins over 55 min at room temperature in SDS-PAGE running buffer (25 mM Tris-base, pH 8.5, 190 mM glycine, and 0.1% w/v SDS). A protein molecular weight ladder (Froggabio, Cat. #PM005-0500) was also run on every gel. Gels were removed from the apparatus, added directly to Coomassie (0.25% w/v Coomassie brilliant blue, 7.5% v/v acetic acid, 50% methanol), and gently rocked overnight. The gels were then removed from the stain solution and rinsed with ddH2O prior to being submerged and gently rocked in destain solution (10% v/v acetic acid, 25% v/v methanol) until protein bands were clearly visible. Gels were then rehydrated in ddH2O and imaged using ChemiGenius Bio Imaging System (Syngene, Frederick, MD).
Western blotting was used to assess the post translational modifications of PK from control, anoxic and frozen tissues as previously described [21]. Following electrophoresis, the proteins were transferred to methanol-equilibrated polyvinylidiene difluoride (PVDF) membranes by a wet transfer procedure. The electroblotting was run at a constant amperage of 160 mA for 90 min in transfer buffer (25 mM Tris, pH 8.5, 192 mM glycine, and 20% v/v methanol) after which membranes were removed from the apparatus and washed in TBST (20 mM Tris-base, pH 7.6, 140 mM NaCl, 0.05% v/v Tween-20) three times for five minutes (3 × 5 min) and blocked in 1.5% (w/v) skim milk protein solution (prepared in TBST) for 20 min at room temperature with gentle rocking. The blocking solution was then removed, and the membranes washed (3 × 5 min) in TBST. Membranes were then incubated overnight at 4 °C with gentle rocking in either of the following primary antibodies which were diluted 1:1000 in TBST: (1) rabbit anti-phosphothreonine (Cat. #718200, Invitrogen, Carlsbad, CA, USA); (2) mouse anti-phosphotyrosine (Cat. #615800, Invitrogen, Carlsbad, CA, USA). After removal of the primary antibodies the following day, the blots were washed (3 × 5 min) in TBST and then probed with 1:5000 TBST-diluted anti-rabbit or anti-mouse horseradish peroxidase-linked antibodies (Bioshop, Cat. #APA007P and Cat. # APA005P.2, respectively) for 45 min at room temperature with gentle rocking. Membranes were washed in TBST (3 × 5 min) and then visualized with the ChemiGenius Bio Imaging System by enhanced chemiluminescence, using peroxide and luminol. Membranes were then stained with Coomassie blue to be used to standardize the chemiluminescence signals to protein amount. Analysis and quantification of blots was performed using Genetools software (Syngene, v 4.02).
2.8 Data and Statistical Analyses
A Microplate analysis program (MPA) [25] was used to analyze all enzyme assays, while Kinetics v. 3.5.1 [26], a nonlinear least squares regression computer program, was used to determine kinetic parameters (Km, Vmax, I50, Ka, t½). Data for all kinetic parameters and western blots were compared using a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test (p < 0.05) or Student’s t-test (p < 0.05), as appropriate. Sigmaplot version 12 statistical analysis software was used for t-tests, ANOVAs, and graph generation.
2.9 Bioinformatics and Phosphorylation Site Prediction
Candidate phosphorylation sites were investigated in silico. Since no sequence for PK is available in L. littorea, all available gastropod PK sequences (5 sequences in total; see Online Resource 1) were aligned to check for putative threonine phosphorylation sites common in this clade (the closest relative to L. littorea being Pomacea canaliculata, common to the subclass of gastropods known as caenogastropoda). PK Sequences were obtained through the NCBI database sorted by taxonomic distribution. These sequences were subject to sequence alignment using the Clustal Omega multiple sequence alignment online tool [27]. NetPhos 3.1 was used to predict potential threonine phosphorylation sites (indicated by a prediction score greater than the threshold of 0.5) [28]. Predicted sites have been highlighted on the aligned sequences to show where these sites are conserved between the species investigated (Online Resource 1).
Predicted threonine phosphorylation sites in the P. canaliculata PK sequence were also compared to known, experimentally determined threonine phosphorylation sites in human PK using the PhosphoSitePlus database [29] in order to assess the possibility of common phosphorylation events between widely divergent species (Online Resource 1).
3 Results
3.1 Purification of Pyruvate Kinase
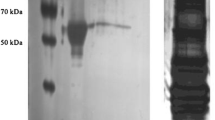
PK was successfully purified from control, frozen and anoxic samples of L. littorea foot muscle using a combination of ion-exchange and affinity column chromatography. PK was eluted from the CM− column by increasing the pH of the elution buffer from pH 6.0 to pH 6.5. This step resulted in 2.2-fold purification with a 40% yield. The second step, a 0–500 mM phosphate gradient elution from an HA column, resulted in a 13-fold purification relative to the crude with a 32% yield (Table 1). The purified PK had a molecular weight of ~ 68 kDa (Fig. 1).

10% SDS-PAGE Coomassie Brilliant Blue stained gel showing purified PK from control foot muscle of L. littorea. Lanes (1): Molecular weight ladder (Froggabio); (2) crude homogenate; (3) pooled peak fractions after elution from hydroxyapatite column; (4) commercial purified rabbit PK (Sigma life sciences)
3.2 Kinetic Activity of PK
The Vmax of PK purified from control (0.78 ± 0.37 U/mg), frozen (0.67 ± 0.08 U/mg) and anoxic (0.67 ± 0.15 U/mg) animals did not differ from each other (Table 2). The Km PEP of PK increased significantly in the anoxic animals (0.40 ± 0.04 mM) when compared to the control (0.15 ± 0.04 mM) and frozen animals (0.14 ± 0.06 mM) (Table 2). No significant change was observed in the Km ADP between the control, frozen and anoxic forms of PK, which averaged 0.500 mM (Table 2). Similarly, the total activity the PK (U/g wet weight) did not differ significantly between the control, frozen and anoxic animals (mean = 2.96 U/g wet weight) (Table 2).
Various allosteric effectors were added to the reaction to investigate the possibility of stress-specific allosteric regulatory patterns. While the inhibitory constants (I50) of urea (1.69 M), NaCl (0.15 M) and ATP (7.13 mM) did not differ significantly between the three conditions, the I50 of alanine was significantly lower in PK from anoxic animals (0.26 ± 0.04) than that from control (0.59 ± 0.05) and frozen (0.58 ± 0.05 mM) animals, as was the I50 of KCl (0.11 ± 0.004 M, 0.54 ± 0.04 M, 0.65 ± 0.04 M from anoxic, control and frozen animals, respectively). The I50 NH4 of PK from control animals (0.19 ± 0.01 M) was lower than that from anoxic animals (0.40 ± 0.02 M) and frozen animals (0.27 ± 0.01 M) (Table 3).
Aspartate and isoleucine were also investigated as activators of PK (Table 4). The Ka (activator constant; concentration of activator resulting in a half maximal increase in activity) values did not change between the three conditions for either of the compounds tested. The fold-activation resulting from aspartate was significantly higher for PK from anoxic animals (2.38 ± 0.09) compared to the that from control animals (1.42 ± 0.08), while that from frozen animals (1.63 ± 0.16) did not differ significantly from either the control or anoxic form. The fold-activation values for isoleucine did not change between the three conditions, and they ranged between 1.35 and 1.68.
Figure 2 shows the effects of alanine (an inhibitor of PK), as well as alanine in combination with various activators of PK, on the activity of the control, frozen and anoxic enzyme. Alanine alone decreased the Vmax by 30% to 40% of uninhibited levels. The combination of alanine and FBP resulted in the activity of PK being activated by 15% to 30% compared to the uninhibited, negative value. The combination of alanine and isoleucine resulted in Vmax values that were 19% to 23% less than the corresponding negative value and were statistically no greater than when alanine alone was added. The addition of aspartate resulted in a recovery of the Vmax of PK from control animals to a value that was 1.1-fold greater than the uninhibited value, however PK from anoxic animals remained completely inhibited. The PK from frozen animals recovered slightly upon the addition of aspartate, but the Vmax still remained 11% less than the uninhibited negative values.

Effect of alanine and combinations of alanine with various activating compounds (FBP, Isoleucine, and Aspartate) on the Vmax of purified PK from the foot muscle of L. littorea. Significant differences in relative Vmax compared to the corresponding negative, uninhibited value of the same animal condition is indicated by an asterisk, Student’s t-test, p < 0.05. Significant differences in relative Vmax compared to the corresponding alanine-inhibited values are indicated by “†”. Student’s t-test, p < 0.05. Data are mean ± SEM, n = 4. All data expressed relative to the corresponding negative value of the same animal condition which was set to 1.0
3.3 Post-translational Modifications of PK
Western blotting was used to assess the differences in the phosphorylation status of purified foot muscle PK from control, frozen and anoxic conditions. Phosphorylation on threonine residues of the anoxic form of PK was 2.8-fold higher than the control or frozen form of PK and 2.4-fold high than the frozen form (Fig. 3). The phosphorylation level of tyrosine residues between PK from control, frozen and anoxic animals did not differ significantly.

Relative levels of threonine and tyrosine phosphorylation of purified PK from control, frozen and anoxic foot muscle tissue of L. littorea determined through western blotting. Chemiluminescence signal intensities were standardized to corresponding coomassie protein amount, and the values for the frozen and anoxic forms were expressed relative to the control value that was set to 1. The same superscript letters indicate no statistical difference determined by one-way ANOVA, Tukeys’s post-hoc, p < 0.05. Data are mean ± SEM, n ≥ 3
3.4 Structural Stability of PK
Structural stability of PK purified from control, anoxic, and frozen L. littorea was investigated by examining stability under various durations of incubation at 60 °C (Table 5). The control form of PK was significantly less stable than the frozen and anoxic forms, with its Vmax being reduced to 50% of its original value after 9.1 ± 0.2 min, compared to 14.4 ± 0.5 min and 12.7 ± 0.9 min for PK from anoxic and frozen animals, respectively.
3.5 Bioinformatic Analysis
Sequence alignment of gastropod PK sequences and subsequent phosphorylation prediction analysis provided three potential threonine sites which were conserved among all of the gastropod species investigated (Online Resource 1). In P. canaliculata, the closest relative to L. littorea, these sites corresponded to T165 (prediction score: 0.797), T392 (prediction score: 0.842) and T453 (prediction score: 0.932). Prediction scores range from 0 to 1, with higher scores indicating a greater likelihood that a given residue is phosphorylated based on the similarity of the sequence to known phosphorylation sites.
Phospho-Site Plus was used to compare the predicted threonine phosphorylation sites of PK in P. canaliculata to known, experimentally determined phosphorylation sites in the human homolog of this protein (Online Resource 1). Three common sites were identified; two of these were predicted in all mollusk species investigated except for Aplysia californica, the most divergent sequence. These two sites corresponded to T135 (prediction score: 0.737) and T564 (prediction score: 0.706) in P. canaliculata.
4 Discussion
Animals living in the intertidal zone often exhibit significant physiological and biochemical adaptations that allow them to survive exposure to two vastly different environmental conditions over the course of the tidal cycle. This is the case for many marine molluscs, which display metabolic rates just 2–20% of their resting values when exposed to anoxic conditions as they often are at low tide [12]. This led to the hypothesis that PK, a key enzyme of carbohydrate metabolism, is regulated during exposure to anoxic and freezing conditions to enable metabolic rate depression. Previous studies suggest that reversible phosphorylation of glycolytic enzymes plays an important role in supporting metabolic arrest in a variety of animals coping with environmental stresses [30,31,32]. The results of this study suggest that this is also the case in L. littorea, with PK playing a key role in mediating survival in anoxic conditions.
The present study demonstrated several differences between the kinetic properties of PK purified from control and anoxic tissues. Significantly, the Km PEP was 2.7-fold higher in the anoxic animal compared to the control animal (Table 2). This significant decrease in substrate affinity is indicative of suppression of PK activity in response to anoxia. This result is consistent with anoxia-induced metabolic rate depression that is well-documented in marine invertebrates, including L. littorea, which supports metabolic reorganization when oxygen for fuel catabolism is limited [9].
PK from anoxic animals was also found to be more sensitive to activation by the fermentative fuel source aspartate, demonstrated by a 1.68-fold greater fold-activation relative to PK from control animals (Table 4). Inhibition by the associated end product of aspartate metabolism, alanine, (Table 3) was also greater in PK from anoxic animals compared to PK from control and frozen animals, exhibited by a 56% decrease in the I50 alanine. Furthermore, while the inhibitory effects of alanine on the control and frozen forms of PK were somewhat alleviated by the addition of the activator aspartate (control PK recovered to its original Vmax; frozen PK recovered to 90% of its original Vmax) this was not the case for the anoxic form of PK which remained strongly inhibited (Fig. 2).
The initial response to anoxia in many marine invertebrates (particularly bivalve or gastropod mollusks) is the coupled fermentation of glycogen and aspartate, as tissues in these animals have a high aspartate content. The breakdown of glycogen and aspartate results in accumulation of alanine and succinate and supports several hours of anaerobiosis through this fermentation [33,34,35]. If anaerobiosis continues aspartate reserves can become depleted and glycolysis will proceed as normal resulting in the formation of PEP which is subsequently metabolized by phosphoenolpyruvate carboxykinase (PEPCK), ultimately leading to the production of primarily succinate and propionate as end products [33, 36]. The results from this study suggest that regulation of PK plays a role in the metabolic transition between early and late stage anaerobiosis. The observed activation of PK by aspartate suggests that, in early anoxia exposure when aspartate reserves are plentiful, PK will be activated and lead to consumption of both aspartate and glycogen producing lactate, alanine and succinate. However, as aspartate is depleted and alanine accumulates, the effects of aspartate activation on PK are lessened and PEP is increasingly pushed through PEPCK instead. This is further supported by the anoxia-induced increase in sensitivity of PK to inhibition by alanine (Table 3), which accumulates early on in anaerobiosis, as well as by the decreased affinity of PK for PEP (Table 2). Pushing PEP through PEPCK during anoxia exposure provides energy by shunting through alternative pathways of energy production. Indeed, one study revealed that the ratio of PEPCK:PK activity, a good marker of anaerobic metabolic capacity, increased in different Littorina spp. that inhabit higher shore environments and therefore undergo more prolonged periods of anoxia exposure [37]. The results of the current study expand on these previous studies by suggesting that increased PEPCK:PK activity is supported by the effects of anaerobic metabolites, in particular aspartate and alanine.
The effects of other inhibitory metabolites on PK activity were also investigated. The anoxic form of PK appeared to be more sensitive to inhibition by KCl than both the control and frozen forms of the enzyme (Table 3). Intertidal gastropods can indeed be exposed to significant hypoosmotic and hyperosmotic stress at low tide as small tidal pools can be diluted by rainwater or evaporated. Unsurprisingly then, L. littorea exhibit strong resistance to osmotic shock and concentrations of key ions, including potassium, appear to remain relatively constant when challenged with osmotic stress [38,39,40]. The relatively constant nature of potassium concentration may make it a reliable regulator of PK activity, especially considering the anoxia-induced increased sensitivity to KCl inhibition observed in the current study. This may serve to further suppress PK activity at low tide during bouts of anoxia exposure. The effects of another potential activating compound, isoleucine, were also compared, though this effect did not vary between the three conditions (Table 4). However, when combined with the inhibitor alanine, both isoleucine and FBP were found to rescue the activity of PK to varying degrees (Fig. 2). Taken together, these results show that the anoxic form of PK is generally more susceptible to allosteric regulation, with alanine and aspartate acting as particularly important regulators in modulating the activity of PK in anaerobic conditions.
The stability of PK increased significantly in the anoxic and frozen animals compared to the non-stressed controls, demonstrated through incubation studies at 60 °C (Table 5). The time required to reduce the enzyme’s activity to 50% of its original value increased 1.6-fold in anoxic animals and 1.4-fold in frozen animals when compared to the control values. This phenomenon may be attributable to the necessity to limit protein turnover during times of environmental and metabolic stress. In the periwinkle, these observations are supported by previous studies showing increased expression of protein chaperone and antioxidant defenses in response to anoxia exposure [9, 41]. This study suggests that PK stability is prioritized during anoxic stress, potentially to reduce the rate of protein turnover and conserve energy.
Kinetically, an interesting pattern emerges when comparing the three conditions investigated. The PK from anoxic animals exhibits numerous kinetic differences compared to the control form (Tables 2, 3 and 4). However, the frozen form exhibits fewer kinetic changes relative to the control (indeed, only sensitivity to inhibition by urea changed between control and frozen PK). This is notable as most freeze tolerant species exposed to freezing conditions are simultaneously exposed to anoxic conditions, accumulating anaerobic end products. This is thought to be due to freezing of the hemolymph inducing ischemia which hinders adequate oxygen supply to tissues [42]. However, the results presented here indicate that the regulation of PK, and perhaps metabolic rate in general, are not prioritized during freezing to the extent that it is during anoxia exposure. It is possible that the low-temperature conditions associated with freezing as a stress occur slowly relative to anoxia. For instance, in the animal experiments performed in this study, whole-body freezing was observed around the first hour of incubation at − 6 °C. As such, oxygen availability would be gradually decreased over the course of this treatment. Anoxia-treated animals, however, were removed from oxygen immediately. Differences in PK regulation may therefore be due to an abrupt versus a progressive exposure to low oxygen, with abrupt changes initiating a more pronounced response. It is also possible that the low temperatures associated with freezing may be sufficient to suppress metabolic rate and therefore direct regulation of metabolic enzymes is no longer necessary. Similar stress-response patterns have been observed in other animals [43] and, considered with the results presented here, suggest that despite some similarities between anoxia and freezing, the metabolic strategies that are required to cope with each stress may differ.
The differences observed in the relative stabilities and kinetic properties of PK purified from control, frozen and anoxic tissues may be explained by structural differences. Often, these changes are due to stable, reversible covalent modifications to the proteins, such as reversible protein phosphorylation (RPP). Phosphorylation levels of threonine and tyrosine residues were therefore assessed by western blotting, which showed a significant increase in threonine phosphorylation of PK from anoxic animals compared to PK from control (2.8-fold increase) or frozen (2.4-fold increase) animals (Fig. 3). Interestingly, PK from control and frozen animals were far more similar to each other in terms of both phosphorylation states and kinetic properties than they were to PK from anoxic animals. This suggests that RPP is responsible for suppression of PK activity during anoxia. Anoxia exposure in intertidal spaces can be considered a relatively brief period of environmental stress, typically lasting for only a few hours. RPP therefore provides an economical regulatory mechanism to suppress metabolic activity instead of degrading enzymes that need to be suppressed during this period only to have them re-synthesized a few hours later.
PK has long been known to be phosphorylated in many mammalian models that possess a liver specific isozyme which is highly regulated by phosphorylation [44, 45]. More recently however, the enzyme has been observed to be regulated by phosphorylation in response to environmental stress, including in other marine invertebrates [18, 19, 21,22,23]. To investigate the mechanism underlying this conserved regulatory strategy, this study identified three potential threonine phosphorylation sites on PK conserved among five sequenced gastropod species using NetPhos3.1. Additionally, two threonine sites, which were conserved and predicted to be phosphorylated in the four most closely related gastropod species, have also been reported to be phosphorylated in the homologous human sequence (Online Resource 1). While we cannot be certain which of these residues is responsible for the stress-dependent PK regulation observed in these animals, the conserved nature of these sites across gastropods, and even humans, makes them plausible candidates.
Interestingly, a previous study on unpurified PK from the foot muscle of L. littorea also suggested that phosphorylation was responsible for kinetic differences observed between control and anoxic forms of PK [13]. However, contrary to the results found here, the previous study concluded that PK was dephosphorylated in response to anoxia, as the enzyme was found to bind less strongly to a positively charged ion-exchange chromatography column following anoxia exposure. These differing results demonstrate the dynamic nature of enzyme regulation by RPP. While the previous study suggested a net dephosphorylation of PK in response to anoxia, this study demonstrates that PK exhibits increased levels of phosphorylation on threonine residues specifically. It is important to note that the results of these two studies are not incompatible; the current study results do not preclude the possibility that PK is also significantly modified in another way such that the enzyme is net less negatively charged following anoxia exposure.
PK purified from L. littorea foot muscle exhibits distinct kinetic properties in response to anoxia compared to when the enzyme is purified from control or frozen animals. Most notably, anoxic PK was found to have significantly lower affinity for PEP than the corresponding control or frozen forms of the enzyme, and it was also significantly more susceptible to allosteric regulation via alanine and aspartate. Western-blotting analysis showed that the anoxic form of the enzyme is significantly more phosphorylated on threonine residues than both the control and frozen forms of the enzyme, providing a possible explanation for the observed changes in kinetic properties. By suppressing the activity of PK, anoxia-exposed animals are able to maintain a lower metabolic rate and support alternative pathways of carbohydrate fermentation during periods of low oxygen availability.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Lutz PL, Storey KB (2011) Adaptations to variations in oxygen tension by vertebrates and invertebrates. Compr Physiol. https://doi.org/10.1002/cphy.cp130221
De Zwaan A, Putzer V (1985) Metabolic adaptations of intertidal invertebrates to environmental hypoxia (a comparison of environmental anoxia to exercise anoxia). Symp Soc Exp Biol 39:33–62
Aarset AV (1982) Freezing tolerance in intertidal invertebrates (a review). Comp Biochem Physiol -Part A Physiol. https://doi.org/10.1016/0300-9629(82)90264-X
English TE, Storey KB (2003) Freezing and anoxia stresses induce expression of metallothionein in the foot muscle and hepatopancreas of the marine gastropod Littorina littorea. J Exp Biol. https://doi.org/10.1242/jeb.00465
Murphy DJ (1983) Freezing Resistance in Intertidal Invertebrates. Annu Rev Physiol. https://doi.org/10.1146/annurev.ph.45.030183.001445
Larade K, Storey KB (2002) Reversible suppression of protein synthesis in concert with polysome disaggregation during anoxia exposure in Littorina littorea. Mol Cell Biochem. https://doi.org/10.1023/A:1014811017753
Larade K, Storey KB (2007) Arrest of transcription following anoxic exposure in a marine mollusc. Mol Cell Biochem. https://doi.org/10.1007/s11010-007-9468-8
Biggar KK, Kornfeld SF, Maistrovski Y, Storey KB (2012) MicroRNA regulation in extreme environments: differential expression of MicroRNAs in the intertidal snail Littorina littorea during extended periods of freezing and anoxia. Genomics Proteomics Bioinforma. https://doi.org/10.1016/j.gpb.2012.09.002
Storey KB, Lant B, Anozie OO, Storey JM (2013) Metabolic mechanisms for anoxia tolerance and freezing survival in the intertidal gastropod Littorina littorea. Comp Biochem Physiol. https://doi.org/10.1016/j.cbpa.2013.03.00
Krivoruchko A, Storey KB (2015) Turtle anoxia tolerance: biochemistry and gene regulation. Biochim Biophys Acta. https://doi.org/10.1016/j.bbagen.2015.02.001
Hand SC, Hardewig I (1996) Downregulation of cellular metabolism during environmental stress: mechanisms and implications. Annu Rev Physiol. https://doi.org/10.1146/annurev.ph.58.030196.002543
Brooks SPJ, Storey KB (1997) Glycolytic controls in estivation and anoxia: a comparison of metabolic arrest in land and marine molluscs. Comp Biochem Physiol 118:1103–1114
Russell EL, Storey KB (1995) Anoxia and freezing exposures stimulate covalent modification of enzymes of carbohydrate metabolism in Littorina littorea. J Comp Physiol B. https://doi.org/10.1007/BF00301477
Lama JL, Bell RAV, Storey KB (2013) Hexokinase regulation in the hepatopancreas and foot muscle of the anoxia-tolerant marine mollusk. Comp Biochem Physiol. https://doi.org/10.1016/j.cbpb.2013.07.001
Shahriari A, Dawson NJ, Bell RAV, Storey KB (2013) Stable suppression of lactate dehydrogenase activity during anoxia in the foot muscle of Littorina littorea and the potential role of acetylation as a novel posttranslational regulatory mechanism. Enzyme Res. https://doi.org/10.1155/2013/461374
Argaud D, Roth H, Wiernsperger N, Leverve XM (1993) Metformin decreases gluconeogenesis by enhancing the pyruvate kinase flux in isolated rat hepatocytes. Eur J Biochem. https://doi.org/10.1111/j.1432-1033.1993.tb17886.x
Rognstad R, Katz J (1977) Role of pyruvate kinase in the regulation of gluconeogenesis from L lactate. J Biol Chem 252:1831–1833
Plaxton WC, Storey KB (1984) Purification and properties of aerobic and anoxic forms of pyruvate kinase from red muscle tissue of the channelled whelk, Busycotypus canaliculatum. Eur J Biochem. https://doi.org/10.1111/j.1432-1033.1984.tb08367.x
Plaxton WC, Storey KB (1985) Purification and properties of aerobic and anoxic forms of pyruvate kinase from the hepatopancreas of the channelled whelk, Busycotypus canaliculatum. Arch Biochem Biophys. https://doi.org/10.1016/0003-9861(85)90788-X
Whitwam RE, Storey KS (1990) Pyruvate kinase from the land snail Otala lactea: regulation by reversible phosphorylation during estivation and anoxia. J Exp Biol 154:321–337
Smolinski MB, Mattice JJL, Storey KB (2017) Regulation of pyruvate kinase in skeletal muscle of the freeze tolerant wood frog, Rana sylvatica. Cryobiology. https://doi.org/10.1016/j.cryobiol.2017.06.002
Mattice AMS, MacLean IA, Childers CL, Storey KB (2018) Characterization of pyruvate kinase from the anoxia tolerant turtle, Trachemys scripta elegans: a potential role for enzyme methylation during metabolic rate depression. PeerJ. https://doi.org/10.7717/peerj.4918
Bell RAV, Storey KB (2018) Purification and characterization of skeletal muscle pyruvate kinase from the hibernating ground squirrel, Urocitellus richardsonii: potential regulation by posttranslational modification during torpor. Mol Cell Biochem. https://doi.org/10.1007/s11010-017-3192-9
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. https://doi.org/10.1016/0003-2697(76)90527-3
Brooks SPJ (1994) A program for analyzing enzyme rate data obtained from a microplate reader. Biotechniques 17:1154–1161
Brooks SPJ (1992) A simple computer program with statistical tests for the analysis of enzyme kinetics. Biotechniques 13:906–911
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol. https://doi.org/10.1038/msb.2011.75
Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. https://doi.org/10.1002/pmic.200300771
Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. https://doi.org/10.1093/nar/gkr1122
Storey KB, Storey JM (1990) Metabolic rate depression and biochemical adaptation in anaerobiosis, hibernation and estivation. Q Rev Biol. https://doi.org/10.1086/416717
Storey KB (1997) Metabolic regulation in mammalian hibernation: Enzyme and protein adaptations. Comp Biochem Physiol. https://doi.org/10.1016/S0300-9629(97)00238-7
Cowan KJ, Storey KB (1999) Reversible phosphorylation control of skeletal muscle pyruvate kinase and phosphofructokinase during estivation in the spadefoot toad, Scaphiopus couchii. Mol Cell Biochem. https://doi.org/10.1023/A:1006932221288
Storey KB, Storey JM (2004) Oxygen limitation and metabolic rate depression. In: Storey KB (ed) Functional metabolism, 1st edn. Wiley, Hoboken, pp 415–442
Churchill TA, Storey KB (1996) Metabolic responses to freezing and anoxia by the periwinkle Littorina littorea. J Therm Biol. https://doi.org/10.1016/0306-4565(95)00022-4
Wieser W (1980) Metabolic end products in three species of marine gastropods. J Mar Biol Assoc UK. https://doi.org/10.1017/S0025315400024231
Storey KB (1986) Aspartate activation of pyruvate kinase in anoxia tolerant molluscs. Comp Biochem Physiol. https://doi.org/10.1016/0305-0491(86)90151-3
Sokolova IM, Pörtner HO (2001) Temperature effects on key metabolic enzymes in Littorina saxatilis and Littorina obtusata from different latitudes and shore levels. Mar Biol. https://doi.org/10.1007/s002270100557
Sokolova IM, Bock C, Pörtner HO (2000) Resistance to freshwater exposure in White Sea Littorina spp. I: anaerobic metabolism and energetics. J Comp Physiol. https://doi.org/10.1007/s003600050264
Hoyaux J, Gilles R, Jeuniaux C (1976) Osmoregulation in molluscs of the intertidal zone. Comp Biochem Physiol Part A. https://doi.org/10.1016/S0300-9629(76)80157-0
Taylor PM, Andrews EB (1988) Osmoregulation in the intertidal gastropod Littorina littorea. J Exp Mar Bio Ecol. https://doi.org/10.1016/0022-0981(88)90210-9
Pannunzio TM, Storey KB (1998) Antioxidant defenses and lipid peroxidation during anoxia stress and aerobic recovery in the marine gastropod Littorina littorea. J Exp Mar Bio Ecol. https://doi.org/10.1016/S0022-0981(97)00132-9
Storey KB, Storey JM (1988) Freeze tolerance in animals. Physiol Rev. https://doi.org/10.1152/physrev.1988.68.1.27
Abboud J, Storey KB (2013) Novel control of lactate dehydrogenase from the freeze tolerant wood frog: role of posttranslational modifications. PeerJ. https://doi.org/10.7717/peerj.12
Titanji VPK, Zetterqvist Ö, Engström L (1976) Regulation in vitro of rat liver pyruvate kinase by phosphorylation-dephosphorylation reactions, catalyzed by cyclic-AMP dependent protein kinases and a histone phosphatase. Biochim Biophys Acta. https://doi.org/10.1016/0005-2744(76)90011-5
Ljungström O, Berglund L, Engström L (1976) Studies on the kinetic effects of adenosine-3′: 5′-monophosphatedependent phosphorylation of purified pig-liver pyruvate kinase type L. Eur J Biochem. https://doi.org/10.1111/j.1432-1033.1976.tb10837.x
Acknowledgements
This study was funded through a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (#6793). K.B.S. holds the Canada Research Chair in Molecular Physiology. M.B.S was funded through an Ontario Graduate Scholarship. The authors thank J.M. Storey for editorial review of this manuscript.
Funding
This study was funded through a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant (#6793). K.B.S. holds the Canada Research Chair in Molecular Physiology. M.B.S was funded through an Ontario Graduate Scholarship.
Author information
Authors and Affiliations
Contributions
MBS carried out experiments, analyzed the data, and helped draft the manuscript. AV analyzed the data and drafted the manuscript. SRG carried out experiments and revised the manuscript. KBS secured funding, conceived the experiments, and helped draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical Approval
Protocols were approved by the Carleton University Animal Care Committee.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Smolinski, M.B., Varma, A., Green, S.R. et al. Purification and Regulation of Pyruvate Kinase from the Foot Muscle of the Anoxia and Freeze Tolerant Marine Snail, Littorina littorea. Protein J 39, 531–541 (2020). https://doi.org/10.1007/s10930-020-09934-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-020-09934-9