
Abstract
Coronary artery disease, the leading cause of death in the developed and developing countries, is prevalent in diabetes mellitus with 68% cardiovascular disease (CVD)-related mortality. Epidemiological studies suggested inverse correlation between HDL and CVD occurrence. Therefore, low HDL concentration observed in diabetic patients compared to non-diabetic individuals was thought to be one of the primary causes of increased risks of CVD. Efforts to raise HDL level via CETP inhibitors, Torcetrapib and Dalcetrapib, turned out to be disappointing in outcome studies despite substantial increases in HDL-C, suggesting that factors beyond HDL concentration may be responsible for the increased risks of CVD. Therefore, recent studies have focused more on HDL function than on HDL levels. The metabolic environment in diabetes mellitus condition such as hyperglycemia-induced advanced glycation end products, oxidative stress, and inflammation promote HDL dysfunction leading to greater risks of CVD. This review discusses dysfunctional HDL as one of the mechanisms of increased CVD risks in diabetes mellitus through adversely affecting components that support HDL function in cholesterol efflux and LDL oxidation. The dampening of reverse cholesterol transport, a key process that removes cholesterol from lipid-laden macrophages in the arterial wall, leads to increased risks of CVD in diabetic patients. Therapeutic approaches to keep diabetes under control may benefit patients from developing CVD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High-density lipoprotein’s role in cardiovascular disease
Coronary artery disease (CAD) remains the leading cause of death in the United States and many developed and developing countries [1]. While elevated levels of low-density lipoprotein cholesterol (LDL-C) and triglycerides are risk factors for developing coronary artery disease [2], the excessive accumulation of cholesterol by macrophages and subsequent conversion to foam cells [3, 4] sets the stage of atherosclerosis progression. Correlation of LDL-C to CAD necessitated statin therapy to prevent atherosclerosis, primarily by inhibiting HMG-CoA reductase, a key enzyme in the de novo cholesterol synthesis, and thereby leading to decreased serum LDL-C [2, 5, 6]. Low levels of high-density lipoprotein cholesterol (HDL-C) is another prominent risk factor for developing premature atherosclerosis [7]. Despite documented benefits of statins [2], a good proportion of individuals still remain at a higher risk of developing CAD [8]. At least in prior clinical studies, it was demonstrated that HDL-C levels inversely correlated with the risk of coronary artery diseases [8,9,10,11] as evidenced by clinical trial results [12,13,14].
The increasing incidence of diabetes worldwide and metabolic derangement and risk factors associated with this, leads to the development of cardiovascular disease (CVD) [15,16,17]. Among those, low HDL-C is characterized as one of the features of metabolic syndrome (MetS). The other risk factors include dyslipidemia, hypertriglyceridemia, hypertension, and impaired glucose tolerance. A recent report suggests that MetS is becoming pandemic and the number of individuals suffering from MetS is likely to double by 2030 worldwide [18]. Therefore, aiming to correct dyslipidemia and increase HDL-C appeared to be a plausible therapeutic approach to reduce the risks of developing atherosclerotic lesion formation. Low HDL-C levels are the most common lipid abnormalities observed in men with CAD [11]. ApoA-I, the major protein component of HDL, determines the blood levels of HDL-C [19] and promotes cholesterol efflux, which in turn promotes reverse cholesterol transport. Therefore, raising HDL-C was thought to have protective effects against developing CAD. HDL’s protective role occurs through inhibition of atherogenesis by promoting cholesterol efflux from peripheral tissues and from lipid-laden macrophages and arterial smooth muscle cells [20]. HDL also inhibits atherogenesis through other pathways like its direct effect on the vessel wall and inhibiting lipoprotein oxidation [21]. The most discussed atheroprotective function of HDL is enhancement of reverse cholesterol transport, a process in which HDL receives excess cholesterol from the peripheral tissues, including macrophages in the arterial wall, which is subsequently delivered to the liver for biliary excretion [19] (Fig. 1). The discovery of scavenger receptor-BI (SR-BI) [22, 23], ATP-binding cassette transporter A1 (ABCA1) [24, 25], and ATP-binding cassette transporter G1 (ABCG1) [26] have further added to our understanding of reverse cholesterol transport.

Cholesterol transport. Apolipoprotein A-I, synthesized from the liver and the gut, forms nascent HDL particles. The nascent discoidal HDL particle accepts cholesterol and phospholipids from the peripheral tissues in ABCA1-dependent manner, and gets converted into cholesteryl ester-rich mature HDL particles. The mature HDL particles are then taken to the liver in a process called reverse cholesterol transport in which SR-B1 plays an important role in docking and accepting the cholesterol esters from HDL particles. The cholesterols delivered to the liver is converted into bile by cholesterol 7-α hydroxylase and excreted to the gut
Key roles of ABCA1 in HDL biogenesis, RCT, and atherosclerosis
A correlation between cholesterol efflux from macrophages and serum apoA1 concentration was first shown by Fournier et al. [27], which suggested a role of apoA-I in cellular cholesterol efflux. Later studies in a variety of cell-types showed that other apoproteins also function as cholesterol acceptors [28]. Marked induction in cholesterol efflux to acceptor lipid-poor apoA-I was observed in macrophages following treatment with cAMP [29, 30]. These observations together with other studies [31, 32] suggested the existence of an interaction between the acceptor apolipoproteins and the cell membrane component(s). In addition to HDL, other players such as SR-BI [23], ABCA1 [25], and hepatic lipase [33] have been shown to be part of the reverse cholesterol transport pathway.
Following the findings in WHAM chickens, the role of ABCA1 gained recognition in HDL biogenesis and reverse cholesterol transport and provided important insights into the correlation between cholesterol efflux and circulating HDL concentration [34]. In these mutant chickens, despite normal secretion rates of apoA-I, they have only 5% of the HDL compared to normal due to rapid catabolism of secreted apoA-I if not assembled into HDL particles. Subsequent studies confirmed the specific role of ABCA1 in the cellular cholesterol trafficking [35,36,37]. In the HDL biogenesis process, the discoidal lipid-poor apoA-I particle functions as a cholesterol acceptor and gets converted into spherical mature HDL particle, which then delivers this cholesterol to the liver and steroidogenic tissues via scavenger receptor class B type 1 (SR-B1)-mediated pathway (Fig. 1). ABCA1 participates in the reverse cholesterol transport by facilitating the efflux of cholesterol in an energy-dependent manner from cells to the acceptor lipid-poor apoA-I-particles (Fig. 2), which are then taken to the liver for excretion as bile salts. Despite other apoproteins being able to induce cholesterol efflux, lipid-poor apoA-I appears to be the preferred acceptor of ABCA1-mediated lipid efflux. Since apoA-I has been shown to specifically bind to ABCA1 [38], the lipid-poor apoA1 (preβ-HDL) functions as an acceptor of cholesterol and phospholipid in an ABCA1-dependent manner resulting the formation of mature cholesterol ester-rich spherical α-HDL particles following the action of LCAT [39]. SR-BI interacts with the mature α-HDL and facilitates the uptake of CE from the HDL particles (Fig. 2). Thus, any compromise in the function of HDL may lead to impaired cholesterol efflux leading to increased risk of CVD (Fig. 3).

A ABCA1-mediated cholesterol efflux. Lipid-poor discoidal HDL particle in the circulation accepts cholesterol and phospholipids from the tissues via ABCA1-mediated pathway. ABCA1 is a membrane protein that facilitates the transport of cholesterol and phospholipids from the tissues to the lipid-poor HDL particles and, as a result, the nascent HDL particles get converted into mature cholesterol ester-rich HDL particles capable of transporting cholesterol to the liver for excretion. Any defect in the membrane-associated ABCA1 protein renders them unable to mediate the cellular cholesterol efflux resulting into the deposition of cholesterol within the tissues. B ABCA1-and ABCG1-mediated cholesterol efflux requires energy in the two-step HDL maturation. Lipid-poor discoidal HDL particle in the circulation becomes lipidated by ABCA1using ATP followed by ATP-dependent maturation of HDL by ABCG1

Schematic representation of atheroprotective effects of ABCA1. A Lipid-laden macrophages in the arterial walls set the stage for the initiation of atherosclerosis via a variety of mechanisms. The removal of lipids from the macrophages is therefore a prerequisite in the process of inhibition of atherosclerosis. B ABCA1-inducing agents and stimuli accelerate the removal of cholesterol and phospholipids from the lipid-laden macrophages, and reduce the risk of predisposition to atherosclerosis
Preβ-HDL particle formation and function
Pre β HDL fraction of HDL constitutes a heterogeneous population generated de novo by interaction between lipid-free apoA-I and ABCA1 [25] and these particles include lipid-free apoA-I to Apo-I-lipid complexes of varying sizes [40]. Thus, the only protein in the preβ-HDL is the apoA-I that has high affinity for lipids at its C-terminus that allows rapid association of apoA-I with phospholipids when preparing reconstituted HDL (rHDL) [41, 42]. The nascent phospholipid-apoA-I complex induces cholesterol efflux [43]. Depending upon the size of apoA-I-lipid complex, these particles are classified into preβ-HDL1 (smaller) and preβ-HDL2 (larger) particles [44]. As depicted in Fig. 2 the lipid-poor apoA-I generated through interaction with ABCA1gets more lipidated via ABCG1-mediated lipid efflux. The maturation of discoidal HDL particles into spherical HDL particles is carried out by LCAT-mediated esterification [39]. The smaller preβ-HDL, preβ1, with 1-2 apoA-I per particle, is suggested to be more efficient in effluxing cholesterol from cells. The steady state level of preβ1 particles is emerging as a biomarker for the protective role of HDL and an independent indicator of CAD risk in humans [45]. Thus, the function and maturation of HDL is linked with the function of ABCA1 that lipidates lipid-poor apoA-I through cholesterol efflux and contributes to the reverse cholesterol transport pathway [46], suggesting that the formation and functionality of HDL is tightly linked to production of apoA-I and membrane-associated transporters, ABCA1 and G1 [47]. Any defect in these proteins lead to dysfunctional HDL [35, 48,49,50].
HDL’s role in preventing LDL oxidation
While the main function of HDL that contributes to the atheroprotective property is its ability to efflux cholesterol from lipid-laden cells and arteries [20] (Fig. 3), the inhibition of LDL oxidation by HDL is another important property of HDL in attenuating progression of atherosclerosis [51]. Thus, HDL on one hand inhibits progression of atherosclerosis and on the other hand promotes plaque regression. HDL acquires antioxidative property by HDL-associated proteins such as paraoxonase I (PON1) and apoA-I. The association of apoA-I, CETP, LCAT, and PON1 has been shown to enhance HDL’s ability to inhibit LDL oxidation [52, 53]. Among these associated proteins, PON1 appears to be the most important player in conferring antioxidant property to HDL particles to protect LDL from undergoing oxidation [54]. Myeloperoxidase (MPO) and PON1 are two key proteins in the promotion and prevention of LDL oxidation, respectively. While MPO is known to cause oxidative modification of lipoproteins [55, 56], PON1 prevents oxidation of lipoproteins [57, 58]. PON1 inhibits oxLDL-induced MCP1 formation by endothelial cells and MCP1 is known to induce recruitment of monocytes into the subendothelial space, a process that sets the stage for the initiation of atherogenesis. Attenuation of oxidative stress in macrophages by PON1 in transfected cells as well as in PON1 transgenic mice [59, 60] reduces atherosclerotic lesion formation [61], and PON1 deficiency was found to be associated with increased macrophage oxidative stress and atherosclerosis [62]. Thus, the ability of HDL to inhibit LDL oxidation appears to be largely dependent on the HDL-associated protein, PON1, which dampens the oxidative stress and confers LDL protection.
Elevated oxidative stress in diabetes mellitus causes oxidative modification of HDL particle and its main protein component, apoA-I, and contributes to the generation of dysfunctional HDL [56, 63,64,65,66]. Hyperglycemia in dyslipidemic non-diabetic individuals induces oxidative modification of HDL resulting in higher oxidized HDL [67]. HDL’s antioxidant property is impaired in Type 1 diabetic individuals [68], suggesting a distinct role of high glucose in impairing HDL’s function as an antioxidant. It is possible to restore HDL function by infusing the apoA-I mimetics, the main protein component of HDL [69]. Thus, the ratio of oxLDL to LDL and oxLDL to HDL are important in determining the risks for developing CVD. Indeed, Motamed et al. [70] determined in type 2 diabetes patients that both oxLDL/LDL and oxLDL/HDL are potent biomarkers for oxidative stress, and support earlier studies by Girona et al. [71] who showed that oxidized lipoprotein ratios are associated with atherosclerotic lesion formation in patients with diabetes. Thus, decreased HDL antioxidant capacity is important in atherosclerosis susceptibility [72]. Pathological and physiological conditions that strip off or decrease HDL-associated proteins, PON-1 [62], apoA-I [73,74,75], or LCAT [76, 77] have been shown to either decrease HDL or make them susceptible to oxidation.
Raising circulating HDL to promote reverse cholesterol transport and attenuate atherosclerosis progression
Epidemiological studies suggested that raising HDL may be beneficial in reducing the risk of CVD [14]. Several approaches have been tried to raise HDL, including CETP inhibition [78], LCAT activation [79, 80], and infusing nascent HDL particles to regress atheroma volume [81]. The very first CETP inhibitor, torcetrapib [82], despite showing massive increase in HDL concentration in ILLUMINATE clinical trial, did not show any benefit in outcome studies, possibly due to high aldosterone levels [83, 84]. Another class of CETP inhibitor, dalcetrapib that covalently binds to CETP, showed enhanced RCT in preclinical models, also failed to show positive results in cardiovascular outcome studies (dal-OUTOMES) [78]. While torcetrapib dampened endothelial function because of increased aldosterone [83], dalcetrapib increased CRP and thereby increased vascular inflammation [85]. These adverse effects may have resulted in lack of positive outcome. Macrophage-to-feces RCT involves ABCA1-dependent cholesterol efflux and may be considered as a RCT biomarker [86]. However, both elevation of CETP as well as absence of CETP increase RCT in this assay [87], thus making it difficult to interpret the data. Anacetrapib is being evaluated in REVEAL clinical trial and the results are expected to be announced in 2017 [78]. A potent and selective CETP inhibitor, evacetrapib, with no effect on aldosterone is being evaluated in ACCENTUATE trial for LDL reduction and in ACCELERATE trial in patients with high risk vascular disease [78]. Because these selective CETP inhibitors massively reduce LDL cholesterol, the clinical data would require careful evaluation to determine contribution of HDL elevation in atheroprotection.
In order to avoid chemotype and class effect of small molecule CETP inhibitors, researchers have explored other approaches like CETP antibody [88] and siRNA [89], albeit only in the preclinical animal models, primarily to show proof-of-concept of these approaches. That the elevation of HDL may not necessarily impart all beneficial effects was demonstrated by mouse genetic models overexpressing SR-BI [90, 91] or lacking SR-BI [92]. Mice lacking SR-BI, although had elevated HDL levels, but showed decreased RCT in macrophage-to-feces assays and increased atherosclerosis [92, 93]. On the other hand, mice overexpressing SR-BI had lower HDL concentrations compared to WT littermates and exhibited higher RCT activity in MS-RCT assay and showed atherosclerosis attenuation [94, 95]. It was observed that HDL functionality was compromised in SR-BI knockout mice despite higher HDL concentration [96], suggesting that SR-BI is atheroprotective and lack of SR-BI while showed increased HDL, but appeared to be dysfunctional that contributed to lack of atheroprotection. Further support to this hypothesis comes from human genetic studies that identified a rare SR-BI variant with higher HDL concentration, but increased risks of coronary artery disease [97]. These findings are consistent with the notion that HDL function is more important than the HDL concentration and any factor that dampens HDL function may have negative effects on CVD outcome. It is quite possible that the lack of benefit in CVD outcome studies with RVX-208 [98] and CER-001 [99] may have to do with the compositional changes in HDL that influences HDL function in a way that does not translate into CVD benefits. RVX-208 is identified as a BET inhibitor [100] and in one study shown to influence glucose metabolism [101]. A clear mechanism of action of BET inhibitors in glucose production or excursion is likely to add further knowledge to our current understanding and to establish a meaningful link between BET inhibition and diabetes.
HDL modification in diabetes mellitus impacts reverse cholesterol transport
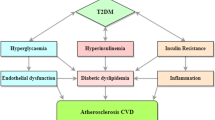
Individuals with diabetes have a greater risk of developing CVD compared to non-diabetic individuals since 2/3rd of CVD-related deaths occur in diabetic population [102]. At least 68% of people age 65 or older with diabetes die from some form of heart disease; and 16% die of stroke. Therefore, adults with diabetes are two to four times more likely to have heart disease or a stroke than adults without diabetes. Given the projected diabetes population worldwide and in the US in particular [103], even larger proportion of population appears to be at risk of developing CVD complications. Type 2 diabetes mellitus and the cluster of pathologies characteristics of metabolic syndrome including insulin resistance, obesity, and high plasma triglycerides are often associated with low HDL [104, 105], and renders them to become dysfunctional as a result of the formation of advanced glycation end products [104, 106,107,108]. Insulin resistance contributes to low HDL cholesterol, and low HDL may promote development of diabetes [109], predict the development of type 2 diabetes in prediabetics [110], and promote progression of glycemia in those with established T2DM [111]. Individuals who do intensive exercise tends to have high HDL and also show improved glucose tolerance [112], suggesting a link between low HDL and energy homeostasis. One of the risk factors of CVD is low level of HDL [8,9,10,11] as seen in individuals with diabetes [104, 105]. Therefore, individuals with diabetes would have higher risks of developing CVD [102] as a result of impaired reverse cholesterol transport through reduced cholesterol efflux capability [107, 113, 114]. Impairment in the RCT may increase CVD risk [115]. Indeed, Rohatgi et al. [116] investigated the cholesterol efflux capacity and its association with incidence of atherosclerotic CVD outcome in a large population cohort. These investigators not only measured the concentration of HDL and number of HDL particles, they also measured cholesterol efflux capacity at baseline in 2924 adults free from CVD from the Dallas Heart Study, a probability-based population sample. The primary endpoint was defined as a first non-fatal myocardial infarction, non-fatal stroke, or coronary revascularization or death from cardiovascular causes, all grouped as atherosclerotic CVD, with a median follow-up period of 9.4 years. The cholesterol efflux capacity, a new biomarker that characterizes a key step in reverse cholesterol transport, was found to be inversely associated with the incidence of cardiovascular events in a population-based cohort. This finding showed the importance of HDL function above plasma HDL cholesterol concentration and received recognition as a surrogate biomarker for CAD risk [117]. Thus, a correlation between cholesterol efflux capability of the serum and incidence of CVD appears to exist. However, this needed to be validated in individuals with diabetes. Kubota et al. [118] carried out serum cholesterol efflux studies in individuals with glucose intolerance. An inverse correlation was found between the cholesterol efflux capability and extent of glucose intolerance in an oral glucose tolerance test in all subjects. Most notably, the serum cholesterol efflux capacity was significantly lower in subjects with glucose intolerance. This study established a link between glucose intolerance and cholesterol efflux and demonstrated that cholesterol efflux capacity is impaired in Japanese-Americans newly diagnosed with glucose intolerance. As suggested [116], the impairment in cholesterol efflux capacity in these individuals may contribute to increased risk of atherosclerotic CVD.
One of the hallmarks of diabetes is increased glycation end products [119,120,121,122]. Since advanced glycation end products promote oxidative stress leading to oxidation of physiologically important biomolecules and increased inflammation, it is possible these biological attributes of glycated proteins in individuals with diabetes may impact the functionality of HDL. To address this, an elegant study was carried out by Mechado-Lima et al. [123]. Basically, these investigators isolated albumin from non-diabetes and type 1 diabetes mellitus individuals and treated J774 cells loaded with 3H-Cholesterol followed by measurement of cholesterol efflux to the media apoA-I, HDL3 or HDL2. Simultaneously, they also measured intracellular ABCA1 protein content and a set of genes by real-time PCR. Both apoA-I and HDL2-mediated cholesterol efflux were found to be impaired in macrophages treated with albumin isolated from diabetic patients compared with non-diabetic albumin-treated cells, which was attributed to intracellular ABCA1 protein content, demonstrating that the advanced glycated albumin isolated from poorly controlled type 1 diabetes mellitus patients alters macrophage gene expression impairing ABCA-1-mediated reverse cholesterol transport that possibly contributes to the increased risk of CVD in diabetic patients. A similar study by Traldi et al. [122] with glycated human serum albumin isolated from poorly controlled diabetic patients showed impairment of cholesterol efflux from macrophages. They treated mouse peritoneal macrophages with human serum albumin isolated from control, type 1 and type 2 diabetic subjects and measured gene expression related to cholesterol efflux as well as cholesterol efflux using J774 macrophages. ABCA-1 protein level and apoA-I mediated cholesterol efflux reduced by 50 and 60%, respectively, in macrophages exposed to HSA from type 1 and type 2 diabetic patients when compared to that exposed to HSA from control subjects. Thus, compromised RCT in diabetes mellitus contributes to atherosclerosis.
A comprehensive clinical study in 1745 diabetic patients and 1749 control patients from the EPIC- Norfolk study of 25,639 individuals were carried out by Saleheen et al. [124]. These investigators quantified cholesterol efflux capacity in 1745 individuals with reported incidence of coronary heart disease and 1749 individuals with no cardiovascular disorder by a widely accepted cholesterol efflux assay using J774 cells loaded with radiolabel cholesterol. Their studies showed a positive correlation of cholesterol efflux with both HDL-C as well as apoA-I, the main apoprotein of HDL that determines HDL concentration and to a great extent HDL function [19]. Interestingly, cholesterol efflux showed an inverse correlation with diabetes, a finding confirming earlier studies [118, 122, 123]. Additionally, cholesterol efflux capacity showed an inverse correlation with incidence of coronary heart disease events in this study [124], suggesting cholesterol efflux capacity of HDL as a predictor of coronary heart disease. A parallel study by Bao et al. [125] provided mechanistic insights into the correlation of cholesterol efflux capacity and type-2 diabetes mellitus. Along with serum cholesterol efflux, these researchers measured expression of CYP7A1, ABCG5, and LXR-beta in the peripheral blood monocytes by realtime PCR and Western blot. Out of 30 type-2 diabetes patients recruited in this study, half of them had complicated heart disease. Fifteen normal control individuals with no diabetes were recruited for comparison. Only CYP7A1 mRNA and protein showed correlation with the cholesterol efflux capacity. A significantly lower rate of macrophage cholesterol efflux was noticed in patients with type 2 diabetes compared to normal control subjects. Since a positive correlation between cholesterol efflux capacity and CYP7A1 existed, it was concluded that the reduction in cholesterol efflux capacity in type 2 diabetes patients is associated with the down-regulation of CYP7A1 expression.
To address impaired cholesterol efflux capacity in type 2 diabetic patients, a quite different approach was undertaken by Apro et al. [126]. These researchers isolated HDL from interstitial fluid as well as from peripheral plasma from type 2 diabetes patients (n = 35) and non-diabetic control individuals (n = 35). Both in normal control individuals as well as in diabetic patients, the cholesterol efflux assay showed lower efflux capacity in interstitial fluid as compared to the peripheral plasma. Whereas, plasma efflux capacity in type 2 diabetic patients were 10% lower compared to normal control individuals, the interstitial fluid cholesterol efflux capacity in type 2 diabetic patients showed a 28% reduction, suggesting that interstitial fluid cholesterol efflux capacity in type 2 diabetes mellitus is severely impaired and may contribute to their increased risk of CAD. In a mouse model of streptozotocin-induced diabetic nephropathy, Tsun et al. [127] studied the role of ABCG1 and SR-BI in renal cellular cholesterol efflux by evaluating expression of cholesterol transporters. In vitro studies established hyperglycemia-induced reduction in cholesterol transporters, ABCA1, G1, and SR-BI. Similar reduction in these three cholesterol transporters were observed in the kidney of streptozotocin-induced mouse model of diabetic nephropathy, suggesting that cholesterol efflux in kidney is compromised in type 1 diabetic conditions, leading to lipid accumulation in the kidney.
In terms of what makes HDL dysfunctional that dampens cholesterol efflux capacity, among other factors, posttranslational modification of HDL has been suggested as one of them [128]. Although other posttranslational modifications of HDL in diabetic patients have been noted [129], the oxidative modification of HDL particle and its main protein component, apoA-I, appears to be the primary cause of rendering HDL dysfunctional [56, 63,64,65,66]. Poor glycemic control in type-1 diabetes is associated with accelerated oxidative damage to apolipoprotein (apo) A-I [130] and advanced glycated albumin diminishes anti-inflammatory properties of HDL [131, 132]. Reconstituted HDL (rHDL) shows anti-inflammatory activity in humans [133,134,135]. ABCA1-mediated cholesterol efflux capability of HDL is compromised in type 2 diabetes patients [136], possibly caused by the oxidatively damaged apoA-I and increased inflammation [137, 138] (Fig. 4). Since antioxidative and anti-inflammatory properties of HDL are impaired in diabetics [138], this may contribute to HDL dysfunction [139]. HDL undergoes modification and multiple structural changes in an inflammatory environment and transforms normal functional HDL into “acute phase HDL” enriched in free fatty acids, triglycerides, serum amyloid A (SAA), and decrease anti-inflammatory enzymes, including paraoxanase [56, 64, 140,141,142,143,144]. In addition, inflammation induces secretion of myeloperoxidase (MPO), which has been shown to modify apolipoprotein A-I and impair its ability to accept cholesterol [64, 66, 142, 145,146,147]. MPO-mediated oxidation of apoA-I makes it proinflammatory [148]. Tryptophan substitution in apoA-I renders it resistance to MPO oxidation [149]. All these studies suggest that oxidative stress-induced HDL modification increases inflammation and contributes to HDL dysfunction.

Oxidative stress and inflammation impair reverse cholesterol transport in DM. Advanced glycation products and reactive oxygen species together with inflammation in DM induce MPO and decrease PON1 leading to oxidative modification of the main protein component of HDL, apoA-I. The resulting dysfunctional and proinflammatory HDL impacts immune response in macrophages through TLR2/4 by repressing the transcription factor ATF3, and impairs ABCA1-mediated cholesterol efflux leading to cholesterol accumulation and oxidation within macrophages entrapped in the subendothelial space. Additionally, oxidative environment in DM induces acute phase proteins like haptoglobin and CRP leading to impaired RCT and increased vascular inflammation that promote lipid deposition in arterial wall
Hyperglycemia causes increased flux through the polyol pathway, formation of advanced glycation end products, activation of protein kinase C isoforms, and increased hexosamine pathway flux, all of which may contribute to increased oxidative stress [150,151,152]. Excessive free fatty acids delivered to nonadipose tissues can lead to reactive oxygen species (ROS) formation through a number of pathways, including oxidative phosphorylation, activation of NADPH oxidase, and alterations in mitochondrial structure leading to ROS production [153,154,155]. In addition to evidence for activation of these pathways in cultured endothelial cells, human studies support the notion of increased systemic oxidative stress in diabetic subjects in whom increased circulating levels of adhesion molecules and oxidized lipids correlate with increases in HbA1c and hypertriglyceridemia [156]. The effects of oxidative stress in diabetes on both the vascular wall and lipoproteins in the circulation may promote atherogenesis. Jaleel et al. [130] provided intriguing evidence that poor glycemic control in type-1 diabetes is associated with accelerated oxidative damage to apoA-I. These investigators labeled newly synthesized proteins with 13C-phenylalanine in human subjects and analyzed various plasma apoA-I isoforms by two-dimensional gel separation and mass spectrometry. Newly synthesized forms of the protein containing the propeptide and in more mature cleaved forms were analyzed. The older forms of apoA-I accumulated significantly more, suggesting damage of apoA-I as a result of a variety of reactions, including deamidation, oxidation, and carbonylation of amino acids that likely contribute to their altered migration in isoelectric focusing.
Given that apoA-I is a major component of HDL that protects against atherosclerosis by facilitating the removal of cholesterol from macrophages in the arterial wall, oxidative damage of apoA-I [130] may impair HDL function. Indeed, recent studies demonstrated the presence of significant amounts of oxidation products of apoA-I in human atherosclerotic plaques [142, 157]. Additionally, Kataoka et al. [158] showed that myeloperoxidase enzyme that participates in the oxidation of apoA-I, predict accelerated progression of atherosclerosis in diabetics. Similarly, Shao et al. [159] quantified site-specific oxidation of apoA-I and measured cholesterol efflux in the HDL isolated from control subjects as well as subjects with stable coronary artery disease or acute coronary syndrome. The two groups of patients, CAD and ACS, had higher levels of chlorinated tyrosine 192 and oxidized methionine 148 compared to control subjects, clearly pointing to the importance of oxidatively damaged apoA-I in rendering HDL dysfunctional (Fig. 4). Interestingly, these researchers found no differences in the MPO level between the groups. Subjects with CAD and ACS showed less cholesterol efflux capacity compared to control group. The concentration of chlorinated tyrosine 192 and oxidized methionine 148 was inversely correlated to ABCA1-mediated cholesterol efflux capacity and positively with the extent of atherosclerosis. This suggests that chlorinated tyrosine and oxidized methionine in circulating HDL may serve as a useful marker of the atherosclerotic CVD. Thus, these studies provide mechanistic insight into the etiology of the oxidative modification of apoA-I, and how the functionality of HDL is linked to increased cardiovascular risks in diabetes. Lu et al. [160] extended this work in diabetic patients and showed that the levels of apoA-I nitration and chlorination were increased, and apoA-I concentrations as well as cholesterol efflux activity were significantly decreased. Specifically, they showed that Tyr 192 was the major nitration and chlorination site in apoA-I from diabetic serum. In addition to decreased cholesterol efflux capacity in patients with diabetes, these investigators further showed loss of antiapoptotic properties of lipoproteins. These findings were corroborated by a recent study by Chen et al. [161] who measured nitrated-apoA-I (NT-apoA-I) in 777 patients with CAD. Additionally, they measured cholesterol efflux capacity in diabetic (n = 327) and non-diabetic (n = 450) individuals. Higher ratio of NT-apoA-I/apoA-I in diabetic patients suggested higher oxidative stress. Indeed, thiobarbituric acid-reactive substances and c-reactive protein levels in diabetes mellitus were independent predictors of elevated NT-apoA-I/apoA-I ratio. Thus, oxidative stress in patients with diabetes leading to oxidative modification of apoA-I renders HDL dysfunctional in carrying out cholesterol efflux function, thus linking dampened cholesterol efflux to coronary artery disease risk in diabetic patients (Figs. 3, 4).
HDL and apoA-I modulate AMPK function and reverse cholesterol transport
Given the higher prevalence of cardiovascular morbidity and mortality in diabetics, this is an important area to pay attention to. Recent cell-based studies suggest that HDL may modulate plasma glucose through both insulin-dependent [162, 163] and -independent and AMPK-mediated mechanisms [164]. The ATP-binding cassette transporter A1 (ABCA1) has been shown to modulate insulin secretion [163], and HDL can reverse the deleterious effects of oxidized low-density lipoprotein (LDL) on insulin secretion by pancreatic beta cells [162]. In addition, HDL may also increase glucose disposal through direct effects in skeletal muscle, the major site of glucose disposal in the body. It was reported that HDL and apoA-I activate the key metabolic regulatory enzyme AMP-activated protein kinase (AMPK) in endothelial cells and are critical for the nitric oxide-mediated vasodilatory effects of HDL [165]. Infusion studies with recombinant and reconstituted HDL (rHDL) demonstrated modest effects on coronary plaque morphology and volume [166, 167] and also showed improved endothelial function in type 2 diabetes mellitus [168].
Diabetic individuals often have higher non-esterified fatty acids that may impact ABCA1-mediated cholesterol efflux. Indeed, unsaturated fatty acids inhibit ABCA1-mediated cholesterol efflux [169]. Given the elevated levels of fatty acids in diabetics [170], this finding is relevant in explaining, at least in part, the dampening of cholesterol efflux capability in diabetic individuals. This together with enhanced apoB secretion by fatty acids as a result of impaired presecretory degradation of apoB [171,172,173] contributes to the CVD risks in diabetic individuals [124, 174,175,176]. It therefore appears that ABCA1 could be important not only in the inhibition of progression of atherosclerosis [25, 34, 177,178,179], but also in metabolic diseases [136]. Wang and Oram [169] studied the effects of fatty acids, ranging in carbon chain length from 8 to 20, on cholesterol and phospholipid efflux in murine J774 and RAW 264.7 cells. The saturated fatty acids, palmitate and stearate, neither inhibited ABCA1-mediated cholesterol and phospholipid efflux nor they influenced ABCA1 protein. However, unsaturated fatty acids, oleate and linoleate, reduced cholesterol efflux as well as ABCA1 protein in a dose-dependent manner. Interestingly, oleate and stearate did not alter ABCA1 mRNA. As determined from ABCA1 turnover studies, it was concluded that unsaturated fatty acids enhanced the degradation of ABCA1 protein. These authors investigated the mechanism of fatty acid-mediated degradation of ABCA1 and carried out elegant studies to demonstrate that unsaturated fatty acids phosphorylate and destabilize ABCA1 through a phospholipase D2 pathway [180]. Further studies revealed that protein kinase C delta pathway is also involved in this process [181]. Thus, it appears that the triggering of the ABCA1 degradation by fatty acids possibly occurs via a mechanism distinct from the one observed with the cAMP withdrawal [38].
Although the role of AMPK in attenuating diabetes through glucose catabolism and energy balance has been well studied [182,183,184], the role of apoA-I on energy and glucose metabolism was first investigated by Han et al. [164] in C2C12 myocytes. These investigators reported AMPK phosphorylation at Thr-172 following treatments with apoA-I, and this effect was found to be specific to apoA-I protein since treatment with apoB did not result into AMPK phosphorylation. ApoA-I also increased glucose uptake by C2C12 cells like AMPK activators [185]. These effects were similar to AMPK activation by adiponectin, leading to increased glucose uptake [186]. Extension of this study in apoA-I−/− mice further supported the hypothesis that apoA-I is involved in glucose and energy metabolism, since apoA-I−/− mice had higher circulating glucose and impaired glucose tolerance compared to the WT littermates; increased HDL in apoA-I Tg mice provided protection against diet-induced hyperglycemia through increased glucose catabolism [187]. Based on these findings, Drew et al. [109] extended these studies in human primary skeletal muscle cells isolated from type 2 diabetic patients infused with either a placebo or reconstituted HDL. There were reductions in the fasting glucose in the rHDL treated group compared to the placebo group. In cultured primary human skeletal muscle cells, apoA-I increased glucose uptake by 50%, which was associated with the activation of AMPK as measured by the AMPK phosphorylation at Thr-172. To further gain insights into the mechanism of apoA-I/HDL-mediated AMPK activation, these investigators examined two primary pathways of AMPK activation, i.e. LKB1 and CaMKK, the two upstream kinases known to phosphorylate AMPK [184, 188, 189]. They found that HDL-mediated induction of AMPK phosphorylation occurs via CaMKK-mediated pathway, since the CaMKK inhibitor STO609 abolished HDL-mediated phosphorylation of AMPK. Interestingly, the HDL-mediated induction of skeletal muscle glucose uptake occurred in ABCA1-dependent manner, since ABCA1 blocking antibody inhibited apoA-I and HDL-mediated uptake of glucose [109].
Low-grade inflammation in diabetes impairs reverse cholesterol transport
Atherosclerosis has been characterized as a chronic inflammatory response to LDL oxidation and deposition in arteries, but the mechanisms linking cholesterol accumulation in macrophage foam cells to inflammation are not completely understood. One of the mechanisms to protect cells from free cholesterol and oxysterol-induced toxicity during progression of atherosclerosis is the macrophage cholesterol efflux [86, 94, 190, 191]. During the cholesterol efflux process, the ATP-binding cassette transporters ABCA1 and ABCG1 are important players responsible for the major part of macrophage cholesterol efflux to HDL in macrophage foam cells [192]. Recent studies have shown that the sterol efflux activities of ABCA1 and ABCG1 modulate macrophage expression of inflammatory cytokines and chemokines as well as lymphocyte proliferative responses [193, 194]. Accumulating evidence suggests that by promoting cholesterol and oxysterol efflux, HDL regulates all these cellular responses in macrophage foam cells [192]. Indeed, several studies demonstrated that native and reconstituted HDL, apoA-I and apoA-I mimetic peptides, all show anti-inflammatory activity [133, 135, 195,196,197,198]. Inflammation modulates HDL composition and function [196, 199] and impairs reverse cholesterol transport [139, 200], and infusion of reconstituted HDL during human endotoxemia exerts anti-inflammatory activity [133]. Thus, native apoA-I and HDL-C show anti-inflammatory activities leading to enhancement in reverse cholesterol transport [201]. Indeed, increased inflammation was observed in mice lacking apoA-I [202], suggesting the role of apoA-I as an anti-inflammatory agent. Thus, accumulating evidence suggests an anti-inflammatory role for native unmodified HDL, but becomes inflammatory when it undergoes modifications [148] (Figs. 4, 5).

Proposed working hypothesis. The biologic sequence of events that leads to HDL dysfunction is shown here. HDL performs its normal function by removing cholesterol from lipid-laden macrophages in the arterial wall, thus causing lesion regression. In diabetic mellitus, high oxidative environment causes inflammation in macrophages entrapped in the subendothelial space resulting in the secretion of proinflammatory cytokines and acute phase proteins like haptoglobin and CRP. The dysfunctional HDL impairs cholesterol efflux from lipid-laden macrophages. Treatment with antidiabetic drugs attenuates hyperglycemia which in turn reduces AGE, oxidative stress, and inflammation, and makes HDL functional and capable of promoting cholesterol efflux. This results in attenuating vascular inflammation leading to atherosclerotic plaque regression
Two proteins, ABCA1 and ABCG1, important in reverse cholesterol transport play distinct roles in macrophages immune response. In macrophages lacking ABCA1 or ABCG1, TLR4 cell surface expression increased, albeit ABCG1 deficiency showed greater macrophage inflammatory response compared to ABCA1 deficiency [203]. These studies demonstrate that the primary function of HDL and ABC transporters in cholesterol efflux and reverse cholesterol transport are linked to anti-inflammatory and immunosuppressive functions of HDL. A recent study [204] demonstrates that dysfunctional HDL from patients with chronic kidney dysfunction (CKD) showed unfavorable physiological functions by increasing superoxide dismutase production and reducing NO. These unfavorable activities were found to be occurring through toll-like receptor-2/4 (TLR-2/4). The HDL isolated from healthy counterpart did not show these unfavorable activities. Thus, the anti-inflammatory properties of HDL is linked to immune response through a number of mechanisms [205, 206], leading to suppression of Toll-like receptor 2 (TLR2) signaling [207] and suggesting that the HDL-mediated cholesterol efflux inhibits cellular inflammatory signaling, including inhibition of MCP-1 expression, a key player in monocyte transmigration. The molecular mechanisms of how HDL can modulate inflammation, particularly in immune cells such as macrophages, were investigated by De Nardo et al. [208]. These researchers found that the transcriptional regulator ATF3 in macrophages downregulates the expression of Toll-like receptor (TLR)-induced proinflammatory cytokines in an HDL-dependent manner, since the protective effects of HDL against TLR-induced inflammation were entirely dependent on ATF3. In LPS-induced animal model, Dandekar et al. [209] demonstrated the role of cAMP-responsive element-binding protein hepatic-specific (CREBH) and TNF receptor-associated factor 6 (TRAF6) in mediating TLR signaling in HDL-dependent manner, suggesting a mechanism of how HDL is involved in inflammation through toll-like receptors. These findings may explain the broad anti-inflammatory and metabolic actions of HDL and provide the basis for predicting the success of new HDL-based therapies.
Lipid raft in the plasma membrane appears to be a key regulator of macrophage inflammation, since one of the mechanisms of enhanced inflammatory responses in ABCA1 or ABCG1 deficiency appears to be through increased lipid raft formation in macrophages [210,211,212,213]. That cholesterol efflux is linked to immune response in ABCA1 or ABCG1 deficient macrophages was further demonstrated by treatment with cyclodextrin that removes cholesterol and attenuates inflammatory response. It was shown [212] that the modulation of membrane cholesterol by cholesterol efflux in ABCA1 or ABCG1 deficiency increased TLR4 cell surface expression.
Recent studies by Bensinger et al. [214] reported that Liver X Receptor (LXR) signaling, that promotes cholesterol efflux via ABCA1 and G1 stimulation, is involved in T-cell lymphocyte proliferation in an ABCG1-dependent fashion. Mice lacking apoA-I, an important component of cholesterol efflux, also stimulates T-cell proliferation and activation and some features of autoimmunity when backcrossed into an LDL receptor-deficient background [215]. These studies strongly suggest that HDL-mediated cholesterol efflux via LXR-regulated ABC transporters plays a key role in dampening lymphocyte proliferation and activation. Regulatory T cells (Tregs) express SR-B1 [216], which facilitates the uptake of HDL from microenvironment. A recent study demonstrates that LDL is not taken up by Treg, and HDL-derived fatty acids serve as fuel for the survival of Tregs [217]. Additionally, these authors further showed that mitochondrial activity was increased in the Tregs that internalized HDL, but not those that did not [217]. Thus, HDL plays an important role in the survival of Tregs, leading to the suppression of proatherogenic effector T cells [218].
To confirm if HDL function is compromised in inflammatory disease, Charles-Schoeman et al. [219] isolated HDL from 40 patients with Rheumatoid Arthritis patients and 40 age and sex matched healthy controls, and found that cholesterol efflux, HDL’s antioxidant function, and paraoxanase-1 (PON-1) activity in RA patients with high disease activity had significantly decreased ability to promote cholesterol efflux compared to HDL from patients with very low disease activity. This was further substantiated by the findings that there was higher plasma MPO activity in patients with dysfunctional HDL. Additionally, cholesterol efflux activity of HDL correlated significantly with its antioxidant activity.
Another study was carried out by Field et al. [220] in patients with Crohn’s disease, which is a tumor necrosis factor-alpha (TNF-alpha)-driven gastrointestinal tract chronic inflammatory condition. These investigators reported dampened basolateral efflux of cholesterol to apolipoprotein A1 (apoA1) through TNF-alpha mediated decreases in HDL cholesterol levels by modulating the expression of intestinal ABCA1 and cholesterol efflux to apoA1. A different approach was pursued by de la Llera Moya et al. [199] to assess the effect of inflammation on HDL and RCT related parameters. They employed low-dose human endotoxemia that induces HDL remodeling through depletion of pre-beta1 HDL particles. Endotoxemia resulted in reduced capacity of HDL to efflux cholesterol. The HDL fraction, isolated following endotoxemia, had reduced capacity to efflux cholesterol in vitro from SR-BI and ABCA1, but not ABCG1 transporter cell models. Thus, inflammatory conditions lead to dysfunctional HDL. Autoimmune disease, systemic lupus erythematosus (SLE), patients have elevated inflammation and have a higher prevalence of subclinical atherosclerosis and higher risk of CV events. The factors causing cardiovascular risks in these patients were investigated by Ammirati et al. [221]. They found that among CV risk factors, only the two important players in the cholesterol efflux, HDL level and ABCA1-dependent cholesterol efflux capability, were markedly reduced, whereas the common carotid artery intima-media thickness (CCA-IMT) significantly increased in SLE patients compared to controls. These and other findings discussed above suggest that reduction in RCT capability, as a result of dysfunctional HDL or ABC transporters, in inflammatory conditions lead to impaired cholesterol efflux and increased risk of CV. In diabetic patients, both oxidative stress and inflammation are elevated that lead to HDL modification and impairs RCT function leading to increased risks of CVD.
HDL-associated proinflammatory protein, haptoglobin, is associated with diabetic atherosclerosis
Haptoglobin (Hp) is an acute phase hemoglobin (Hb) binding serum protein primarily synthesized in the liver [222,223,224]. Hp binds to free Hb in the serum and forms Hp/Hb complex. The endocytosis of this Hp/Hb complex by monocyte-macrophages is mediated by the scavenger receptor CD163 [225, 226]. The main function of haptoglobin is in infection and inflammation, where it acts as a natural antagonist for receptor-ligand activation of the immune system [224]. The level of haptoglobin increases in proinflammatory conditions [224, 227]. It is now well established that HDL function is impaired in proinflammatory conditions [133, 139, 202, 228]. Both the anti-inflammatory properties as well as the cholesterol efflux capabilities of HDL are significantly dampened [193, 229]. Increased levels of haptoglobin in proinflammatory condition preferentially associates with proinflammatory HDL in animal models as well as in humans [230, 231]. Elevated level of haptoglobin has been observed in humans with CVD [232,233,234], and HDL isolated from mice lacking this protein show anti-inflammatory activity compared to WT mice [231]. These findings together with the observations that the endocytosis of Hp/Hb complex [235, 236] is mediated by a receptor, CD163, exclusively expressed in the macrophages [225, 226], and the involvement of macrophages and inflammatory cytokines in the progression of atherosclerosis [237], suggest a potential role of Hp in macrophage cholesterol efflux and the progression of atherosclerosis [238,239,240]. Indeed, HDL-raising agent in apoE-deficient mice decreases haptoglobin, which was associated with attenuation of aortic lipid deposition [241]. Several studies carried out in diabetic patients with CVD show a strong association between diabetic CVD and haptoglobin [233, 234].
Hp gene exists as Hp-1 and Hp-2 alleles and the phenotypes show important molecular heterogeneity. In individuals with DM, the Hp2-2 genotype is suggested as a contributor for increased cardiovascular events compared with Hp1-1 or Hp2-1 genotype [233, 234]. Indeed, haptoglobin genotype was found to be associated with compromised reverse cholesterol transport in Hp2 diabetic mice because of increased oxidative stress [242]. Levy et al. [234] tested this hypothesis in a case–control sample from the Strong Heart study, a population-based longitudinal study of CVD in American Indians. These investigators determined haptoglobin phenotype in 206 CVD cases and 206 matched controls and followed-up for 6 years. In multivariate analyses, DM patients with haptoglobin phenotype were highly statistically significant and independent predictor of CVD. The odds ratio of having CVD in DM with the haptoglobin 2-2 phenotype was found to be 5.0 times greater than in DM with the haptoglobin 1-1 phenotype. An intermediate risk of CVD was associated with the haptoglobin 2-1 phenotype. In another study, Lioupis et al. [243] investigated iron burden of carotid atherosclerotic plaques removed from patients treated for carotid disease and examined correlation with haptoglobin genotype and common cardiovascular risk factors. Twenty seven plaques from diabetic patients (16 with the Hp 1-1 or 2-1 genotype and 11 with the Hp 2-2 genotype) and 43 plaques from non-diabetic patients (20 with the Hp 1-1 or 2-1 genotype and 23 with the Hp 2-2 genotype) were evaluated. They found that the density of Perl’s iron stain was significantly higher in plaques from diabetic patients with the Hp 2-2 group compared with that in the Hp 1-1 or 2-1 group. The correlation and regression analysis of all possible clinical and laboratory predictors of intraplaque iron deposition showed that four factors were independently associated with intraplaque iron deposition; these were male gender, serum homocysteine, Hp 2-2 genotype and diabetes mellitus treatment. To further corroborate the relation between Hp genotype and CV risks in DM, Purushothaman et al. [244] carried an elegant study in 40 diabetic patients. These patients were genotyped for haptoglobin allele, Hp-1 and Hp-2 and after atherectomy, several parameters like plaque hemorrhage, hemoglobin-binding protein, CD163, and heme-oxygenase 1 were measured. To evaluate oxidative and inflammatory pattern, these investigators also quantitated myeloperoxidase, IL-10, and VCAM1. Consistent with earlier findings, it was reported that plaques with Hp2-2 allele had increased hemorrhage, increased heme-oxygenase, decreased CD163 protein, increased MPO, and decreased IL-10. Some of these unfavorable changes appear to be associated with oxidative stress, since patients with Hp2-2 genotype had greater oxidative stress [245]. Thus, these independent studies demonstrated that haptoglobin, an HDL-associated proinflammatory protein and a risk factor for CVD, is elevated in diabetes; especially the Hp2-2 allele show stronger correlation with CVD risk factors.
Conclusion
As a working hypothesis, the biologic sequence of events that leads to HDL dysfunction is shown in Fig. 5. HDL performs its normal function by removing cholesterol from lipid-laden macrophages in the arterial wall, thus causing lesion regression. In high oxidative environment, the inflamed macrophages entrapped in the subendothelial space secrete proinflammatory cytokines and haptoglobin, and activates wnt signaling through LRP5/6 as a result of uptake of aggregated LDL [246] in hyperlipidemic conditions. Wnt/β-catanin signaling has been shown to induce proliferation of vascular smooth muscle cells [247], which may lead to narrowing of artery lumen and eventually causing occlusion. High oxidative stress, diabetes, and proinflammatory proteins, including acute phase proteins like haptoglobin and CRP cause dysfunctional HDL, leading to dampening of cholesterol efflux capability and impaired arterial cholesterol removal. Treatments that attenuates hyperlipidemia, oxidative stress and inflammation, at least in animal models of diabetes and hyperlipidemia, improves HDL function [248] and promotes removal of cholesterol from lipid-laden macrophages entrapped in the subendothelial space leading to atherosclerotic lesion regression [248]. Thus, systemic metabolic disturbances of diabetes, including hyperglycemia and hyperlipidemia, likely play a central role in the pathogenesis of diabetes-associated atherosclerosis through the generation of oxidative stress and inflammation, and aggressive treatment of diabetes mellitus offer promise to reduce progression of CVD in this highly susceptible group of individuals with diabetes.
References
American Heart Association HDass-u (2007) A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 115:e69–e71
Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM (2004) Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 350:1495–1504. doi:10.1056/NEJMoa040583
Ross R (1993) The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362:801–809. doi:10.1038/362801a0
Ross R (1999) Atherosclerosis: an inflammatory disease. N Engl J Med 340:115–126. doi:10.1056/nejm199901143400207
Fonarow GC, Watson KE (2003) Effective strategies for long-term statin use. Am J Cardiol 92:27i–34i
Kastelein JJ (2003) The future of lipid-lowering therapy: the big picture. Neth J Med 61:35–39
Linsel-Nitschke P, Tall AR (2005) HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov 4:193–205. doi:10.1038/nrd1658
Gordon DJ, Knoke J, Probstfield JL, Superko R, Tyroler HA (1986) High-density lipoprotein cholesterol and coronary heart disease in hypercholesterolemic men: the Lipid Research Clinics Coronary Primary Prevention Trial. Circulation 74:1217–1225
Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR Jr, Bangdiwala S, Tyroler HA (1989) High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 79:8–15
Hopkins PN, Heiss G, Ellison RC, Province MA, Pankow JS, Eckfeldt JH, Hunt SC (2003) Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case-control comparison from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation 108:519–523. doi:10.1161/01.cir.0000081777.17879.85
Genest JJ, McNamara JR, Salem DN, Schaefer EJ (1991) Prevalence of risk factors in men with premature coronary artery disease. Am J Cardiol 67:1185–1189
Investigators DAIS (2001) Effect of fenofibrate on progression of coronary-artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet 357:905–910
Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, Huttunen JK, Kaitaniemi P, Koskinen P, Manninen V et al (1987) Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med 317:1237–1245. doi:10.1056/nejm198711123172001
Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J (1999) Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med 341:410–418. doi:10.1056/nejm199908053410604
Reaven GM (1995) Pathophysiology of insulin resistance in human disease. Physiol Rev 75:473–486
Moller DE, Kaufman KD (2005) Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med 56:45–62. doi:10.1146/annurev.med.56.082103.104751
Srivastava RA, Srivastava N (2004) Search for obesity drugs: targeting central and peripheral pathways. Curr Med Chem 4:75–90
Hossain P, Kawar B, El Nahas M (2007) Obesity and diabetes in the developing world: a growing challenge. N Engl J Med 356:213–215. doi:10.1056/NEJMp068177
Srivastava RA, Srivastava N (2000) High density lipoprotein, apolipoprotein A-I, and coronary artery disease. Mol Cell Biochem 209:131–144
Choudhury RP, Rong JX, Trogan E, Elmalem VI, Dansky HM, Breslow JL, Witztum JL, Fallon JT, Fisher EA (2004) High-density lipoproteins retard the progression of atherosclerosis and favorably remodel lesions without suppressing indices of inflammation or oxidation. Arterioscler Thromb Vasc Biol 24:1904–1909. doi:10.1161/01.atv.0000142808.34602.25
Farbstein D, Levy AP (2012) HDL dysfunction in diabetes: causes and possible treatments. Expert Rev Cardiovasc Ther 10:353–361. doi:10.1586/erc.11.182
Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271:518–520
Van Eck M, Pennings M, Hoekstra M, Out R, Van Berkel TJ (2005) Scavenger receptor BI and ATP-binding cassette transporter A1 in reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol 16:307–315
Joyce CW, Amar MJ, Lambert G, Vaisman BL, Paigen B, Najib-Fruchart J, Hoyt RF Jr, Neufeld ED, Remaley AT, Fredrickson DS, Brewer HB Jr, Santamarina-Fojo S (2002) The ATP binding cassette transporter A1 (ABCA1) modulates the development of aortic atherosclerosis in C57BL/6 and apoE-knockout mice. Proc Natl Acad Sci USA 99:407–412. doi:10.1073/pnas.012587699
Srivastava N (2002) ATP binding cassette transporter A1–key roles in cellular lipid transport and atherosclerosis. Mol Cell Biochem 237:155–164
Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA (2005) ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab 1:121–131. doi:10.1016/j.cmet.2005.01.002
Fournier N, de la Llera Moya M, Burkey BF, Swaney JB, Paterniti J Jr, Moatti N, Atger V, Rothblat GH (1996) Role of HDL phospholipid in efflux of cell cholesterol to whole serum: studies with human apoA-I transgenic rats. J Lipid Res 37:1704–1711
Bielicki JK, Johnson WJ, Weinberg RB, Glick JM, Rothblat GH (1992) Efflux of lipid from fibroblasts to apolipoproteins: dependence on elevated levels of cellular unesterified cholesterol. J Lipid Res 33:1699–1709
Smith JD, Miyata M, Ginsberg M, Grigaux C, Shmookler E, Plump AS (1996) Cyclic AMP induces apolipoprotein E binding activity and promotes cholesterol efflux from a macrophage cell line to apolipoprotein acceptors. J Biol Chem 271:30647–30655
Sakr SW, Williams DL, Stoudt GW, Phillips MC, Rothblat GH (1999) Induction of cellular cholesterol efflux to lipid-free apolipoprotein A-I by cAMP. Biochim Biophys Acta 1438:85–98
Li Q, Czarnecka H, Yokoyama S (1995) Involvement of a cellular surface factor(s) in lipid-free apolipoprotein-mediated cellular cholesterol efflux. Biochim Biophys Acta 1259:227–234
Mendez AJ, Oram JF (1997) Limited proteolysis of high density lipoprotein abolishes its interaction with cell-surface binding sites that promote cholesterol efflux. Biochim Biophys Acta 1346:285–299
Srivastava N, Chowdhury PR, Averna M, Srivastava RA (2001) Estrogen increases hepatic lipase levels in inbred strains of mice: a possible mechanism for estrogen-dependent lowering of high density lipoprotein. Mol Cell Biochem 220:87–93
Attie AD, Kastelein JP, Hayden MR (2001) Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res 42:1717–1726
Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet 22:336–345. doi:10.1038/11905
Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet 22:347–351. doi:10.1038/11914
Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, Assmann G (1999) Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet 22:352–355. doi:10.1038/11921
Oram JF, Lawn RM, Garvin MR, Wade DP (2000) ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem 275:34508–34511. doi:10.1074/jbc.M006738200
Francone OL, Royer L, Haghpassand M (1996) Increased prebeta-HDL levels, cholesterol efflux, and LCAT-mediated esterification in mice expressing the human cholesteryl ester transfer protein (CETP) and human apolipoprotein A-I (apoA-I) transgenes. J Lipid Res 37:1268–1277
Wroblewska M (2011) The origin and metabolism of a nascent pre-beta high density lipoprotein involved in cellular cholesterol efflux. Acta Biochim Pol 58:275–285
Chroni A, Koukos G, Duka A, Zannis VI (2007) The carboxy-terminal region of apoA-I is required for the ABCA1-dependent formation of alpha-HDL but not prebeta-HDL particles in vivo. Biochemistry 46:5697–5708. doi:10.1021/bi602354t
Troutt JS, Alborn WE, Mosior MK, Dai J, Murphy AT, Beyer TP, Zhang Y, Cao G, Konrad RJ (2008) An apolipoprotein A-I mimetic dose-dependently increases the formation of prebeta1 HDL in human plasma. J Lipid Res 49:581–587. doi:10.1194/jlr.M700385-JLR200
Avdulov NA, Chochina SV, Igbavboa U, Wood WG (2000) Cholesterol efflux to high-density lipoproteins and apolipoprotein A-I phosphatidylcholine complexes is inhibited by ethanol: role of apolipoprotein structure and cooperative interaction of phosphatidylcholine and cholesterol. Biochemistry 39:10599–10606
Rye KA, Barter PJ (2004) Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol 24:421–428. doi:10.1161/01.ATV.0000104029.74961.f5
Kane JP, Malloy MJ (2012) Prebeta-1 HDL and coronary heart disease. Curr Opin Lipidol 23:367–371. doi:10.1097/MOL.0b013e328353eef1
Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC (2007) Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem 282:25123–25130. doi:10.1074/jbc.M704590200
Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, Cartland S, Packianathan M, Kritharides L, Jessup W (2006) ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler Thromb Vasc Biol 26:534–540. doi:10.1161/01.ATV.0000200082.58536.e1
Dastani Z, Dangoisse C, Boucher B, Desbiens K, Krimbou L, Dufour R, Hegele RA, Pajukanta P, Engert JC, Genest J, Marcil M (2006) A novel nonsense apolipoprotein A-I mutation (apoA-I(E136X)) causes low HDL cholesterol in French Canadians. Atherosclerosis 185:127–136. doi:10.1016/j.atherosclerosis.2005.05.028
Koukos G, Chroni A, Duka A, Kardassis D, Zannis VI (2007) LCAT can rescue the abnormal phenotype produced by the natural ApoA-I mutations (Leu141Arg)Pisa and (Leu159Arg)FIN. Biochemistry 46:10713–10721. doi:10.1021/bi7003203
Savel J, Lafitte M, Pucheu Y, Pradeau V, Tabarin A, Couffinhal T (2012) Very low levels of HDL cholesterol and atherosclerosis, a variable relationship: a review of LCAT deficiency. Vasc Health Risk Manag 8:357–361. doi:10.2147/vhrm.s29985
Persegol L, Brindisi MC, Rageot D, Pais de Barros JP, Monier S, Verges B, Duvillard L (2015) Oxidation-induced loss of the ability of HDL to counteract the inhibitory effect of oxidized LDL on vasorelaxation. Heart Vessels 30:845–849. doi:10.1007/s00380-014-0543-2
Hine D, Mackness B, Mackness M (2011) Cholesteryl-ester transfer protein enhances the ability of high-density lipoprotein to inhibit low-density lipoprotein oxidation. IUBMB Life 63:772–774. doi:10.1002/iub.508
Hine D, Mackness B, Mackness M (2012) Coincubation of PON1, APO A1, and LCAT increases the time HDL is able to prevent LDL oxidation. IUBMB Life 64:157–161. doi:10.1002/iub.588
Huang Y, Wu Z, Riwanto M, Gao S, Levison BS, Gu X, Fu X, Wagner MA, Besler C, Gerstenecker G, Zhang R, Li XM, DiDonato AJ, Gogonea V, Tang WH, Smith JD, Plow EF, Fox PL, Shih DM, Lusis AJ, Fisher EA, DiDonato JA, Landmesser U, Hazen SL (2013) Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest 123:3815–3828. doi:10.1172/jci67478
Smith JD (2010) Myeloperoxidase, inflammation, and dysfunctional high-density lipoprotein. J Clin Lipidol 4:382–388. doi:10.1016/j.jacl.2010.08.007
Shao B, Oda MN, Oram JF, Heinecke JW (2010) Myeloperoxidase: an oxidative pathway for generating dysfunctional high-density lipoprotein. Chem Res Toxicol 23:447–454. doi:10.1021/tx9003775
Blatter Garin MC, Moren X, James RW (2006) Paraoxonase-1 and serum concentrations of HDL-cholesterol and apoA-I. J Lipid Res 47:515–520. doi:10.1194/jlr.M500281-JLR200
Garcia-Heredia A, Marsillach J, Rull A, Triguero I, Fort I, Mackness B, Mackness M, Shih DM, Joven J, Camps J (2013) Paraoxonase-1 inhibits oxidized low-density lipoprotein-induced metabolic alterations and apoptosis in endothelial cells: a nondirected metabolomic study. Mediators Inflamm 2013:156053. doi:10.1155/2013/156053
Rozenberg O, Shih DM, Aviram M (2005) Paraoxonase 1 (PON1) attenuates macrophage oxidative status: studies in PON1 transfected cells and in PON1 transgenic mice. Atherosclerosis 181:9–18. doi:10.1016/j.atherosclerosis.2004.12.030
Mackness B, Quarck R, Verreth W, Mackness M, Holvoet P (2006) Human paraoxonase-1 overexpression inhibits atherosclerosis in a mouse model of metabolic syndrome. Arterioscler Thromb Vasc Biol 26:1545–1550. doi:10.1161/01.ATV.0000222924.62641.aa
Tward A, Xia YR, Wang XP, Shi YS, Park C, Castellani LW, Lusis AJ, Shih DM (2002) Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation 106:484–490
Rozenberg O, Rosenblat M, Coleman R, Shih DM, Aviram M (2003) Paraoxonase (PON1) deficiency is associated with increased macrophage oxidative stress: studies in PON1-knockout mice. Free Radic Biol Med 34:774–784
Shao B, Oda MN, Vaisar T, Oram JF, Heinecke JW (2006) Pathways for oxidation of high-density lipoprotein in human cardiovascular disease. Curr Opin Mol Ther 8:198–205
Heinecke JW (2007) The role of myeloperoxidase in HDL oxidation and atherogenesis. Curr Atheroscler Rep 9:249–251
Shao B, Cavigiolio G, Brot N, Oda MN, Heinecke JW (2008) Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc Natl Acad Sci USA 105:12224–12229. doi:10.1073/pnas.0802025105
Shao B, Tang C, Heinecke JW, Oram JF (2010) Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J Lipid Res 51:1849–1858. doi:10.1194/jlr.M004085
Kotani K, Sakane N, Ueda M, Mashiba S, Hayase Y, Tsuzaki K, Yamada T, Remaley AT (2012) Oxidized high-density lipoprotein is associated with increased plasma glucose in non-diabetic dyslipidemic subjects. Clin Chim Acta 414:125–129. doi:10.1016/j.cca.2012.08.021
Sampaio E, Barbosa DS, Mazzuco TL, Nunes VS, Passarelli M, Nakandakare ER, Carrilho AJ (2013) Impaired antioxidant action of high density lipoprotein in patients with type 1 diabetes with normoalbuminuria and microalbuminuria. Diabetes Res Clin Pract 99:321–326. doi:10.1016/j.diabres.2012.12.012
Kaysen GA (2009) Potential restoration of HDL function with apolipoprotein A-I mimetic peptide in end-stage renal disease. Kidney Int 76:359–361. doi:10.1038/ki.2009.205
Motamed M, Nargesi AA, Heidari B, Mirmiranpour H, Esteghamati A, Nakhjavani M (2016) Oxidized low-density lipoprotein (ox-LDL) to LDL ratio (ox-LDL/LDL) and ox-LDL to high-density lipoprotein ratio (ox-LDL/HDL). Clin Lab 62:1609–1617. doi:10.7754/Clin.Lab.2016.150412
Girona J, Manzanares JM, Marimon F, Cabre A, Heras M, Guardiola M, Ribalta J, Masana L (2008) Oxidized to non-oxidized lipoprotein ratios are associated with arteriosclerosis and the metabolic syndrome in diabetic patients. Nutr Metab Cardiovasc Dis 18:380–387. doi:10.1016/j.numecd.2007.04.002
Jurek A, Turyna B, Kubit P, Klein A (2006) LDL susceptibility to oxidation and HDL antioxidant capacity in patients with renal failure. Clin Biochem 39:19–27. doi:10.1016/j.clinbiochem.2005.08.009
Ng DS, Leiter LA, Vezina C, Connelly PW, Hegele RA (1994) Apolipoprotein A-I Q[-2]X causing isolated apolipoprotein A-I deficiency in a family with analphalipoproteinemia. J Clin Invest 93:223–229. doi:10.1172/jci116949
Miller M, Aiello D, Pritchard H, Friel G, Zeller K (1998) Apolipoprotein A-I(Zavalla) (Leu159→Pro): HDL cholesterol deficiency in a kindred associated with premature coronary artery disease. Arterioscler Thromb Vasc Biol 18:1242–1247
Santos RD, Schaefer EJ, Asztalos BF, Polisecki E, Wang J, Hegele RA, Martinez LR, Miname MH, Rochitte CE, Da Luz PL, Maranhao RC (2008) Characterization of high density lipoprotein particles in familial apolipoprotein A-I deficiency. J Lipid Res 49:349–357. doi:10.1194/jlr.M700362-JLR200
Gigante M, Ranieri E, Cerullo G, Calabresi L, Iolascon A, Assmann G, Morrone L, Pisciotta L, Schena FP, Gesualdo L (2006) LCAT deficiency: molecular and phenotypic characterization of an Italian family. J Nephrol 19:375–381
Holleboom AG, Kuivenhoven JA, Peelman F, Schimmel AW, Peter J, Defesche JC, Kastelein JJ, Hovingh GK, Stroes ES, Motazacker MM (2011) High prevalence of mutations in LCAT in patients with low HDL cholesterol levels in The Netherlands: identification and characterization of eight novel mutations. Hum Mutat 32:1290–1298. doi:10.1002/humu.21578
Kosmas CE, DeJesus E, Rosario D, Vittorio TJ (2016) CETP inhibition: past failures and future hopes. Clin Med Insights Cardiol 10:37–42. doi:10.4137/cmc.s32667
Chen Z, Wang SP, Krsmanovic ML, Castro-Perez J, Gagen K, Mendoza V, Rosa R, Shah V, He T, Stout SJ, Geoghagen NS, Lee SH, McLaren DG, Wang L, Roddy TP, Plump AS, Hubbard BK, Sinz CJ, Johns DG (2012) Small molecule activation of lecithin cholesterol acyltransferase modulates lipoprotein metabolism in mice and hamsters. Metabolism 61:470–481. doi:10.1016/j.metabol.2011.08.006
Freeman LA, Demosky SJ Jr, Konaklieva M, Kuskovsky R, Aponte A, Ossoli AF, Gordon SM, Koby RF, Manthei KA, Shen M, Vaisman BL, Shamburek RD, Jadhav A, Calabresi L, Gucek M, Tesmer JJG, Levine RL, Remaley AT (2017) Lecithin: cholesterol acyltransferase activation by sulfhydryl-reactive small molecules: role of cysteine-31. J Pharmacol Exp Ther 362:306–318. doi:10.1124/jpet.117.240457
Chenevard R, Hurlimann D, Spieker L, Bechir M, Enseleit F, Hermann M, Flammer AJ, Sudano I, Corti R, Luscher TF, Noll G, Ruschitzka F (2012) Reconstituted HDL in acute coronary syndromes. Cardiovasc Ther 30:e51–e57. doi:10.1111/j.1755-5922.2010.00221.x
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B (2007) Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 357:2109–2122. doi:10.1056/NEJMoa0706628
Connelly MA, Parry TJ, Giardino EC, Huang Z, Cheung WM, Chen C, Cools F, Van der Linde H, Gallacher DJ, Kuo GH, Sarich TC, Demarest KT, Damiano BP (2010) Torcetrapib produces endothelial dysfunction independent of cholesteryl ester transfer protein inhibition. J Cardiovasc Pharmacol 55:459–468. doi:10.1097/FJC.0b013e3181cf03cb
Simic B, Hermann M, Shaw SG, Bigler L, Stalder U, Dorries C, Besler C, Luscher TF, Ruschitzka F (2012) Torcetrapib impairs endothelial function in hypertension. Eur Heart J 33:1615–1624. doi:10.1093/eurheartj/ehr348
Luscher TF, Taddei S, Kaski JC, Jukema JW, Kallend D, Munzel T, Kastelein JJ, Deanfield JE (2012) Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: the dal-VESSEL randomized clinical trial. Eur Heart J 33:857–865. doi:10.1093/eurheartj/ehs019
Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, Rothblat GH (2007) The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res 48:2453–2462. doi:10.1194/jlr.M700274-JLR200
Annema W, Tietge UJ (2012) Regulation of reverse cholesterol transport: a comprehensive appraisal of available animal studies. Nutr Metab (Lond) 9:25. doi:10.1186/1743-7075-9-25
Whitlock ME, Swenson TL, Ramakrishnan R, Leonard MT, Marcel YL, Milne RW, Tall AR (1989) Monoclonal antibody inhibition of cholesteryl ester transfer protein activity in the rabbit. Effects on lipoprotein composition and high density lipoprotein cholesteryl ester metabolism. J Clin Invest 84:129–137. doi:10.1172/jci114132
Liu M, Chen Y, Zhang L, Wang Q, Ma X, Li X, Xiang R, Zhu Y, Qin S, Yu Y, Jiang XC, Duan Y, Han J (2015) Regulation of hepatic cholesteryl ester transfer protein expression and reverse cholesterol transport by inhibition of DNA topoisomerase II. J Biol Chem 290:14418–14429. doi:10.1074/jbc.M115.643015
Arai T, Wang N, Bezouevski M, Welch C, Tall AR (1999) Decreased atherosclerosis in heterozygous low density lipoprotein receptor-deficient mice expressing the scavenger receptor BI transgene. J Biol Chem 274:2366–2371
Webb NR, de Beer MC, Yu J, Kindy MS, Daugherty A, van der Westhuyzen DR, de Beer FC (2002) Overexpression of SR-BI by adenoviral vector promotes clearance of apoA-I, but not apoB, in human apoB transgenic mice. J Lipid Res 43:1421–1428
Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M (1999) Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci USA 96:9322–9327
Van Eck M, Twisk J, Hoekstra M, Van Rij BT, Van der Lans CA, Bos IS, Kruijt JK, Kuipers F, Van Berkel TJ (2003) Differential effects of scavenger receptor BI deficiency on lipid metabolism in cells of the arterial wall and in the liver. J Biol Chem 278:23699–23705. doi:10.1074/jbc.M211233200
Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ (2007) Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 117:2216–2224. doi:10.1172/jci32057
Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ (2005) Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest 115:2870–2874. doi:10.1172/jci25327
Van Eck M, Hoekstra M, Hildebrand RB, Yaong Y, Stengel D, Kruijt JK, Sattler W, Tietge UJ, Ninio E, Van Berkel TJ, Pratico D (2007) Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol 27:2413–2419. doi:10.1161/atvbaha.107.145474
Zanoni P, Khetarpal SA, Larach DB, Hancock-Cerutti WF, Millar JS, Cuchel M, DerOhannessian S, Kontush A, Surendran P, Saleheen D, Trompet S, Jukema JW, De Craen A, Deloukas P, Sattar N, Ford I, Packard C, Majumder A, Alam DS, Di Angelantonio E, Abecasis G, Chowdhury R, Erdmann J, Nordestgaard BG, Nielsen SF, Tybjaerg-Hansen A, Schmidt RF, Kuulasmaa K, Liu DJ, Perola M, Blankenberg S, Salomaa V, Mannisto S, Amouyel P, Arveiler D, Ferrieres J, Muller-Nurasyid M, Ferrario M, Kee F, Willer CJ, Samani N, Schunkert H, Butterworth AS, Howson JM, Peloso GM, Stitziel NO, Danesh J, Kathiresan S, Rader DJ (2016) Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351:1166–1171. doi:10.1126/science.aad3517
Di Bartolo BA, Scherer DJ, Nicholls SJ (2016) Inducing apolipoprotein A-I synthesis to reduce cardiovascular risk: from ASSERT to SUSTAIN and beyond. Arch Med Sci 12:1302–1307. doi:10.5114/aoms.2016.62906
Balder JW, Staels B, Kuivenhoven JA (2013) Pharmacological interventions in human HDL metabolism. Curr Opin Lipidol 24:500–509. doi:10.1097/mol.0000000000000018
McLure KG, Gesner EM, Tsujikawa L, Kharencko OA, Attwell S, Campeau E, Wasiak S, Stein A, White A, Fontano E, Suto RK, Wong NC, Wagner GS, Hansen HC, Young PR (2013) RVX-208, An Inducer of apoA-I in Humans, is a BET Bromodomain Antagonist. PLoS ONE 8:e83190
Siebel AL, Trinh SK, Formosa MF, Mundra PA, Natoli AK, Reddy-Luthmoodoo M, Huynh K, Khan AA, Carey AL, van Hall G, Cobelli C, Dalla-Man C, Otvos JD, Rye KA, Johansson J, Gordon A, Wong NC, Sviridov D, Barter P, Duffy SJ, Meikle PJ, Kingwell BA (2016) Effects of the BET-inhibitor, RVX-208 on the HDL lipidome and glucose metabolism in individuals with prediabetes: a randomized controlled trial. Metabolism 65:904–914. doi:10.1016/j.metabol.2016.03.002
Kleinman JC, Donahue RP, Harris MI, Finucane FF, Madans JH, Brock DB (1988) Mortality among diabetics in a national sample. Am J Epidemiol 128:389–401
Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF (2010) Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 8:29. doi:10.1186/1478-7954-8-29
Ford ES, Giles WH, Dietz WH (2002) Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287:356–359
Gatti A, Maranghi M, Bacci S, Carallo C, Gnasso A, Mandosi E, Fallarino M, Morano S, Trischitta V, Filetti S (2009) Poor glycemic control is an independent risk factor for low HDL cholesterol in patients with type 2 diabetes. Diabetes Care 32:1550–1552. doi:10.2337/dc09-0256
Abou-Seif MA, Youssef AA (2004) Evaluation of some biochemical changes in diabetic patients. Clin Chim Acta 346:161–170. doi:10.1016/j.cccn.2004.03.030
Ohgami N, Miyazaki A, Sakai M, Kuniyasu A, Nakayama H, Horiuchi S (2003) Advanced glycation end products (AGE) inhibit scavenger receptor class B type I-mediated reverse cholesterol transport: a new crossroad of AGE to cholesterol metabolism. J Atheroscler Thromb 10:1–6
Hoang A, Murphy AJ, Coughlan MT, Thomas MC, Forbes JM, O’Brien R, Cooper ME, Chin-Dusting JP, Sviridov D (2007) Advanced glycation of apolipoprotein A-I impairs its anti-atherogenic properties. Diabetologia 50:1770–1779. doi:10.1007/s00125-007-0718-9
Drew BG, Duffy SJ, Formosa MF, Natoli AK, Henstridge DC, Penfold SA, Thomas WG, Mukhamedova N, de Courten B, Forbes JM, Yap FY, Kaye DM, van Hall G, Febbraio MA, Kemp BE, Sviridov D, Steinberg GR, Kingwell BA (2009) High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 119:2103–2111. doi:10.1161/circulationaha.108.843219
Haffner SM, Valdez RA, Hazuda HP, Mitchell BD, Morales PA, Stern MP (1992) Prospective analysis of the insulin-resistance syndrome (syndrome X). Diabetes 41:715–722
Waldman B, Jenkins AJ, Davis TM, Taskinen MR, Scott R, O’Connell RL, Gebski VJ, Ng MK, Keech AC (2014) HDL-C and HDL-C/ApoA-I predict long-term progression of glycemia in established type 2 diabetes. Diabetes Care 37:2351–2358. doi:10.2337/dc13-2738
Kraus WE, Houmard JA, Duscha BD, Knetzger KJ, Wharton MB, McCartney JS, Bales CW, Henes S, Samsa GP, Otvos JD, Kulkarni KR, Slentz CA (2002) Effects of the amount and intensity of exercise on plasma lipoproteins. N Engl J Med 347:1483–1492. doi:10.1056/NEJMoa020194
Quintao EC, Medina WL, Passarelli M (2000) Reverse cholesterol transport in diabetes mellitus. Diabetes Metab Res Rev 16:237–250
Cavallero E, Brites F, Delfly B, Nicolaiew N, Decossin C, De Geitere C, Fruchart JC, Wikinski R, Jacotot B, Castro G (1995) Abnormal reverse cholesterol transport in controlled type II diabetic patients. Studies on fasting and postprandial LpA-I particles. Arterioscler Thromb Vasc Biol 15:2130–2135
Capaldo B, Di Bonito P, Iaccarino M, Roman MJ, Lee ET, Devereux RB, Riccardi G, Howard BV, de Simone G (2012) Cardiovascular characteristics in subjects with increasing levels of abnormal glucose regulation: the Strong Heart Study. Diabetes Care. doi:10.2337/dc12-1501
Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW (2014) HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med 371:2383–2393. doi:10.1056/NEJMoa1409065
Rohatgi A, Grundy SM (2015) Cholesterol efflux capacity as a therapeutic target: rationale and clinical implications. J Am Coll Cardiol 66:2211–2213. doi:10.1016/j.jacc.2015.09.012
Kubota M, Nakanishi S, Hirano M, Maeda S, Yoneda M, Awaya T, Yamane K, Kohno N (2014) Relationship between serum cholesterol efflux capacity and glucose intolerance in Japanese-Americans. J Atheroscler Thromb 21:1087–1097
Passarelli M, Tang C, McDonald TO, O’Brien KD, Gerrity RG, Heinecke JW, Oram JF (2005) Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes 54:2198–2205
Walcher D, Marx N (2009) Advanced glycation end products and C-peptide-modulators in diabetic vasculopathy and atherogenesis. Semin Immunopathol 31:103–111. doi:10.1007/s00281-009-0144-9
Pu LJ, Lu L, Zhang RY, Du R, Shen Y, Zhang Q, Yang ZK, Chen QJ, Shen WF (2012) Glycation of apoprotein A-I is associated with coronary artery plaque progression in type 2 diabetic patients. Diabetes Care. doi:10.2337/dc12-1411
Traldi P, Castilho G, Sartori CH, Machado-Lima A, Nakandakare ER, Correa-Giannella ML, Roverso M, Porcu S, Lapolla A, Passarelli M (2015) Glycated human serum albumin isolated from poorly controlled diabetic patients impairs cholesterol efflux from macrophages: an investigation by mass spectrometry. Eur J Mass Spectrom (Chichester, Eng) 21:233–244. doi:10.1255/ejms.1322
Machado-Lima A, Iborra RT, Pinto RS, Castilho G, Sartori CH, Oliveira ER, Okuda LS, Nakandakare ER, Giannella-Neto D, Machado UF, Correa-Giannella ML, Traldi P, Porcu S, Roverso M, Lapolla A, Passarelli M (2015) In type 2 diabetes mellitus glycated albumin alters macrophage gene expression impairing ABCA1-mediated cholesterol efflux. J Cell Physiol 230:1250–1257. doi:10.1002/jcp.24860
Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, Mucksavage ML, Luben R, Billheimer J, Kastelein JJ, Boekholdt SM, Khaw KT, Wareham N, Rader DJ (2015) Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol 3:507–513. doi:10.1016/s2213-8587(15)00126-6
Bao LD, Li CQ, Peng R, Ren XH, Ma RL, Wang Y, Lv HJ (2015) Correlation between the decrease of cholesterol efflux from macrophages in patients with type II diabetes mellitus and down-regulated CYP7A1 expression. Genet Mol Res 14:8716–8724. doi:10.4238/2015.July.31.20
Apro J, Tietge UJ, Dikkers A, Parini P, Angelin B, Rudling M (2016) Impaired cholesterol efflux capacity of high-density lipoprotein isolated from interstitial fluid in type 2 diabetes mellitus-brief report. Arterioscler Thromb Vasc Biol 36:787–791. doi:10.1161/atvbaha.116.307385
Tsun JG, Yung S, Chau MK, Shiu SW, Chan TM, Tan KC (2014) Cellular cholesterol transport proteins in diabetic nephropathy. PLoS ONE 9:e105787. doi:10.1371/journal.pone.0105787
Manjunatha S, Distelmaier K, Dasari S, Carter RE, Kudva YC, Nair KS (2016) Functional and proteomic alterations of plasma high density lipoproteins in type 1 diabetes mellitus. Metabolism 65:1421–1431. doi:10.1016/j.metabol.2016.06.008
Attia N, Nakbi A, Smaoui M, Chaaba R, Moulin P, Hammami S, Hamda KB, Chanussot F, Hammami M (2007) Increased phospholipid transfer protein activity associated with the impaired cellular cholesterol efflux in type 2 diabetic subjects with coronary artery disease. Tohoku J Exp Med 213:129–137
Jaleel A, Henderson GC, Madden BJ, Klaus KA, Morse DM, Gopala S, Nair KS (2010) Identification of de novo synthesized and relatively older proteins: accelerated oxidative damage to de novo synthesized apolipoprotein A-1 in type 1 diabetes. Diabetes 59:2366–2374. doi:10.2337/db10-0371
Nobecourt E, Tabet F, Lambert G, Puranik R, Bao S, Yan L, Davies MJ, Brown BE, Jenkins AJ, Dusting GJ, Bonnet DJ, Curtiss LK, Barter PJ, Rye KA (2010) Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler Thromb Vasc Biol 30:766–772. doi:10.1161/atvbaha.109.201715
Okuda LS, Castilho G, Rocco DD, Nakandakare ER, Catanozi S, Passarelli M (2012) Advanced glycated albumin impairs HDL anti-inflammatory activity and primes macrophages for inflammatory response that reduces reverse cholesterol transport. Biochim Biophys Acta 1821:1485–1492. doi:10.1016/j.bbalip.2012.08.011
Pajkrt D, Doran JE, Koster F, Lerch PG, Arnet B, van der Poll T, ten Cate JW, van Deventer SJ (1996) Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J Exp Med 184:1601–1608
Bursill CA, Castro ML, Beattie DT, Nakhla S, van der Vorst E, Heather AK, Barter PJ, Rye KA (2010) High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol 30:1773–1778. doi:10.1161/atvbaha.110.211342
Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye KA, Chin-Dusting J, Hoang A, Sviridov D, Celermajer DS, Kingwell BA (2009) Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol 53:962–971. doi:10.1016/j.jacc.2008.12.008
Patel DC, Albrecht C, Pavitt D, Paul V, Pourreyron C, Newman SP, Godsland IF, Valabhji J, Johnston DG (2011) Type 2 diabetes is associated with reduced ATP-binding cassette transporter A1 gene expression, protein and function. PLoS ONE 6:e22142. doi:10.1371/journal.pone.0022142
Shao B, Pennathur S, Pagani I, Oda MN, Witztum JL, Oram JF, Heinecke JW (2010) Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J Biol Chem 285:18473–18484. doi:10.1074/jbc.M110.118182
Morgantini C, Natali A, Boldrini B, Imaizumi S, Navab M, Fogelman AM, Ferrannini E, Reddy ST (2011) Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 60:2617–2623. doi:10.2337/db11-0378
McGillicuddy FC, de la Llera Moya M, Hinkle CC, Joshi MR, Chiquoine EH, Billheimer JT, Rothblat GH, Reilly MP (2009) Inflammation impairs reverse cholesterol transport in vivo. Circulation 119:1135–1145. doi:10.1161/circulationaha.108.810721
Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen SL (2004) Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest 114:529–541. doi:10.1172/jci21109
Jornayvaz FR, Brulhart-Meynet MC, James RW (2009) Myeloperoxidase and paraoxonase-1 in type 2 diabetic patients. Nutr Metab Cardiovasc Dis 19:613–619. doi:10.1016/j.numecd.2008.12.005
Shao B, Pennathur S, Heinecke JW (2012) Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J Biol Chem 287:6375–6386. doi:10.1074/jbc.M111.337345
Kappelle PJ, Bijzet J, Hazenberg BP, Dullaart RP (2011) Lower serum paraoxonase-1 activity is related to higher serum amyloid a levels in metabolic syndrome. Arch Med Res 42:219–225. doi:10.1016/j.arcmed.2011.05.002
Murakami H, Tanabe J, Tamasawa N, Matsumura K, Yamashita M, Matsuki K, Murakami H, Matsui J, Suda T (2013) Reduction of paraoxonase-1 activity may contribute the qualitative impairment of HDL particles in patients with type 2 diabetes. Diabetes Res Clin Pract 99:30–38. doi:10.1016/j.diabres.2012.10.022
Annema W, Nijstad N, Tolle M, de Boer JF, Buijs RV, Heeringa P, van der Giet M, Tietge UJ (2010) Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2). J Lipid Res 51:743–754. doi:10.1194/jlr.M000323
Shao B, Oda MN, Oram JF, Heinecke JW (2006) Myeloperoxidase: an inflammatory enzyme for generating dysfunctional high density lipoprotein. Curr Opin Cardiol 21:322–328. doi:10.1097/01.hco.0000231402.87232.aa
Nicholls SJ, Zheng L, Hazen SL (2005) Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc Med 15:212–219. doi:10.1016/j.tcm.2005.06.004
Undurti A, Huang Y, Lupica JA, Smith JD, DiDonato JA, Hazen SL (2009) Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem 284:30825–30835. doi:10.1074/jbc.M109.047605
Peng DQ, Brubaker G, Wu Z, Zheng L, Willard B, Kinter M, Hazen SL, Smith JD (2008) Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler Thromb Vasc Biol 28:2063–2070. doi:10.1161/atvbaha.108.173815
Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M (2003) Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112:1049–1057. doi:10.1172/jci18127
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790. doi:10.1038/35008121
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414:813–820. doi:10.1038/414813a
Cacicedo JM, Benjachareowong S, Chou E, Ruderman NB, Ido Y (2005) Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 54:1838–1845
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H (2000) High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49:1939–1945
Ostrander DB, Sparagna GC, Amoscato AA, McMillin JB, Dowhan W (2001) Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem 276:38061–38067. doi:10.1074/jbc.M107067200
Ceriello A, Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, Esposito K, Giugliano D (2004) Effect of postprandial hypertriglyceridemia and hyperglycemia on circulating adhesion molecules and oxidative stress generation and the possible role of simvastatin treatment. Diabetes 53:701–710
Huang Y, Didonato JA, Levison BS, Schmitt D, Li L, Wu Y, Buffa J, Kim T, Gerstenecker GS, Gu X, Kadiyala CS, Wang Z, Culley MK, Hazen JE, Didonato AJ, Fu X, Berisha SZ, Peng D, Nguyen TT, Liang S, Chuang CC, Cho L, Plow EF, Fox PL, Gogonea V, Tang WH, Parks JS, Fisher EA, Smith JD, Hazen SL (2014) An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med 20:193–203. doi:10.1038/nm.3459
Kataoka Y, Shao M, Wolski K, Uno K, Puri R, Murat Tuzcu E, Hazen SL, Nissen SE, Nicholls SJ (2014) Myeloperoxidase levels predict accelerated progression of coronary atherosclerosis in diabetic patients: insights from intravascular ultrasound. Atherosclerosis 232:377–383. doi:10.1016/j.atherosclerosis.2013.11.075
Shao B, Tang C, Sinha A, Mayer PS, Davenport GD, Brot N, Oda MN, Zhao XQ, Heinecke JW (2014) Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ Res 114:1733–1742. doi:10.1161/circresaha.114.303454
Lu N, Xie S, Li J, Tian R, Peng YY (2015) Myeloperoxidase-mediated oxidation targets serum apolipoprotein A-I in diabetic patients and represents a potential mechanism leading to impaired anti-apoptotic activity of high density lipoprotein. Clin Chim Acta 441:163–170. doi:10.1016/j.cca.2014.12.014
Chen X, Bakillah A, Zhou L, Pan X, Hoepfner F, Jacob M, Jiang XC, Lazar J, Schlitt A, Hussain MM (2016) Nitrated apolipoprotein AI/apolipoprotein AI ratio is increased in diabetic patients with coronary artery disease. Atherosclerosis 245:12–21. doi:10.1016/j.atherosclerosis.2015.11.021
Abderrahmani A, Niederhauser G, Favre D, Abdelli S, Ferdaoussi M, Yang JY, Regazzi R, Widmann C, Waeber G (2007) Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 50:1304–1314. doi:10.1007/s00125-007-0642-z
Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS, Verchere CB, Hayden MR (2007) Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 13:340–347. doi:10.1038/nm1546
Han R, Lai R, Ding Q, Wang Z, Luo X, Zhang Y, Cui G, He J, Liu W, Chen Y (2007) Apolipoprotein A-I stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia 50:1960–1968. doi:10.1007/s00125-007-0752-7
Drew BG, Fidge NH, Gallon-Beaumier G, Kemp BE, Kingwell BA (2004) High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc Natl Acad Sci USA 101:6999–7004. doi:10.1073/pnas.0306266101
Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R (2003) Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA 290:2292–2300. doi:10.1001/jama.290.17.2292
Tardif JC, Gregoire J, L’Allier PL, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodes-Cabau J (2007) Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA 297:1675–1682. doi:10.1001/jama.297.15.jpc70004
Nieuwdorp M, Vergeer M, Bisoendial RJ, Roodt J, Levels H, Birjmohun RS, Kuivenhoven JA, Basser R, Rabelink TJ, Kastelein JJ, Stroes ES (2008) Reconstituted HDL infusion restores endothelial function in patients with type 2 diabetes mellitus. Diabetologia 51:1081–1084. doi:10.1007/s00125-008-0975-2
Wang Y, Oram JF (2002) Unsaturated fatty acids inhibit cholesterol efflux from macrophages by increasing degradation of ATP-binding cassette transporter A1. J Biol Chem 277:5692–5697. doi:10.1074/jbc.M109977200
Reaven GM, Chen YD (1988) Role of abnormal free fatty acid metabolism in the development of non-insulin-dependent diabetes mellitus. Am J Med 85:106–112
Srivastava RA, Srivastava N, Averna M, Cefalu AB, Schonfeld G (1999) Molecular bases of low production rates of apolipoprotein B-100 and truncated apoB-82 in a mutant HepG2 cell line generated by targeted modification of the apolipoprotein B gene. J Lipid Res 40:901–912
Adeli K, Wettesten M, Asp L, Mohammadi A, Macri J, Olofsson SO (1997) Intracellular assembly and degradation of apolipoprotein B-100-containing lipoproteins in digitonin-permeabilized HEP G2 cells. J Biol Chem 272:5031–5039
Srivastava N, Cefalu AB, Noto D, Schonfeld G, Averna M, Srivastava RA (2010) The production of 85 kDa N-terminal fragment of apolipoprotein B in mutant HepG2 cells generated by targeted modification of apoB gene occurs by ALLN-inhibitable protease cleavage during translocation. Biochem Biophys Res Commun 398:665–670. doi:10.1016/j.bbrc.2010.06.130
Wu KK, Huan Y (2007) Diabetic atherosclerosis mouse models. Atherosclerosis 191:241–249. doi:10.1016/j.atherosclerosis.2006.08.030
Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364:127–135. doi:10.1056/NEJMoa1001689
Yamamoto S, Narita I, Kotani K (2016) The macrophage and its related cholesterol efflux as a HDL function index in atherosclerosis. Clin Chim Acta 457:117–122. doi:10.1016/j.cca.2016.04.012
Aiello RJ, Brees D, Bourassa PA, Royer L, Lindsey S, Coskran T, Haghpassand M, Francone OL (2002) Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arterioscler Thromb Vasc Biol 22:630–637
Ou X, Dai X, Long Z, Tang Y, Cao D, Hao X, Hu Y, Li X, Tang C (2008) Liver X receptor agonist T0901317 reduces atherosclerotic lesions in apoE−/− mice by up-regulating NPC1 expression. Sci. China C 51:418–429. doi:10.1007/s11427-008-0054-4
Brunham LR, Singaraja RR, Duong M, Timmins JM, Fievet C, Bissada N, Kang MH, Samra A, Fruchart JC, McManus B, Staels B, Parks JS, Hayden MR (2009) Tissue-specific roles of ABCA1 influence susceptibility to atherosclerosis. Arterioscler Thromb Vasc Biol 29:548–554. doi:10.1161/atvbaha.108.182303
Wang Y, Oram JF (2005) Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a phospholipase D2 pathway. J Biol Chem 280:35896–35903. doi:10.1074/jbc.M506210200
Wang Y, Oram JF (2007) Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a protein kinase C delta pathway. J Lipid Res 48:1062–1068. doi:10.1194/jlr.M600437-JLR200
Hawley SA, Gadalla AE, Olsen GS, Hardie DG (2002) The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 51:2420–2425
Hardie DG (2008) AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 32(Suppl 4):S7–S12. doi:10.1038/ijo.2008.116
Srivastava RA, Pinkosky SL, Filippov S, Hanselman JC, Cramer CT, Newton RS (2012) AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J Lipid Res 53:2490–2514. doi:10.1194/jlr.R025882
Park KG, Min AK, Koh EH, Kim HS, Kim MO, Park HS, Kim YD, Yoon TS, Jang BK, Hwang JS, Kim JB, Choi HS, Park JY, Lee IK, Lee KU (2008) Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Hepatology 48:1477–1486. doi:10.1002/hep.22496
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8:1288–1295. doi:10.1038/nm788
Lehti M, Donelan E, Abplanalp W, Al-Massadi O, Habegger KM, Weber J, Ress C, Mansfeld J, Somvanshi S, Trivedi C, Keuper M, Ograjsek T, Striese C, Cucuruz S, Pfluger PT, Krishna R, Gordon SM, Silva RA, Luquet S, Castel J, Martinez S, D’Alessio D, Davidson WS, Hofmann SM (2013) High-density lipoprotein maintains skeletal muscle function by modulating cellular respiration in mice. Circulation 128:2364–2371. doi:10.1161/circulationaha.113.001551
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13:2004–2008
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG (2005) Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2:9–19. doi:10.1016/j.cmet.2005.05.009
Vaughan AM, Oram JF (2006) ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J Lipid Res 47:2433–2443. doi:10.1194/jlr.M600218-JLR200
Son SH, Goo YH, Choi M, Saha PK, Oka K, Chan LC, Paul A (2016) Enhanced atheroprotection and lesion remodelling by targeting the foam cell and increasing plasma cholesterol acceptors. Cardiovasc Res 109:294–304. doi:10.1093/cvr/cvv241
Lorenzi I, von Eckardstein A, Radosavljevic S, Rohrer L (2008) Lipidation of apolipoprotein A-I by ATP-binding cassette transporter (ABC) A1 generates an interaction partner for ABCG1 but not for scavenger receptor BI. Biochem Biophys Acta 1781:306–313. doi:10.1016/j.bbalip.2008.04.006
Yin K, Liao DF, Tang CK (2010) ATP-binding membrane cassette transporter A1 (ABCA1): a possible link between inflammation and reverse cholesterol transport. Mol Med 16:438–449. doi:10.2119/molmed.2010.00004
Yvan-Charvet L, Wang N, Tall AR (2010) Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol 30:139–143. doi:10.1161/atvbaha.108.179283
Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hui EK, Nayak DP, Fogelman AM (2004) D-4F, an apolipoprotein A-I mimetic peptide, inhibits the inflammatory response induced by influenza A infection of human type II pneumocytes. Circulation 110:3252–3258. doi:10.1161/01.CIR.0000147232.75456.B3
Barter PJ, Puranik R, Rye KA (2007) New insights into the role of HDL as an anti-inflammatory agent in the prevention of cardiovascular disease. Curr Cardiol Rep 9:493–498
Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SP, Remaley AT, Sviridov D, Chin-Dusting J (2008) High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol 28:2071–2077. doi:10.1161/atvbaha.108.168690
Tabet F, Remaley AT, Segaliny AI, Millet J, Yan L, Nakhla S, Barter PJ, Rye KA, Lambert G (2010) The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arterioscler Thromb Vasc Biol 30:246–252. doi:10.1161/atvbaha.109.200196
de la Llera Moya M, McGillicuddy FC, Hinkle CC, Byrne M, Joshi MR, Nguyen V, Tabita-Martinez J, Wolfe ML, Badellino K, Pruscino L, Mehta NN, Asztalos BF, Reilly MP (2012) Inflammation modulates human HDL composition and function in vivo. Atherosclerosis. doi:10.1016/j.atherosclerosis.2012.02.032
Majdalawieh A, Ro HS (2009) LPS-induced suppression of macrophage cholesterol efflux is mediated by adipocyte enhancer-binding protein 1. Int J Biochem Cell Biol 41:1518–1525. doi:10.1016/j.biocel.2009.01.003
Yvan-Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR (2010) Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol 30:1430–1438. doi:10.1161/atvbaha.110.207142
Moore RE, Navab M, Millar JS, Zimetti F, Hama S, Rothblat GH, Rader DJ (2005) Increased atherosclerosis in mice lacking apolipoprotein A-I attributable to both impaired reverse cholesterol transport and increased inflammation. Circ Res 97:763–771. doi:10.1161/01.RES.0000185320.82962.F7
Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, Tall AR (2010) ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res 106:1861–1869. doi:10.1161/circresaha.110.217281
Speer T, Rohrer L, Blyszczuk P, Shroff R, Kuschnerus K, Krankel N, Kania G, Zewinger S, Akhmedov A, Shi Y, Martin T, Perisa D, Winnik S, Muller MF, Sester U, Wernicke G, Jung A, Gutteck U, Eriksson U, Geisel J, Deanfield J, von Eckardstein A, Luscher TF, Fliser D, Bahlmann FH, Landmesser U (2013) Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 38:754–768. doi:10.1016/j.immuni.2013.02.009
Flegel WA, Baumstark MW, Weinstock C, Berg A, Northoff H (1993) Prevention of endotoxin-induced monokine release by human low- and high-density lipoproteins and by apolipoprotein A-I. Infect Immun 61:5140–5146
Parker TS, Levine DM, Chang JC, Laxer J, Coffin CC, Rubin AL (1995) Reconstituted high-density lipoprotein neutralizes gram-negative bacterial lipopolysaccharides in human whole blood. Infect Immun 63:253–258
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17:1–14. doi:10.1093/intimm/dxh186
De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, Vogelhuber J, Kraut M, Ulas T, Kerksiek A, Krebs W, Bode N, Grebe A, Fitzgerald ML, Hernandez NJ, Williams BR, Knolle P, Kneilling M, Rocken M, Lutjohann D, Wright SD, Schultze JL, Latz E (2014) High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol 15:152–160. doi:10.1038/ni.2784
Dandekar A, Qiu Y, Kim H, Wang J, Hou X, Zhang X, Zheng Z, Mendez R, Yu FS, Kumar A, Fang D, Sun F, Zhang K (2016) Toll-like receptor (TLR) signaling interacts with CREBH to modulate high-density lipoprotein (HDL) in response to bacterial endotoxin. J Biol Chem 291:23149–23158. doi:10.1074/jbc.M116.755728
Francone OL, Royer L, Boucher G, Haghpassand M, Freeman A, Brees D, Aiello RJ (2005) Increased cholesterol deposition, expression of scavenger receptors, and response to chemotactic factors in Abca1-deficient macrophages. Arterioscler Thromb Vasc Biol 25:1198–1205. doi:10.1161/01.atv.0000166522.69552.99
Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS (2008) Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem 283:22930–22941. doi:10.1074/jbc.M801408200
Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR (2008) Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 118:1837–1847. doi:10.1161/circulationaha.108.793869
Baldan A, Gomes AV, Ping P, Edwards PA (2008) Loss of ABCG1 results in chronic pulmonary inflammation. J Immunol 180:3560–3568
Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA, Jamieson BD, Tontonoz P (2008) LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134:97–111. doi:10.1016/j.cell.2008.04.052
Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, Bharadwaj M, Walzem R, Chan L, Oka K, Thomas MJ, Sorci-Thomas MG (2009) Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol 29:843–849. doi:10.1161/atvbaha.108.183442
Feng H, Guo L, Wang D, Gao H, Hou G, Zheng Z, Ai J, Foreman O, Daugherty A, Li XA (2011) Deficiency of scavenger receptor BI leads to impaired lymphocyte homeostasis and autoimmune disorders in mice. Arterioscler Thromb Vasc Biol 31:2543–2551. doi:10.1161/atvbaha.111.234716
Rueda CM, Rodriguez-Perea AL, Moreno-Fernandez M, Jackson CM, Melchior JT, Davidson WS, Chougnet CA (2017) High density lipoproteins selectively promote the survival of human regulatory T-cells. J Lipid Res. doi:10.1194/jlr.M072835
Pastrana JL, Sha X, Virtue A, Mai J, Cueto R, Lee IA, Wang H, Yang XF (2012) Regulatory T cells and atherosclerosis. J Clin Exp Cardiol 2012:2. doi:10.4172/2155-9880.s12-002
Charles-Schoeman C, Lee YY, Grijalva V, Amjadi S, FitzGerald J, Ranganath VK, Taylor M, McMahon M, Paulus HE, Reddy ST (2012) Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann Rheum Dis 71:1157–1162. doi:10.1136/annrheumdis-2011-200493
Field FJ, Watt K, Mathur SN (2010) TNF-alpha decreases ABCA1 expression and attenuates HDL cholesterol efflux in the human intestinal cell line Caco-2. J Lipid Res 51:1407–1415. doi:10.1194/jlr.M002410
Ammirati E, Bozzolo EP, Contri R, Baragetti A, Palini AG, Cianflone D, Banfi M, Uboldi P, Bottoni G, Scotti I, Pirillo A, Grigore L, Garlaschelli K, Monaco C, Catapano AL, Sabbadini MG, Manfredi AA, Norata GD (2014) Cardiometabolic and immune factors associated with increased common carotid artery intima-media thickness and cardiovascular disease in patients with systemic lupus erythematosus. Nutr Metab Cardiovasc Dis 24:751–759. doi:10.1016/j.numecd.2014.01.006
Altruda F, Poli V, Restagno G, Argos P, Cortese R, Silengo L (1985) The primary structure of human hemopexin deduced from cDNA sequence: evidence for internal, repeating homology. Nucleic Acids Res 13:3841–3859
Katnik I, Jadach J (1996) Haptoglobin concentration in serum and other body fluids measured by comparison of its reactivity with hemoglobin and concanavalin A. Arch Immunol Ther Exp (Warsz) 44:45–50
Dobryszycka W (1997) Biological functions of haptoglobin—new pieces to an old puzzle. Eur J Clin Chem Clin Biochem 35:647–654
Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G (2000) Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol 67:97–103
Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK (2001) Identification of the haemoglobin scavenger receptor. Nature 409:198–201. doi:10.1038/35051594
Engstrom G, Hedblad B, Tyden P, Lindgarde F (2009) Inflammation-sensitive plasma proteins are associated with increased incidence of heart failure: a population-based cohort study. Atherosclerosis 202:617–622. doi:10.1016/j.atherosclerosis.2008.05.038
Navab M, Anantharamaiah GM, Fogelman AM (2005) The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med 15:158–161. doi:10.1016/j.tcm.2005.05.008
Ye D, Lammers B, Zhao Y, Meurs I, Van Berkel TJ, Van Eck M (2011) ATP-binding cassette transporters A1 and G1, HDL metabolism, cholesterol efflux, and inflammation: important targets for the treatment of atherosclerosis. Curr Drug Targets 12:647–660
Watanabe J, Chou KJ, Liao JC, Miao Y, Meng HH, Ge H, Grijalva V, Hama S, Kozak K, Buga G, Whitelegge JP, Lee TD, Farias-Eisner R, Navab M, Fogelman AM, Reddy ST (2007) Differential association of hemoglobin with proinflammatory high density lipoproteins in atherogenic/hyperlipidemic mice. A novel biomarker of atherosclerosis. J Biol Chem 282:23698–23707. doi:10.1074/jbc.M702163200
Watanabe J, Grijalva V, Hama S, Barbour K, Berger FG, Navab M, Fogelman AM, Reddy ST (2009) Hemoglobin and its scavenger protein haptoglobin associate with apoA-1-containing particles and influence the inflammatory properties and function of high density lipoprotein. J Biol Chem 284:18292–18301. doi:10.1074/jbc.M109.017202
Matuszek MA, Aristoteli LP, Bannon PG, Hendel PN, Hughes CF, Jessup W, Dean RT, Kritharides L (2003) Haptoglobin elutes from human atherosclerotic coronary arteries—a potential marker of arterial pathology. Atherosclerosis 168:389–396
Asleh R, Levy AP (2005) In vivo and in vitro studies establishing haptoglobin as a major susceptibility gene for diabetic vascular disease. Vasc Health Risk Manag 1:19–28
Levy AP, Hochberg I, Jablonski K, Resnick HE, Lee ET, Best L, Howard BV (2002) Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: the Strong Heart Study. J Am Coll Cardiol 40:1984–1990
Graversen JH, Madsen M, Moestrup SK (2002) CD163: a signal receptor scavenging haptoglobin-hemoglobin complexes from plasma. Int J Biochem Cell Biol 34:309–314
Kaempfer T, Duerst E, Gehrig P, Roschitzki B, Rutishauser D, Grossmann J, Schoedon G, Vallelian F, Schaer DJ (2011) Extracellular hemoglobin polarizes the macrophage proteome toward Hb-clearance, enhanced antioxidant capacity and suppressed HLA class 2 expression. J Proteome Res 10:2397–2408. doi:10.1021/pr101230y
Ross R (1999) Atherosclerosis is an inflammatory disease. Am Heart J 138:S419–S420
Wallberg-Jonsson S, Cvetkovic JT, Sundqvist KG, Lefvert AK, Rantapaa-Dahlqvist S (2002) Activation of the immune system and inflammatory activity in relation to markers of atherothrombotic disease and atherosclerosis in rheumatoid arthritis. J Rheumatol 29:875–882
Morita T (2005) Heme oxygenase and atherosclerosis. Arterioscler Thromb Vasc Biol 25:1786–1795. doi:10.1161/01.atv.0000178169.95781.49
Smeets MB, Pasterkamp G, Lim SK, Velema E, van Middelaar B, de Kleijn DP (2002) Nitric oxide synthesis is involved in arterial haptoglobin expression after sustained flow changes. FEBS Lett 529:221–224
Jahagirdar R, Zhang H, Azhar S, Tobin J, Attwell S, Yu R, Wu J, McLure KG, Hansen HC, Wagner GS, Young PR, Srivastava RA, Wong NC, Johansson J (2014) A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 236:91–100. doi:10.1016/j.atherosclerosis.2014.06.008
Asleh R, Miller-Lotan R, Aviram M, Hayek T, Yulish M, Levy JE, Miller B, Blum S, Milman U, Shapira C, Levy AP (2006) Haptoglobin genotype is a regulator of reverse cholesterol transport in diabetes in vitro and in vivo. Circ Res 99:1419–1425. doi:10.1161/01.res.0000251741.65179.56
Lioupis C, Barbatis C, Drougou A, Koliaraki V, Mamalaki A, Klonaris C, Georgopoulos S, Andrikopoulos V, Bastounis E (2011) Association of haptoglobin genotype and common cardiovascular risk factors with the amount of iron in atherosclerotic carotid plaques. Atherosclerosis 216:131–138. doi:10.1016/j.atherosclerosis.2011.01.028
Purushothaman M, Krishnan P, Purushothaman KR, Baber U, Tarricone A, Perez JS, Wiley J, Kini A, Sharma SK, Fuster V, Moreno PR (2012) Genotype-dependent impairment of hemoglobin clearance increases oxidative and inflammatory response in human diabetic atherosclerosis. Arterioscler Thromb Vasc Biol 32:2769–2775. doi:10.1161/atvbaha.112.252122
Purushothaman KR, Purushothaman M, Levy AP, Lento PA, Evrard S, Kovacic JC, Briley-Saebo KC, Tsimikas S, Witztum JL, Krishnan P, Kini A, Fayad ZA, Fuster V, Sharma SK, Moreno PR (2012) Increased expression of oxidation-specific epitopes and apoptosis are associated with haptoglobin genotype: possible implications for plaque progression in human atherosclerosis. J Am Coll Cardiol 60:112–119. doi:10.1016/j.jacc.2012.04.011
Borrell-Pages M, Romero JC, Juan-Babot O, Badimon L (2011) Wnt pathway activation, cell migration, and lipid uptake is regulated by low-density lipoprotein receptor-related protein 5 in human macrophages. Eur Heart J 32:2841–2850. doi:10.1093/eurheartj/ehr062
Tsaousi A, Williams H, Lyon CA, Taylor V, Swain A, Johnson JL, George SJ (2011) Wnt4/beta-catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ Res 108:427–436. doi:10.1161/circresaha.110.233999
Srivastava R, Cefalu, AB, Davide, A, Averna MR (2013) A combination of metformin, quercetin, and curcumin restores HDL function and improves atherosclerosis burden in LDLr−/−/ob.ob leptin−/− and LDLr−/− mice by attenuating insulin resistance, hyperglycemia, and low-grade inflammation. ATVB Scientific Session: Abstract
Acknowledgements
The author would like to thank Maurizio Averna, University of Palermo, Palermo, Italy and Charles L Bisgaier, Gemphire Therapeutics, Livonia, MI, USA for many stimulating discussions relating to dysfunctional HDL, diabetes, and atherosclerosis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
During the preparation of this manuscript, the Rai Ajit K. Srivastava served as a consultant to Gemphire Therapeutics and currently employed at Gemphire Therapeutics Inc. The author has no conflict of interest.
Rights and permissions
About this article

Cite this article
Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol Cell Biochem 440, 167–187 (2018). https://doi.org/10.1007/s11010-017-3165-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3165-z