
Abstract
Isorhamnetin, a flavonoid compound extracted from the Chinese herb Hippophae rhamnoides L., is well known for its anti-inflammatory, anti-oxidative, anti-adipogenic, anti-proliferative, and anti-tumor activities. However, the role of isorhamnetin in cardiac hypertrophy has not been reported. The aims of the present study were to find whether isorhamnetin could alleviate cardiac hypertrophy and to define the underlying molecular mechanisms. Here, we investigated the effects of isorhamnetin (100 mg/kg/day) on cardiac hypertrophy induced by aortic banding in mice. Cardiac hypertrophy was evaluated by echocardiographic, hemodynamic, pathological, and molecular analyses. Our data demonstrated that isorhamnetin could inhibit cardiac hypertrophy and fibrosis 8 weeks after aortic banding. The results further revealed that the effect of isorhamnetin on cardiac hypertrophy was mediated by blocking the activation of phosphatidylinositol 3-kinase–AKT signaling pathway. In vitro studies performed in neonatal rat cardiomyocytes confirmed that isorhamnetin could attenuate cardiomyocyte hypertrophy induced by angiotensin II, which was associated with phosphatidylinositol 3-kinase–AKT signaling pathway. In conclusion, these data indicate for the first time that isorhamnetin has protective potential for targeting cardiac hypertrophy by blocking the phosphatidylinositol 3-kinase–AKT signaling pathway. Thus, our study suggests that isorhamnetin may represent a potential therapeutic strategy for the treatment of cardiac hypertrophy and heart failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiac hypertrophy is an adaptive response of the heart to pressure overload, and is characterized by an increase in myocardial mass and accumulation of extracellular matrix [1, 2]. Although the hypertrophic response is initially a compensatory mechanism that augments cardiac output, prolonged hypertrophy can lead to ventricular dilatation and heart failure, which is increasing in prevalence and is a debilitating disease with high rates of mortality and morbidity [3]. In the previous researches, a series of studies have illuminated the signaling transduction pathways that induce myocardial hypertrophy [4, 5]. These signaling pathways including the PKC pathway [6], the mitogen-activated protein kineses (MAPK) pathway [7], and the phosphatidylinositol 3-kinase (PI3K)–AKT pathway [8], which directly modify transcriptional regulatory factors promoting alterations in cardiac gene expression and result in cardiac hypertrophy. However, the molecular modulators that antagonize the development of cardiac remodeling and the transition to heart failure remain incompletely defined. Therefore, promoting or blocking the signals and their transduction processes will be a key strategy for preventing the development of heart failure resulting from cardiac hypertrophy.
Chinese medicine extracts, such as polyphenols from grapes and grape products, have recently attracted considerable attention for their protective effect on cardiomyocytes hypertrophy [9, 10]. Isorhamnetin (molecular formula: C16H12O7), a flavonol aglycone, isolated from the traditional Chinese medicine H. rhamnoides L., was frequently used in traditional medicine to prevent and treat diverse diseases [11]. Isorhamnetin has been shown to play a variety of roles in anti-oxidation, anti-inflammation, anti-tumor, anti-viral, and neurodegenerative injury protection and is also considered to have health benefits in humans [12–14]. It has been reported that isorhamnetin could protect rat ventricular myocytes from ischemia and reperfusion injury [15]. Yun Luo et al. have demonstrated that isorhamnetin could inhibit the H2O2-induced activation of the intrinsic apoptotic pathway by scavenging free ROS and extracellular signal-regulated kinase 1/2 (ERK1/2) inactivation [16]. Yun Luo et al. also found that isorhamnetin could protect heart against doxorubicin-induced cardiotoxicity in vivo and in vitro [17]. In addition, Bao and colleagues found that isorhamnetin had protective effects on ox-LDL-induced endothelial cell injuries by increasing antioxidant activity and activating p38 MAPK signaling pathway [18]. In the type 2 diabetic rat model, isorhamnetin was found to be able to inhibit the NF-κB signaling activity, attenuate oxidative stress, and decrease the production of inflammatory mediators [19]. Besides, present studies indicate that isorhamnetin can suppress skin cancer [20], breast cancer [21], colon cancer [22], and atherosclerosis [23] by inhibiting PI3K/AKT activation. Seeing that isorhamnetin can regulate multiple hypertrophy-related signalings like ERK1/2, MAPKs, NF-κB, and PI3K/AKT in different experimental models, and it also plays a protective role in the cardiovascular diseases, we fully speculated that isorhamnetin may have a protective effect on cardiac hypertrophy. Therefore, this study was designed to determine whether isorhamnetin has the protective properties against cardiac hypertrophy and to explore the possible molecular mechanisms.
In the present study, we investigate whether isorhamnetin could ameliorate cardiac hypertrophy induced by pressure overload in mice. Our results show that isorhamnetin mitigated cardiac hypertrophy and fibrosis induced by pressure overload and preserved cardiac function. Mechanistically, we discovered that isorhamnetin-prevented maladaptive remodeling was partially dependent on the regulation of the PI3K–Akt signaling pathway. Based on our results, we believe that isorhamnetin would become a potential therapeutic strategy for the treatment of cardiac hypertrophy and heart failure.
Methods and materials
Reagents
Isorhamnetin (purity, 98%) was purchased from Shanghai Winherb Medical S&T Development (Shanghai, China). The following primary antibodies were used in our experiments: Anti-GAPDH (MB001) was purchased from Bioworld Technology; LY294002 (#9901), anti-AKT (#4691), anti-phospho-AKT (#4060), anti-GSK3β (#9315), anti-phospho-GSK3β (#9322), anti-mTOR (#2983), anti-phospho-mTOR (#2971), anti-eIF-4E (#2067), anti-phospho-eIF-4E (#9741), anti-PI3K (#4257), anti-phospho-PI3K (#4228),anti-P70S6K (#2708), and anti-phospho-P70S6K (#9208) were purchased from Cell Signaling Technology. The BCA protein assay kit was purchased from Pierce (Rockford, IL, USA). Peroxidase-conjugated secondary antibodies (Jackson Immuno Research Laboratories, West Grove, PA, USA) were utilized for the visualization of primary antibody binding. The primary antibodies were diluted at the ratio of 1:1000, and the dilution ratio of the second antibody was 1:10,000. Fetal calf serum was purchased from HyClone (Waltham, MA, USA). The cell culture reagents and all of the other reagents were purchased from Sigma (St. Louis, MO, USA).
Animals and animal models
All of the animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and approved by the Animal Care and Use Committee of Department of Cardiology, Zhengzhou University. The male C57B/L6J mice used in the experiment were obtained from Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). Mice were randomly assigned into four groups. Isorhamnetin suspension was prepared using 0.5% carboxy methylcellulose solution for animal experiments. Suspensions were freshly prepared and administered at a constant volume of 1 ml/100 g body weight by oral gavage once a day. The control group of these animal experiments was given the same volume of liquid comprising solely of the vehicle solution (0.5% carboxy methylcellulose).
Aortic banding (AB) was conducted as described previously [24]. Treatment with 100 mg/kg/day of isorhamnetin or vehicle for 8 weeks after AB surgery or sham operation allowed for critical evaluation. Adequate constriction of the aorta was determined by Doppler analysis. A similar procedure, without constricting the aorta, was performed in the sham-operated group. The wall thickness and internal diameter of the left ventricle (LV) were assessed using echocardiography at the indicated time after surgery. At the end of these procedures, the hearts, lungs, and tibiae from the sacrificed mice were harvested, weighed, and analyzed to compare the heart weight/body weight (HW/BW, mg/g), lung weight/body weight (LW/BW, mg/g), and heart weight/tibia length (HW/TL, mg/mm) ratios between the isorhamnetin-treated mice and vehicle-treated mice.
Echocardiography and hemodynamic evaluations
Echocardiography was performed in mice anesthetized with 1.5% isoflurane using a MyLab 30CV (Esaote SpA) with a 10-MHz linear-array ultrasound transducer in accordance with previously described methods [25]. The LV dimensions were assessed in the parasternal long-axis and the short-axis views at a frame rate of 120 Hz. The LV end-systolic diameter (LVESD) and LV end-diastolic diameter (LVEDD) were measured from the M-mode tracing with a sweep speed of 50 mm/s at the mid-papillary muscle level.
For the hemodynamic measurements, a microtip catheter transducer (SPR-839, Millar Instruments, Houston, TX, USA) was inserted into the right carotid artery and advanced into the left ventricle of mice anesthetized with 1.5% isoflurane. After stabilization for 15 min, pressure, volume signals, and heart rate were continuously recorded using a Millar Pressure–Volume System (MPVS-400, Millar Instruments, Houston, TX, USA). The results were analyzed with Chart 5.0 software.
Histological analysis
The animals were sacrificed 8 weeks after AB or sham surgery. The hearts were arrested with a 10% potassium chloride solution at end-diastole and then fixed in 10% formalin. Paraffin-embedded hearts were cut transversely into 4–5-µm sections. Heart sections were stained with HE and PSR (to detect collagen). Cross-sectional areas of the myocytes were visualized with FITC-conjugated WGA (Invitrogen) staining, and the cell size was measured using a quantitative digital image analysis system (Image-Pro Plus 6.0).
Neonatal rat cardiomyocytes (NRCMs) culture
Primary cultured NRCMs were prepared as previously described. In brief, the hearts of 1–2-day-old Sprague–Dawley rats were excised and digested with PBS containing 0.03% trypsin and 0.04% collagenase type II to isolate the cardiomyocytes from the fibroblasts. The cardiomyocytes were then seeded at a density of 1 × 106 cells/well in six-well culture plates coated with fibronectin in plating medium, which consisted of F10 medium supplemented with 10% fetal calf serum and penicillin/streptomycin.
Immunofluorescence analysis
Immunofluorescence analysis was performed using standard immunocytochemical techniques. Briefly, NRCMs were pretreated with LY294002 for 1 h and then treated the cells with angiotensin (Ang) II for 48 h after treat with Isorhamnetin or vehicle for 24 h. Subsequently, the cardiomyocytes were fixed with 3.7% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 in PBS for 40 min, blocked with a 10% BSA solution for 1 h at room temperature and then incubated with an anti-α-actinin antibody (1:100 dilution). The surface areas were measured using Image-Pro Plus 6.0 software.
Western blotting and quantitative real-time PCR
Whole cell lysates were obtained by homogenizing the hearts or NRCMs in RIPA lysis buffer. The proteins (50 μg) were resolved via SDS–PAGE (Invitrogen) and transferred to a PVDF membrane (Millipore). The membrane was blocked with 10% non-fat milk and then incubated with the indicated primary antibodies overnight at 4 °C. After incubation with secondary antibodies for 1 h at room temperature, the membranes were treated with ECL reagents (170–5061, Bio-Rad) prior to visualization using a Fluor Chem E imager (Cell Biosciences) according to the manufacturer’s instructions. The specific protein expression levels were normalized to GAPDH on the same nitrocellulose membrane.
Total RNA was isolated from heart tissues or NRCMs using TRIzol Reagent (Invitrogen), and the Transcriptor First Strand cDNA Synthesis Kit (Roche) was used to synthesize cDNA. The mRNA levels of the indicated genes were quantified with real-time PCR using SYBR Green (Roche). The real-time PCR primers that were used are shown in Table 2 (in supplementary material).
Statistical analysis
The data are represented as the mean ± SD. Student’s two-tailed t test was used to compare the means of two groups of samples, and two-way analysis of variance with general linear model procedures using a univariate approach was applied for more than two groups. A value of P < 0.05 was considered a statistically significant difference. All of the statistical analysis was performed with SPSS software (version 17.0, SPSS Inc.).
Results
Isorhamnetin inhibited cardiac hypertrophy in vitro
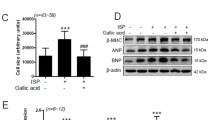
To determine the possible role of isorhamnetin in cardiac remodeling, we first evaluated the effect of isorhamnetin on cardiomyocyte hypertrophy in vitro, using primary cultured NRCMs in a well-controlled experimental setting. Cardiac myocytes were pretreated with isorhamnetin at the indicated concentrations for 24 h and subsequently stimulated with Ang II for 48 h, followed by immunostaining with α-actinin to measure the cell size. Our results revealed isorhamnetin treatment markedly attenuated the increase in cardiac myocyte size seen in the presence of Ang II after 48 h of culture (Fig. 1a, b). In addition, isorhamnetin markedly reduced the increased mRNA levels of atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC) induced by Ang II, especially in isorhamnetin (50 μM) group (Fig. 1c). However, the inhibitory effect of isorhamnetin did not occur in a dose-dependent manner. These data suggested that isorhamnetin could inhibit cardiac hypertrophy in vitro.

Isorhamnetin inhibited cardiac hypertrophy in vitro. a Representative images of the cardiomyocytes showed the inhibitory effect of isorhamnetin on the enlargement of cardiomyocytes in response to AngII for 48 h. b Quantification of cell cross-sectional area by measuring 100 random cells. c Real-time PCR analysis of the hypertrophy markers atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC) incubated with different doses of isorhamnetin (5–100 μM) for 48 h subsequently treated with 1 μM Ang II for 24 h.*P < 0.05 versus control. #P < 0.05 versus AngII-treated cells
Isorhamnetin protected against cardiac hypertrophy induced by pressure overload
After exploring the functional contribution of isorhamnetin on cardiomyocyte hypertrophy in vitro, we next sought to identify whether isorhamnetin could antagonize the hypertrophic response induced by pressure overload. Mice were randomly assigned to four groups: isorhamnetin-treated mice and vehicle-treated mice were subjected to either AB or sham surgery for 8 weeks. It is important to pay attention to that under basal conditions, mice treated with isorhamnetin did not show any pathological/physiological alterations in cardiac structure or function with vehicle-treated mice. However, compared to the AB-induced vehicle-treated mice, the myocardial hypertrophic response was significantly blocked in isorhamnetin-treated mice after 8 weeks of AB, as shown by direct examination of the gross heart, the cardiomyocyte cross-sectional area (CSA) accessed by hematoxylin–eosin (HE), and wheat germ agglutinin–fluorescein (WGA) isothiocyanate staining (Fig. 2a, b). In parallel, the ratios of heart weight/body weight, heart weight/tibia length, and lung weight/body weight were significantly decreased in isorhamnetin-treated mice compared with the vehicle-treated mice after 8 weeks of AB (Fig. 2c and Table 1 in supplementary material). Together, the above data demonstrated that isorhamnetin could retard the progression of cardiac hypertrophy induced by pressure overload.

Isorhamnetin protected against cardiac hypertrophy induced by pressure overload. a Histological analyses of the hematoxylin and eosin (H&E) staining and the WGA staining of each group of mice at 8 weeks after AB or sham surgery (n = 5–6). b Statistical results for the cardiomyocyte cross-sectional area (CSA, n = 100 cells). c Statistical results for the ratios of heart weight (HW)/body weight (BW), lung weight (LW)/BW, and HW/tibia length (TL) in the indicated groups (n = 11–14).*P < 0.05 versus vehicle/Sham. # P < 0.05 versus vehicle/AB after AB
Isorhamnetin improved the impaired cardiac function after AB
To examine the protective effect of isorhamnetin on hypertrophy induced by pressure overload, we assessed the cardiac function by echocardiography. Although the LV dimensions and wall thickness were significantly increased in both the vehicle-treated and isorhamnetin-treated mice after AB, vehicle-treated mice exhibited deteriorated cardiac hypertrophy and dysfunction compared with isorhamnetin-treated mice, as measured by echocardiographic parameters, LVEDD, LVESD, and fractional shortening (FS) (Fig. 3a, b and Table 1 in supplementary material). Moreover, we measured the expression of hypertrophy markers induced by pressure overload with quantitative real-time PCR. The mRNA levels of ANP, BNP, and β-MHC were obviously increased in both isorhamnetin-treated mice and vehicle-treated mice after AB. However, these increased levels were more significant in vehicle-treated mice than that in the isorhamnetin-treated mice 8 weeks after AB (Fig. 3c). Collectively, these data suggest that isorhamnetin is responsible for the preservation of the cardiac function induced by pressure overload.

Isorhamnetin improved the impaired cardiac function after AB. a Representative echo images of each group of mice at 8 weeks after AB or sham surgery (n = 5–6). b Echocardiographic parameters of four groups of mice after 8 weeks of AB or sham operations (n = 6 mice per experimental group). c Real-time PCR analyses of the hypertrophy markers ANP, BNP, and β-MHC induced by AB or sham surgery in each group of mice (n = 4). *P < 0.05 versus vehicle/Sham. # P < 0.05 versus vehicle/AB after AB
Isorhamnetin attenuated fibrosis in pressure overload hearts
Pathological cardiac hypertrophy is accompanied by increased fibrosis, which is characterized by the overproduction of extracellular matrix proteins in the heart2. To further define the effect of isorhamnetin on maladaptive cardiac remodeling, we assessed the effect of isorhamnetin on cardiac fibrosis. The extent of fibrosis was detected by both picrosirius red staining and quantitative analysis. Our results showed that interstitial and perivascular fibrosis was dramatically increased in the vehicle-treated hearts subjected to chronic AB, which was remarkably limited in isorhamnetin-treated hearts (Fig. 4a, b). Finally, we measured the synthesis of collagen by analyzing the mRNA expression levels of fibrotic markers [eg, collagen I, collagen III, and connective tissue growth factor (CTGF)] (Fig. 4c), which also demonstrated a blunted fibrotic response in isorhamnetin-treated mice, compared with vehicle-treated mice. Together, our results suggested that isorhamnetin could alleviate myocardial fibrosis induced by pressure overload.

Isorhamnetin ameliorated fibrotic response induced by pressure overload. a Picrosirius red staining of left ventricular sections in the indicated groups after 8 weeks of AB (n = 6, scale bar = 50 μm). b Fibrotic areas were quantified using an image-analyzing system (n = 33–36 fields). c Real-time PCR analyses of the fibrotic markers [collagen I, collagen III, and connective tissue growth factor (CTGF)] in the indicated mice (n = 4). *P < 0.05 versus vehicle/Sham. # P < 0.05 versus vehicle/AB after AB
Isorhamnetin inhibits PI3K–AKT signaling pathway in response to hypertrophic stimuli
The above results suggest that isorhamnetin might attenuate pressure overload-induced cardiac remodeling. However, the molecular mechanism by which isorhamnetin regulates the hypertrophic response remains unknown. Given that the PI3K–AKT cascade signaling pathway has been proved to play an important part in the development of cardiac hypertrophy, we first examined whether isorhamnetin affected the AB-induced activation of PI3K–AKT signaling pathway. As expected, we observed that PI3K, AKT, glycogen synthase kinase3β (GSK3β), mTOR, p70 ribosomal S6 protein kinase (p70S6K), and eIF-4E were significantly phosphorylated in AB mice. However, isorhamnetin dramatically reduced the phosphorylation levels of Akt, GSK3b, mTOR, PI3K, P70S6K, and eIF-4E, compared with those of vehicle-treated hearts after AB (Fig. 5a, b). Although MAPK signaling plays an important role in the conformation and the development of cardiac hypertrophy, there is not much difference in the assessment of MAPK activation between the groups (data not shown). The above results suggest that the regulation of isorhamnetin on hypertrophy might be associated with PI3K–AKT rather than MAPK signaling pathway.

Isorhamnetin inhibited PI3K–AKT signaling pathway in response to hypertrophic stimuli. a, b The levels of total and phosphorylated AKT, GSK3β, mTOR, PI3K, P70S6K, and eIF-4E expression in heart tissues of mice in the indicated groups (n = 4). a Representative blots, b Quantitative results. *P < 0.05 versus vehicle/Sham. # P < 0.05 versus vehicle/AB after AB
Protective effect of isorhamnetin against cardiac hypertrophy is largely dependent on the regulation of PI3K–AKT signaling pathway
To further examine whether PI3K–AKT signaling has a causative role in isorhamnetin-protected cardiac hypertrophy, additional in vitro experiments were performed. We pretreated NRCMs with LY294002 (a PI3K inhibitor that prevents PI3K phosphorylation) for 1 h and then with Ang II for 48 h after being treated with isorhamnetin or vehicle for 24 h. As expected, cells treated with vehicle showed pronounced hypertrophy induced by Ang II as assessed by surface area measurements and the expression of hypertrophic hallmarks. Moreover, the hypertrophic response was strongly blunted in the LY294002-treated cells compared with cells treated with Ang II alone (Fig. 6a, b). However, LY294002 did not affect the decreased hypertrophic response in isorhamnetin-treated cells (Fig. 6c). Collectively, these figures suggest that the protective effect of isorhamnetin against pathological cardiac hypertrophy, at least partly, is by blocking the activation of PI3K–AKT signaling pathway.

Isorhamnetin-mediated cardiac hypertrophy is largely dependent on the regulation of PI3K–AKT signaling pathway. a Representative images of cardiomyocytes that were pretreated with LY294002 for 1 h and subsequently treated with 1 μM Ang II for 48 h after being treated with Isorhamnetin for 24 h. b Quantitation of the cell surface area (n = 50 cells). c Real-time PCR analysis of the hypertrophy markers β-MHC and ANP
Discussion
Heart failure has become one of the most important causes of illness and death in modern times. Although many studies have been carried out on the process of cardiac hypertrophy to heart failure, mechanisms that inhibit the process have not been well defined. Therefore, it is vital for us to identify the molecular mechanisms of cardiac hypertrophy and the functional clarification of the anti-hypertrophic targets. In recent decades, multiple signaling pathways mediating the hypertrophic development process have been identified, such as the MAPKs, calcineurin/NFAT, and the PI3K/AKT pathway. Effective blockading of these signaling pathways might provide promising approaches for inhibiting cardiac hypertrophy and heart failure. Thus, the current challenge will be to find promising pharmacological agents that selectively modulate the specific signaling pathways and thus to prevent pathological cardiac hypertrophy. Unfortunately, up to now, no effective drugs targeting the molecular changes involved in cardiac hypertrophy have been found. In our study, we used a model of cardiac remodeling following mechanical overload. In the present study, our data showed that isorhamnetin prevents myocardial hypertrophy and fibrosis in response to stress. Furthermore, we demonstrated that isorhamnetin ameliorates cardiac hypertrophy partially through the regulation of PI3K–AKT signaling transduction pathway. To our knowledge, our study results are the first to demonstrate that isorhamnetin could alleviate adverse cardiac remodeling and regulate hypertrophic signaling pathway due to chronic pressure overload.
The underlying mechanisms by which isorhamnetin regulates cardiac remodeling remain largely unknown. Although the hypertrophic response is just an accommodation response at first, sustained myocardium hypertrophy may cause ventricular arrhythmias, heart failure, and subsequent cardiovascular mortality, which is one of the most important causes for cardiovascular diseases death. It is generally accepted that MAPK cascade is a key pathway in the process of cardiac hypertrophy [26, 27]. MAPK signaling transduction contains a series of kinases, including p38, c-Jun N-terminal kinase 1/2 (JNK1/2), and ERK1/2. The activation of these kinases could directly regulate transcription factors, modulate cardiac gene expression, and lead to cardiac hypertrophy. A recent study reported that isorhamnetin could inhibit cell proliferation and induce apoptosis through MAPK signaling pathway in breast cancer [21]; however, the phosphorylation of p38, JNK1/2, and ERK1/2 that were significantly affected in isorhamnetin-treated mice could not be observed. These results demonstrate that the regulatory effect of isorhamnetin on MAPK cascades might be tissue/cell dependent.
We then examined the PI3K–AKT signaling pathway, the role of which in cardiac hypertrophy is well established. Chronic PI3K activation in the heart accentuated cardiac hypertrophy and myocardial dysfunction, whereas the hearts lacking PI3K showed blunted hypertrophic response to physiological stimuli [28]. The hypertrophic response following PI3K activation is apparently associated with PI3K downstream AKT [29], which is a serine/threonine kinase involved in the regulation of a variety of cellular functions including metabolism, glucose uptake, proliferation, and protein synthesis, all assigned towards a single goal of cell survival. Targeted overexpression of constitutively active AKT in the heart resulted in increased myocyte size and cardiac hypertrophy. Activated AKT subsequently stimulates cell protein synthetic machinery by inhibition of GSK3β, which could inhibit both normal size of the heart and pressure overload-induced hypertrophy [30]. mTOR is a large serine–threonine protein kinase, which is activated by phosphorylation of TSC2 by AKT. Activation of mTOR and its downstream targets results in increased cell size which is associated with cardiac hypertrophy [31]. Furthermore, mTOR is believed to act primarily through p70S6K- and eIF-4E-binding proteins, which are both important regulators of protein synthesis in cardiac hypertrophy [32, 33]. An important finding of the current study is that PI3K and AKT activation were almost entirely blunted in isorhamnetin-treated mice. Consistent with the activation of PI3K and AKT, the phosphorylation of GSK3β, mTOR, p70S6K, and eIF-4E after AB were significantly attenuated by isorhamnetin treatment.
To further examine whether PI3K–AKT signaling has a causative role in isorhamnetin-protected cardiac hypertrophy, we pretreated NRCMs with LY294002. As a PI3K inhibitor, though LY294002 has been found to inhibit other protein kinases, like CK2 (casein kinase 2) and Pim-1 [34, 35], LY294002 was commonly used as a PI3K inhibitor in scientific research and has helped to validate pathway inhibition [36, 37]. Interestingly, the results showed that isorhamnetin could not further blunt LY294002, which induced decreased hypertrophic response. These data indicate that the protective effect of isorhamnetin against pathological cardiac hypertrophy may be dependent, at least partly, on the blockade of PI3K–AKT signaling pathway. However, further investigations are needed to determine the molecular mechanism by which isorhamnetin inhibits PI3K–AKT pathway.
Fibrosis is another classical feature of pathological cardiac hypertrophy, which is characterized by the deposition of the extracellular matrix. Previous study reported that isorhamnetin could attenuate CCl4-induced liver fibrosis by inhibiting TGF-β/Smad signaling. To further investigate the mechanism by which isorhamnetin ameliorates cardiac hypertrophy, we examined the effect of isorhamnetin on cardiac fibrosis. In our study, we found that isorhamnetin could retard the development of fibrosis in pressure overload-induced hearts by detecting LV collagen volume. In addition, our results showed that the mRNA expression levels of known mediators of fibrosis markers were almost normalized by isorhamnetin treatment. This study is the first to report that isorhamnetin could ameliorate cardiac fibrosis and reduce the expression of fibrotic mediators induced by pressure overload.
In conclusion, the present study evidences that isorhamnetin mitigates cardiac hypertrophy and fibrosis induced by pressure overload through the inhibition of PI3K–AKT signaling pathway, which indicates there is a great possibility that isorhamnetin can be applied clinically for the treatment of cardiac hypertrophy and heart failure. However, future study and clinical trials are needed to confirm the new potential clinical prospect of isorhamnetin.
Change history
06 November 2021
A Correction to this paper has been published: https://doi.org/10.1007/s11010-021-04288-x
References
Chen K, Gao L, Liu Y, Zhang Y, Jiang DS, Wei X et al (2013) Vinexin-β protects against cardiac hypertrophy by blocking the Akt-dependent signalling pathway. Basic Res Cardiol 108:338
Tamargo J, Lopez-Sendon J (2011) Novel therapeutic targets for the treatment of heart failure. Nat Rev Drug Discov 10:536–555
Koitabashi N, Kass DA (2012) Reverse remodeling in heart failure–mechanisms and therapeutic opportunities. Nat Rev Cardiol 9:147–157
Heineke J, Molkentin JD (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7:589–600
Lorell BH, Carabello BA (2000) Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 102:470–479
Dorn GW 2nd, Force T (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115:527–537
Gao L, Huang K, Jiang DS, Liu X, Huang D, Li H et al (2015) Novel role for caspase-activated DNase in the regulation of pathological cardiac hypertrophy. Hypertension 65:871–881
Tang Q, Cai J, Shen D, Bian Z, Yan L, Wang YX et al (2009) Lysosomal cysteine peptidase cathepsin L protects against cardiac hypertrophy through blocking AKT/GSK3beta signaling. J Mol Med 87:249–260
Yan L, Huang H, Tang QZ, Zhu LH, Wang L, Liu C et al (2010) Breviscapine protects against cardiac hypertrophy through blocking PKC-alpha-dependent signaling. J Cell Biochem 109:1158–1171
Ai W, Zhang Y, Tang QZ, Yan L, Bian ZY, Liu C et al (2010) Silibinin attenuates cardiac hypertrophy and fibrosis through blocking EGFR-dependent signaling. J Cell Biochem 110:1111–1122
Park JC, Young HS, Yu YB, Lee JH (1995) Isorhamnetin sulphate from the leaves and stems of Oenanthe javanica in Korea. Planta Med 61:377–378
Yang JH, Kim SC, Shin BY, Jin SH, Jo MJ, Jegal KH et al (2013) O-Methylated flavonol isorhamnetin prevents acute inflammation through blocking of NF-kappaB activation. Food Chem Toxicol 59:362–372
Teng BS, Lu YH, Wang ZT, Tao XY, Wei DZ (2006) In vitro anti-tumor activity of isorhamnetin isolated from Hippophae rhamnoides L. against BEL-7402 cells. Pharmacol Res 54:186–194
Yang JH, Shin BY, Han JY, Kim MG, Wi JE, Kim YW et al (2014) Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol Appl Pharmacol 274:293–301
Zhang N, Pei F, Wei H, Zhang T, Yang C, Ma G et al (2011) Isorhamnetin protects rat ventricular myocytes from ischemia and reperfusion injury. Exp Toxicol Pathol 63:33–38
Sun B, Sun GB, Xiao J, Chen RC, Wang X, Wu Y et al (2012) Isorhamnetin inhibits H(2)O(2)-induced activation of the intrinsic apoptotic pathway in H9c2 cardiomyocytes through scavenging reactive oxygen species and ERK inactivation. J Cell Biochem 113:473–485
Sun J, Sun G, Meng X, Wang H, Luo Y, Qin M et al (2013) Isorhamnetin protects against doxorubicin-induced cardiotoxicity in vivo and in vitro. PLoS ONE 8:e64526
Bao M, Lou Y (2006) Isorhamnetin prevent endothelial cell injuries from oxidized LDL via activation of p38MAPK. Eur J Pharmacol 547:22–30
Qiu S, Sun G, Zhang Y, Li X, Wang R (2016) Involvement of the NF-kappaB signaling pathway in the renoprotective effects of isorhamnetin in a type 2 diabetic rat model. Biomed Rep 4:628–634
Kim JE, Lee DE, Lee KW, Son JE, Seo SK, Li J et al (2011) Isorhamnetin suppresses skin cancer through direct inhibition of MEK1 and PI3-K. Cancer Prev Res 4:582–591
Hu S, Huang L, Meng L, Sun H, Zhang W, Xu Y (2015) Isorhamnetin inhibits cell proliferation and induces apoptosis in breast cancer via Akt and mitogen-activated protein kinase kinase signaling pathways. Mol Med Rep 12:6745–6751
Li C, Yang X, Chen C, Cai S, Hu J (2014) Isorhamnetin suppresses colon cancer cell growth through the PI3KAktmTOR pathway. Mol Med Rep 9:935–940
Luo Y, Sun G, Dong X, Wang M, Qin M, Yu Y, et al (2015) Isorhamnetin attenuates atherosclerosis by inhibiting macrophage apoptosis via pi3k/akt activation and HO-1 induction. PloS ONE 10: e0120259
Jiang DS, Luo YX, Zhang R, Zhang XD, Chen HZ, Zhang Y et al (2014) Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin. Hypertension 63:119–127
Jiang DS, Bian ZY, Zhang Y, Zhang SM, Liu Y, Zhang R et al (2013) Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension 61:1193–1202
Konhilas JP, Boucek DM, Horn TR, Johnson GL, Leinwand LA (2010) The role of MEKK1 in hypertrophic cardiomyopathy. Int Heart J 51:277–284
Zhang Y, Zhang XF, Gao L, Liu Y, Jiang DS, Chen K et al (2014) Growth/differentiation factor 1 alleviates pressure overload-induced cardiac hypertrophy and dysfunction. Biochim Biophys Acta 1842:232–244
Aoyagi T, Matsui T (2011) Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr Pharm Des 17:1818–1824
DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ (2006) Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem 281:32841–32851
Sugden PH, Fuller SJ, Weiss SC, Clerk A (2008) Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br J Pharmacol 153:S137–S153
Clemente CF, Xavier-Neto J, Dalla Costa AP, Consonni SR, Antunes JE, Rocco SA et al (2012) Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J Mol Cell Cardiol 52:493–501
Yang Z, Ming XF (2012) mTOR signalling: the molecular interface connecting metabolic stress, aging and cardiovascular diseases. Obes Rev 13(Suppl 2):58–68
Kenessey A, Ojamaa K (2006) Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J Biol Chem 281:20666–20672
Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105
Jacobs MD, Black J, Futer O, Swenson L, Hare B, Fleming M et al (2005) Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J Biol Chem 280:13728–13734
Chen L, Han L, Shi Z, Zhang K, Liu Y, Zheng Y, et al (2012) LY294002 enhances cytotoxicity of temozolomide in glioma by down-regulation of the PI3K/Akt pathway. Mol Med Rep 5:575–579
Liu P, Xu B, Li J, Lu H (2008) LY294002 inhibits leukemia cell invasion and migration through early growth response gene 1 induction independent of phosphatidylinositol 3-kinase-Akt pathway. Biochem Biophys Res Commun 377:187–190
Acknowledgements
This work was supported by a Grant from the The First Affiliated Hospital of Zhengzhou University.
Author contributions
Lu Gao, Rui Yao, and Yanzhou Zhang conceived and designed the research; Lu Gao, Yuzhou Liu, and Zheng Wang performed experiments; Lu Gao, Zhen Huang, and Zhen Huang analyzed data; Lu Gao and Binbin Du interpreted the results of experiments; Dianhong Zhang, and Leiming Wu prepared figures; Lu Gao drafted the manuscript; Rui Yao and Yanzhou Zhang edited and revised the manuscript; Lu Gao, Rui Yao, Yuzhou Liu, Zheng Wang, Zhen Huang, Binbin Du, Dianhong Zhang, Leiming Wu, Lili Xiao, and Yanzhou Zhang approved the final version of manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Lu Gao, Rui Yao, and Yuzhou Liu are the co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gao, L., Yao, R., Liu, Y. et al. Isorhamnetin protects against cardiac hypertrophy through blocking PI3K–AKT pathway. Mol Cell Biochem 429, 167–177 (2017). https://doi.org/10.1007/s11010-017-2944-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-2944-x